

Abstract
The food-borne pathogen Campylobacter jejuni possesses a single-domain globin (Cgb) whose role in detoxifying nitric oxide has been unequivocally demonstrated through genetic and molecular approaches. The x-ray structure of cyanide-bound Cgb has been solved to a resolution of 1.35 Å. The overall fold is a classic three-on-three α-helical globin fold, similar to that of myoglobin and Vgb from Vitreoscilla stercoraria. However, the D region (defined according to the standard globin fold nomenclature) of Cgb adopts a highly ordered α-helical conformation unlike any previously characterized members of this globin family, and the GlnE7 residue has an unexpected role in modulating the interaction between the ligand and the TyrB10 residue. The proximal hydrogen bonding network in Cgb demonstrates that the heme cofactor is ligated by an imidazolate, a characteristic of peroxidase-like proteins. Mutation of either proximal hydrogen-bonding residue (GluH23 or TyrG5) results in the loss of the high frequency νFe-His stretching mode (251 cm−1), indicating that both residues are important for maintaining the anionic character of the proximal histidine ligand. Cyanide binding kinetics for these proximal mutants demonstrate for the first time that proximal hydrogen bonding in globins can modulate ligand binding kinetics at the distal site. A low redox midpoint for the ferrous/ferric couple (−134 mV versus normal hydrogen electrode at pH 7) is consistent with the peroxidase-like character of the Cgb active site. These data provide a new insight into the mechanism via which Campylobacter may survive host-derived nitrosative stress.
Keywords: Heme, Nitric Oxide, Oxygen Binding, Peroxidase, Protein Structure, Raman Spectroscopy, Globin
Introduction
Globins are an ancient and diverse superfamily of proteins. The first report of a microbial globin was in yeast over half a century ago (1), but only in the past 15 years have molecular studies of their structure, function, and regulation of their biosynthesis been pursued. This has resulted in a dramatic increase in our understanding of the roles of microbial globins in bacterial respiration and physiology, pathogenesis, and biotechnological opportunities, fuelled by the recognition that a major role of certain microbial globins is in protection from nitric oxide (NO)2 (2, 3).
At least three classes of bacterial globin are recognized. Members of the best understood class, the flavohemoglobins, are distinguished by the presence of an N-terminal globin domain with an additional C-terminal domain with binding sites for FAD and NAD(P)H (4–7). Widely distributed in bacteria, these proteins undoubtedly confer protection from NO and nitrosative stresses by direct consumption of NO (8). The truncated globins are the most recently discovered and appear widely distributed in bacteria, microbial eukaryotes, and plants (9). They are characterized by a polypeptide 20–40 residues shorter than myoglobin, folded into a two-over-two, more compact, helical structure (10) while retaining the essential features of the globin superfamily. Roles in oxygen and NO metabolism have been proposed (11, 12). Truncated globins may be further divided into three subgroups on the basis of phylogenetic analyses (9).
The third class of bacterial globin is the single-domain globins. These comprise a three-on-three α-helical fold similar to myoglobin. This is typified by the globin of Vitreoscilla (named Vgb (V. stercoraria single-domain globin), VtHb, or Vhb), an obligate aerobic bacterium that grows in low oxygen environments, such as stagnant ponds and rotten vegetables. The ferric homodimeric Vgb was the first bacterial hemoglobin to be crystallized (13, 14), and the three-dimensional structure conforms to the well defined globin fold. However, the region following the C-helix is disordered, residues E7–E10 do not adopt the usual α-helical conformation, and GlnE7 is located out of the heme pocket.3 Instead, residues PheCD1, ProE8, and LeuE11 fill the distal site of the heme pocket. Binding of azide causes conformational rearrangement at the heme pocket distal site (i.e. residue ProE8 moves out of the heme pocket, and there are shifts of GlnE7 to AlaE10 by 2–4 Å). The three-dimensional structures of the thiocyanate and imidazole derivatives of recombinant ferric Vgb have also been determined (15). In agreement with the crystallographic studies, site-directed mutagenesis shows that GlnE7 does not appear to stabilize the heme iron-bound dioxygen through hydrogen bonding (14). The quaternary structure of Vgb and of its azide, thiocyanate, and imidazole derivatives is unique in that the protein is dimeric in the crystal lattice. The association of the two subunits appears to be weak; they interact through van der Waals contacts at a very small molecular interface area of only 434 Å2. It has been suggested that the disordered CD region is a potential site of interaction with the FAD/NADH reductase partner (14), although this has not been demonstrated, and cognate partner reductases for this class of globin remain elusive.
Considerable interest has been directed at Vgb because its physiological role has long been thought to be similar to that of myoglobin in transporting oxygen to terminal respiratory oxidases. In agreement with this proposal, Vgb is up-regulated under microaerobic conditions (16) and shows promise for biotechnological application by enhancing product formation under oxygen limitation in heterologous hosts (17). Interestingly, a chimeric protein comprising the Vitreoscilla hemoglobin and the flavoreductase domain from the Ralstonia eutropha flavohemoglobin Fhp was found to be able to relieve nitrosative stress in Escherichia coli. It was proposed that when Vgb is in the dimeric form, it participates in oxygen transport; when it is in a monomeric form, it could associate with a reductase and relieve nitrosative stress (18).
Vitreoscilla is not amenable to genetic analysis, frustrating understanding of the role of Vgb in vivo. The globin most similar to Vgb is from an obligately anaerobic pathogen Clostridium perfringens (19), and the next closest relative is Cgb from the microaerophilic food-borne pathogen Campylobacter jejuni, a bacterium in which genetic analysis of gene and protein function is possible. C. jejuni is exposed to NO and other nitrosating species during host infection (20). The single-domain globin, Cgb, and a truncated globin, Ctb, are both up-regulated by the transcription factor NssR in response to nitrosative stress (21). Cgb has been shown to detoxify NO (2), whereas Ctb is thought to have a role in oxygen metabolism (22) and has been shown to possess a peroxidase-like heme-binding cleft (11). It has been suggested that inhibition of respiration by NO increases the intracellular oxygen tension and that Ctb may deliver oxygen to Cgb for the dioxygenase reaction with NO to produce nitrate (23). In marked contrast to Vitreoscilla Vgb, there is no evidence to date that Cgb functions in oxygen delivery. It has previously been proposed that Cgb can provide an electronic push from the proximal ligand and an electronic pull from the distal binding pocket, creating a favorable environment for the isomerization of a putative peroxynitrite intermediate in the NO dioxygenase reaction (24). Unlike many bacterial hemoglobins whose structures have been probed intensively, Cgb has a clear physiological function, defined both by gene deletion and gene regulation studies (2, 21). Therefore, a structure study of Cgb is timely. In this paper, we report the crystal structure of Cgb, functional analysis of the hydrogen bonding network in the proximal pocket, and a preliminary characterization of the redox properties of the heme cofactor.
EXPERIMENTAL PROCEDURES
Purification of Cgb
For the crystallization work, Cgb was purified from cells disrupted via sonication, resulting in cyanide-bound ferric Cgb (24). For the solution studies, Cgb was purified from cells disrupted via a French pressure cell, resulting in oxyferrous protein (24). The Y94F (G5) and E134A (H23) mutants were prepared using the Stratagene QuikChange site-directed mutagenesis kit. The absorption spectra of the Cgb crystal and Cgb solutions are shown in supplemental Fig. S1A. Primers were designed with the codon for the mutated amino acid in the center: Y94F (G5) forward primer (5′-GTTAATTTAGGAGTTAAAGAAGAACATTTTCCAATAGTTGGAGCTTGCCTTTTAAAG-3′) and reverse primer (5′-CTTTAAAAGGCAAGCTCCAACTATTGGAAAATGTTCTTCTTTAACTCCTAAATTAAC-3′); E134A (H23) forward primer (5′- GCTAAATTTTATATCGATATCGCAAAAAAGCTCTATGATAAATAA-3′) and reverse primer (5′- TTATTTATCATAGAGCTTTTTTGCGATATCGATATAAAATTTAGC-3′). The mutagenesis was carried out according to the manufacturer's instructions, and mutations were confirmed with sequencing. The mutant constructs were transformed into E. coli BL21 pLysS cells, and protein expression and purification were carried out in the same way as for wild type Cgb (24).
Crystallization
Crystals of Cgb were grown at 17 °C using vapor diffusion techniques against a reservoir solution containing ammonium sulfate (Sigma) and buffer. Hanging drops contained 5 μl of 15 mg/ml protein solution and 5 μl of reservoir solution. The best crystals were prepared using a mixture of 1.8 m ammonium sulfate and Tris buffer (0.1 m, pH 8.0) as precipitant, growing to a maximal size of 0.4 × 0.1 × 0.1 mm within 2 weeks. These crystals belong to the monoclinic space group P21 with cell parameters a = 39.55 Å, b = 37.98 Å, c = 43.76 Å, β = 102.68°. The asymmetric unit contains one protein molecule, corresponding to a solvent content of 39%.
X-ray Data Collection
A data set at 1.35 Å resolution was collected at 100 K on Station PX 14.2 at the Daresbury Laboratory (UK) using an ADSC Quantum4 CCD detector. A solution of 30% glucose, 2.2 m ammonium sulfate, and 0.1 m Tris-HCl, pH 8.0, was used as cryoprotectant. Data scaling and reduction were performed by the programs DENZO and SCALEPACK (25). The details of data collection statistics are presented in supplemental Table S1. The Matthews number, Vm (26), was calculated using the program MATTHEWS_COEF (CCP4 suite (27)).
Solving the Cgb Structure
Subsequent calculations were performed using the CCP4 suite of crystallographic programs (27). The structure was solved by molecular replacement using the program MOLREP from CCP4 (27), the initial Cgb model being based on the model of homodimeric bacterial hemoglobin from Vitreoscilla stercoraria (Protein Data Bank code 1VHB (14)). The initial model was fitted with the known Cgb sequence by the program O (28). The asymmetric unit contains a single chain of 139 protein residues, and all main chain atoms were unambiguously defined.
Refinement of the Structure and Model Validation
Five rounds of single-subunit model building and refinement were performed to obtain a complete model of 139 residues. This model of Cgb was refined by the program REFMAC (29) with isotropic atomic B values at resolution limits 19.3–1.35 Å. 6% of the reflections were randomly assigned to calculate Rfree (30). When the R-factor dropped to 25.3% (Rfree was 27.2%), 109 water molecules were added using the program ARP (31) coupled with REFMAC. Cyanide ligand and multiple side chain conformations were modeled at this stage during inspection of waters. Anisotropic B-factor refinement by REFMAC (32) was performed in the final stages of refinement with hydrogen atoms included in standard positions. Three extra manual corrections of the model were performed during the process of complete model refinement. The structure was refined to an R-factor of 18.5% (Rfree = 21.4%) for all data in the resolution range 20.9–1.35 Å excluding 6% randomly distributed reflections taken to calculate Rfree = 21.4%. The validity of the model was assessed using the program PROCHECK (33). The final model, as defined by PROCHECK, has 95.2% of the residues in the most favored regions of the Ramachandran plot and 4.8% in the additionally allowed regions (33). Some characteristics of the refined models are presented in supplemental Table S1.
Resonance Raman Spectroscopy
The resonance Raman measurements were carried out using previously described instrumentation (34). Briefly, the sample was excited with a krypton ion laser (Spectra Physics) set to 413 nm. The laser was focused on a quartz cuvette that was constantly rotating at 6000 rpm to avoid photodamage of the sample. The scattered light was detected with a liquid nitrogen charge cooled coupled device (Princeton Instruments), and a halographic notch filter (Kaiser, Ann Arbor, MI) was used to eliminate Rayleigh scattering. The Raman spectra were calibrated with indene (Sigma) for the 200–1700 cm−1 spectral region. The deoxy-Cgb sample was prepared by first flushing the sample with argon, followed by reduction with a 5-fold molar excess of sodium dithionite.
Stopped-flow Measurements
The reactions of Cgb with cyanide were performed in 100 mm phosphate buffer (pH 7.0) with a π*180 stopped-flow instrument (Applied Photophysics, Leatherhead, UK). The final concentration of Cgb was 1.5 μm. The reaction kinetics were monitored using a photodiode array detector, and these data were analyzed with the ProK software from Applied Photophysics. Potassium cyanide was obtained from Sigma.
Potentiometry
Titrations were performed inside a Belle Technology anaerobic glove box ([O2] < 10 ppm) as described previously (35). The titration solution contained 1.5 ml of 15 μm Cgb and a 5 μm concentration of each mediator (supplemental Table S2) buffered with 50 mm Bistris propane, 50 mm 2-amino-2-methyl-1-propanol. Reductive titrations were performed with titanium(III) citrate, and oxidative titrations were performed with potassium peroxodisulfate. The potentials were measured using a combination platinum wire-Ag/AgCl electrode calibrated with quinhydrone (87 mV versus Ag/AgCl at pH 7). Absorption spectra were collected using an Ocean Optics USB2000+ fiber optic spectrometer and DT-MINI-2-GS deuterium/tungsten light source, both located outside the glove box and connected by fiber optic cables to a CUV-UV cell stand within the glove box. The solutions were stirred continuously with a Rank model 300 magnetic stirrer, and the potentials were measured with a Hanna Instruments model 8417 meter. Absorbance (A) at 430 nm was plotted against potential (E), and the data were fitted to Equation 1,
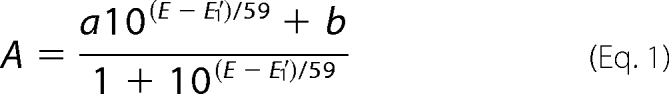 |
where a and b are limiting absorbance values contributed by the heme cofactor in the ferric and ferrous states, respectively, and E′1 is the midpoint potential for the ferrous/ferric couple.
RESULTS
Globin Fold of Cgb
Cgb has a classic three-on-three α-helical globin fold (Fig. 1A), similar to that of myoglobin, Vgb, and the globin domain of Hmp (E. coli flavohemoglobin) (Fig. 1, B–D). The C and D regions of Cgb adopt 310- and α-helical conformations, respectively. This is similar to sperm whale myoglobin (swMb) (36), although the D-helix in Cgb is closer to the heme-binding cleft than that of swMb (Fig. 1D). The N-terminal region of the E-helix of Vgb is distorted (14), and the D region of E. coli flavohemoglobin Hmp (6) lacks the α-helical structure of Cgb and myoglobin (Fig. 1). In the case of Vgb, part of the D region is disordered and is missing in the crystal structure (Fig. 1B), whereas Hmp was crystallized with the D region as a loop projecting away from the heme-binding cleft (Fig. 1C). This region is adjacent to the adenine portion of the FAD cofactor in Hmp, and the CD region in Vgb is proposed to interact with an NADH-dependent FAD-containing redox partner (14).
FIGURE 1.

Backbone topology of Cgb. A, Cα chain tracing of Cgb with heme cofactor (black). Helices/regions are labeled according to conventional globin nomenclature. B–D, overlays of the Cgb backbone (red) with Vgb from Vitreoscilla stercoraria (Protein Data Bank entry 1VHB (14)) (B), N-terminal domain of Hmp from E. coli (Protein Data Bank entry 1GVH (6)) (C), and sperm whale myoglobin (Protein Data Bank entry 2JHO (36)) (D).
Monomeric Structure
To understand why Cgb exists as a monomer, the Vgb dimer was overlaid with two molecules of Cgb (Fig. 2A). In Vgb, the intersubunit contact region is a loose four-helix bundle based on the juxtaposition of helices F and H from each subunit (Fig. 2B) and is held together by van der Waals contacts in a hydrophobic zipper (14). The subunits are also held together by two water molecules acting as hydrogen-bonding bridges between Ala-73 and Asp-139 and between Pro-72 and Asp-139. Fig. 2A demonstrates that several large charged residues occupy this region in Cgb, and the resulting steric clashes are the likely reason that Cgb does not adopt the homodimeric structure of Vgb.
FIGURE 2.

Charged residues near F and H helices prevent dimerization in Cgb. A, a Cgb dimer was modeled by overlaying the Vgb dimer with two Cgb monomers. Only the Cgb subunits are shown for clarity. Subunit A is shown in magenta, and subunit B is in yellow. B, the intersubunit hydrophobic zipper in Vgb. Subunit A is shown in green, and subunit B is in cyan.
Distal Site and D-helix
The distal site in Cgb resembles that of Hmp and Vgb, with the Tyr and Gln in positions B10 and E7, respectively (supplemental Fig. S2). The TyrB10 hydroxyl is 2.44 Å from the bound CN− and stabilizes this ligand via a hydrogen bond. However, the GlnE7 residue adopts an unexpected conformation, where the amine moiety is 3.53 Å from the CN− ligand and 2.95 Å from the TyrB10 hydroxyl group (Fig. 3A). This side chain projects into the solvent in the ligand-free ferric and azide-bound structures of Vgb (14) and points toward the heme propionate in Hmp (Fig. 4). The proximity of the GlnE7 to the TyrB10 in Cgb (Figs. 3 and 4) is consistent with a novel role in modulating ligand coordination by the B10 residue.
FIGURE 3.

Stereoviews of the distal pocket and D-helix of Cgb. A, the distances between residues are as follows: TyrB10–CN−, 2.52 Å; GlnE7–CN−, 3.53 Å; GlnE7–TyrB10, 2.95 Å. B, the distances are as follows: Lys-46(N)–Gln-52(O), 2.90 Å; Lys-46–H2O70, 2.84 Å; Lys-46–H2O68, 3.04 Å; Asn-43(ND2)–Hem(O2D), 2.99 Å; Asn-43(N)–Hem(O1D), 2.82 Å; Hem(OD2)–H2O68, 2.58 Å. Hydrogen bonding interactions inferred from the structure are indicated by dotted lines.
FIGURE 4.

The active site structure of Cgb (A), Vgb (Protein Data Bank entry 1VHB) (B), azide-bound Vgb (Protein Data Bank entry 2VHB) (C), and Hmp (Protein Data Bank entry 1GVH) (D). The B, E, G, and H helices are colored green, yellow, orange, and salmon, respectively. Bound cyanide and azide are labeled CN and Az, respectively, and hydrogen bonding interactions inferred from the structures are indicated by dotted lines.
In Cgb, GlnE7 is also stabilized by the D-helix, where the main chain amine and side chain oxygen are hydrogen-bonded to the Lys-46 residue (Fig. 3B). The D-helix is also stabilized by a hydrogen bonding network involving two water molecules and Asn-43, where both the main chain and side chain amine groups are hydrogen-bonded to the adjacent carboxylate group of the heme propionate (Fig. 3B). The electron density of the D-helix is well defined (supplemental Fig. S3), except for the side chain of Glu-45, which projects into the solvent and is disordered after the Cβ atom.
Proximal Site
The imidazole ring of HisF8 provides the proximal ligand to the heme iron (2.09 Å) and is maintained in an azimuthal orientation with respect to the pyrrole atoms of the heme cofactor. The conformation of HisF8 is similar to that of Vgb and Hmp (6, 14), where the imidazolates are stabilized by hydrogen bonding of HisF8 ND1 to two atoms: TyrG5 OH and GluH23 OE2 (Fig. 4). Both TyrG5 and GluH23 residues are invariant in bacterial globins and are connected in Cgb by a hydrogen bond between atoms TyrG5 OH and GluH23 OE1 (2.62 Å; Fig. 5). Like that of Hmp and Vgb, the HisF8 imidazole ring forms an angle of ∼5° with respect to the heme pyrrole NB-ND direction, which is rotated ∼90° relative to commonly adopted conformations of vertebrate and invertebrate globin axial histidines. The G-helix and the FG region provide the heme-contacting residues: ValFG4, TyrG5, and ValG8. TrpH8 is the only tryptophan residue in Cgb, is conserved among Vgb and Hmp, and is hydrogen-bonded (via the NE1 atom) to the SG atom of CysA11 (3.39 Å; Fig. 5). The A11 residue is Thr-13 in Vgb and is hydrogen-bonded to TrpH8 (14). Additionally, TyrH27 OH of Cgb is hydrogen-bonded to the peptide oxygen atom of ValFG4 (2.64 Å; Fig. 5), and TyrH12 OH is hydrogen-bonded to the peptide oxygen atom of TyrG5 (2.75 Å; Fig. 5). Elimination of the hydrogen bond between the TyrH12 hydroxyl and the TyrG5 peptide oxygen had little effect in Vgb (19).
FIGURE 5.

Stereoview of the proximal pocket of Cgb. The distances between residues are as follows: TyrG5–HisF8, 3.20 Å; GluH23–HisF8, 2.70 Å; GluH23–TyrG5, 2.62 Å; TyrH12–TyrG5, 2.75 Å; TyrH27–ValFG4, 2.64 Å; TrpH8–CysA11, 3.39 Å. Hydrogen bonding interactions inferred from the structure are indicated by dotted lines.
Perturbation of the Proximal Hydrogen Bonding Network
E134A (H23) and Y94F (G5) single mutations were introduced into Cgb, and the absorption spectra indicate that these proteins were purified mainly in the oxyferrous state (supplemental Fig. S1B). The resonance Raman spectra demonstrate that loss of either proximal hydrogen-bonding residue results in a shift of the νFe-His stretching mode from 251 to 225 cm−1 (Fig. 6A). This indicates that the contribution of hydrogen bonds from both GluH23 and TyrG5 are required to maintain the anionic character of the axial histidine. The ν3 and ν4 modes of the wild type spectrum (1470 and 1353 cm−1, respectively; Fig. 6B) are typical of a five-coordinate high spin deoxy species. The E134A (H23) spectrum shows a similar five-coordinate high spin deoxy spectrum, indicating that loss of this hydrogen-bonding residue does not alter the coordination or spin state of the heme cofactor. However, mutation of TyrG5 results in the appearance of the new ν3 and ν2 modes at 1493 and 1582 cm−1, respectively, indicating partial conversion of the five-coordinate high spin protein to a six-coordinate low spin species. It accounts for the relatively low intensity of the νFe-His mode, which is observable only in the five-coordinated ferrous heme. The presence of a small absorption peak at 560 nm (supplemental Fig. S1B) is consistent with a partial cytochrome-like six-coordinate low spin heme. To determine if these proximal residues modulate ligand binding in the distal pocket, CN− binding kinetics were performed on wild type, E134A (H23), and Y94F (G5) Cgb (Fig. 7). These data demonstrate that kon for wild type Cgb is 431 m−1 s−1, and the E134A (H23) mutation results in a 2-fold increase in kon to 824 m−1 s−1. The Y94F (G5) mutation results in a slight decrease in kon (316 m−1 s−1).
FIGURE 6.

Resonance Raman spectra of deoxy derivatives of Cgb proximal mutants in the low (A) and high (B) frequency regions at pH 7.4. Wild type, E134A (H23), and Y94F (G5) proteins were used. Optical absorption spectra are presented in supplemental Fig. S1.
FIGURE 7.

CN− binding kinetics for ferric derivatives of Cgb proximal mutants. Shown are the observed rate constants of the wild type Cgb (black), E134A (H23) (light gray), and Y94F (G5) (dark gray) as a function of cyanide concentration. The observed rate constants were measured by mixing 1.5 μm protein with various concentrations of KCN in a stopped-flow instrument. Inset, kinetic traces obtained by mixing 3.3 mm CN with the wild type (black), E134A (H23) (light gray), and Y94F (G5) (dark gray) protein.
Redox Properties of the Heme Cofactor
The present structural analysis demonstrates a hydrogen bonding network involving the proximal histidine residue. Such interactions are likely to impose a negative charge on this residue. For example, horseradish peroxidase has a redox midpoint of −250 mV (37) as a result of a strong hydrogen bond to the proximal histidine (38). The redox midpoint for the ferrous/ferric couple in Cgb was measured anaerobically and was found to be −134 ± 3 mV at pH 7.0 (Fig. 8). The spectra of the five-coordinate ferric and ferrous species (Fig. 8A) agree with previous studies (24), and the reductive and oxidative titrations overlay, confirming the reversibility of the system. Decreasing the pH resulted in a concomitant small rise in the midpoint potential: pH 8.0, −140 ± 5 mV; pH 7.0, −134 ± 3 mV; pH 6.0, −110 ± 4 mV (Fig. 8B). We attribute this to the protonation of GluH23 or TyrG5, because the loss of this hydrogen bond would confer a more neutral axial histidine. The change in redox potential with pH over this range is too small to be indicative of a proton-coupled electron transfer on the heme cofactor.
FIGURE 8.

Potentiometric titration of Cgb. A, spectrophotometric changes of the unliganded heme cofactor during reductive and oxidative titrations at pH 7.0. B, the redox midpoints for the ferrous/ferric transition at pH 6.0 (black), pH 7.0 (light gray), and pH 8.0 (dark gray) are −110 ± 4 mV, −134 ± 3 mV, and −140 ± 5 mV, respectively. The data sets are offset for clarity, and both the reductive and oxidative data for each titration are fitted to Equation 1.
DISCUSSION
The current work reports the first structure of a monomeric bacterial single-domain globin. Significantly, this protein, Cgb, is one of a relatively small number of microbial globins whose cellular function has been demonstrated genetically (2), biochemically (24), and through expression analyses (23). Although the general fold is similar to that of Vgb, Hmp, and swMb, there are a number of notable differences. Of particular importance is the D-helix; this is the predicted site of redox partner interaction and is not resolved in previous structures of Vgb (14). This region in Cgb is stabilized via hydrogen bonding interactions with GlnE7 and the heme carboxylate adjacent to Asn-43 (Fig. 3B). Structure predictions suggest that this loop in Vgb projects away from the active site (39) in a manner similar to that of Hmp. However, substitution of Glu-49 for Gln resulted in a predicted structure similar to that of Cgb, where an α-helical structure is formed in the middle of the D-loop, albeit sterically crowding the heme cofactor (39). The corresponding residues in Cgb and Hmp are Ile-48 and Arg-49, respectively, which suggests that the presence of a charged residue at this position perturbs helix formation in bacterial globin D regions. The E49Q mutation was shown to diminish the binding affinity of Vgb for the flavin domain of 2,4-dinitrotoluene dioxygenase from Burkholderia, indicating that the lack of secondary structure in the D-loop may be an adaptation of bacterial globins to enhance interaction with flavin-containing oxidoreductases (39). Indeed, this region in Hmp interacts with the adenine moiety of the FAD cofactor in the reductase domain (Fig. 1). Hence, the α-helical D region in Cgb implies that the likely interaction with a redox partner will differ from that of Vgb. A putative methemoglobin reductase has been reported for Vgb (40) but not for Cgb.
Another novel aspect of this work is that Cgb purifies and crystallizes as a monomer. The overlay of Cgb monomers onto the Vgb dimer clearly demonstrates the presence of large charged residues, particularly Asp-71 and Arg-74, likely to prevent Cgb from self-association at this interface (Fig. 2A). In Vgb, these residues are Pro-72 and Leu-75 (Fig. 2B), which permit the formation of a loose four-helix bundle held together by van der Waals contacts in a hydrophobic zipper (14). The formation of a dimer might be critical for oxygen binding via cooperative subunit interactions in Vgb (14) but not for nitric oxide detoxification by Cgb.
Recent spectroscopic studies on Cgb report that ligand-bound Cgb can exist in an open or a closed conformation, where either the diatomic ligand is stabilized by a hydrogen bond from the Tyr-28 (B10)/Gln-52 (E7) pair (closed) or the ligand remains bound without a hydrogen bond (open) (24). The current cyanide-bound structure displays a closed conformation where the TyrB10-CN− hydrogen bond is stabilized by a second hydrogen bond between the GlnE7 and TyrB10 residues (Fig. 3). Mutation of the conserved GlnE7 in Vgb to histidine resulted in a slight increase in oxygen affinity (41), which the authors attribute to a direct role for E7 in ligand stabilization. However, structures of Vgb or Hmp display GlnE7 projecting away from the bound ligand (Fig. 4), and there is no evidence to suggest that the native GlnE7 performs such a role in Vgb or Hmp, whereas the current structure and previous spectroscopic evidence (24) demonstrate a direct role in ligand binding for GlnE7 of Cgb. In this respect, Cgb is more similar to myoglobin, where the E7 residue (His-64 in swMb) forms a hydrogen bond directly to the bound ligand (Protein Data Bank code 2JHO (36)), and mutation of HisE7 results in an increase in koff for oxygen by at least 2 orders of magnitude (42).
The proximal pocket of Cgb contains a hydrogen bonding network that stabilizes the proximal histidine ligand (Fig. 5), as previously predicted by resonance Raman spectroscopy (24); the high νFe-His mode value of 251 cm−1 is consistent with a proximal ligand with imidazolate (anionic) character. This same hydrogen bonding network involving GluH23 and TyrG5 is present in Hmp and Vgb (Fig. 4), which have νFe-His modes of 244 cm−1 (43) and 250 cm−1 (34), respectively. This compares to much lower values of 200–220 cm−1 for most globins (34), which lack this hydrogen bonding network, resulting in a less polar proximal ligand. Perturbation of this hydrogen bonding network via the introduction of either E134A (H23) or Y94F (G5) mutations diminishes the magnitude of this high νFe-His mode at pH 7.4 (Fig. 6) and, in the case of Y94F (G5), the partial formation of a six-coordinate low spin species. Because no exogenous ligands are present, we attribute this spectral signal to the coordination of heme by the TyrB10 residue (24). Mutation of TyrG5 in Vgb perturbed oxygen binding and diminished NO dioxygenase activity (19), indicating a crucial role for this residue in protein function.
To further probe the role of GluH23 and TyrG5 in Cgb, the CN− on rates for wild type, E134A (H23), and Y94F (G5) were measured (Fig. 7). Substitution of GluH23 for Ala results in a 2-fold increase in the on rate for CN−, which is at first surprising because one might expect mutation of such a residue to perturb ligand binding. It has been suggested that the cyanide binding reaction in Mb involves three steps: (i) the movement of neutral HCN into the heme pocket; (ii) the deprotonation of HCN to CN− in the distal site; and (iii) the binding of CN− to the heme iron (44). Our data suggest that cyanide binding in Cgb is not rate-limited by ligand migration or deprotonation and that the mutation of GluH23 to Ala in Cgb promotes CN− binding by raising the formal charge of the heme iron, where the “electronic push” from the proximal side is diminished.
Although the proximal hydrogen bonding network has previously been proposed to affect the redox properties of the heme cofactor in Vgb (14), the current work represents the first potentiometric analysis of a bacterial single-domain globin. The current method employs a series of stable transition metal-containing compounds as redox mediators (35), which offer negligible interference with the spectral features of ferrous and ferric Cgb. At pH 7.0, the redox midpoint of Cgb is −134 mV versus normal hydrogen electrode (Fig. 8), which is similar to that of the globin domain of Hmp (−121 ± 3 mV (45)). These midpoint potentials are considerably lower than those of myoglobin (58 mV (46)) or cytochrome b562 (168 mV). Such low redox midpoints for globins with histidyl proximal ligands are typical of peroxidases; the midpoint potential of horseradish peroxidase is −250 mV (37), where a strong hydrogen bond is present between the proximal imidazole ring and a carboxylate group (38). Like the νFe-His stretching modes, these differences can be explained in terms of the difference in character of the proximal ligand (i.e. the hydrogen bonds to the axial ligands in Cgb and horseradish peroxidase confer a partial negative charge to the imidazole rings, whereas the absence of these hydrogen bonds in myoglobin and cytochrome b562 results in essentially neutral proximal ligands). Decreasing the pH has previously been shown to perturb this hydrogen bonding network and decrease the νFe-His stretching mode of Cgb, conferring a more “myoglobin-like” character on the proximal ligand (24). Based on the low pKa of this transition (pH < 4), this was attributed to the protonation of Glu-134 (H23), which diminishes the anionic character of the Cgb proximal histidine. The protonation of GluH23 would also explain the current observation that decreasing the pH results in a concomitant increase the redox midpoint of Cgb. Unfortunately, neither the E134A (H23) nor the Y94F (G5) mutant were stable enough for potentiometric analysis; the protein precipitated under the conditions used for titration of the wild type protein. The redox responses of Cgb exhibit a pH dependence of far less than −59 mV/pH unit, indicating that the pH-induced variations in redox midpoint are unlikely to be due to proton-coupled electron transfer at the heme and are more likely a consequence of GluH23 protonation.
Taken together, the TyrB10 and GlnE7 residues provide a positively polar distal environment, and the proximal hydrogen bonding network confers a greater “electron-donating” capability to the proximal histidine. In peroxidases, these properties have been shown to be crucial for catalyzing the O–O bond cleavage reaction of heme-bound peroxides on the basis of the archetypal “push-pull” model (47). These properties have been implicated in the isomerization of the presumed peroxynitrite intermediate to nitrate as suggested for the NO dioxygenase reaction catalyzed by both Hmp (43) and Cgb (24); the anionic proximal histidine provides the electronic “push,” and the positively polar distal environment provides the electronic “pull.” However, Hmp has been shown to bind preferentially to NO (Kd = 8 pm) compared with oxygen (Kd = 12 nm) and can also catalyze a denitrosylase reaction (48), where heme-nitrosyl complex formation precedes a reaction with dioxygen to form nitrate. This requires cleavage of the N–O bond of the nitroxyl moiety, a more thermodynamically unfavorable process than cleavage of bound dioxygen. Measurement of the Kd for NO and a quantitative understanding of intracellular oxygen and nitric oxide concentrations during infection is necessary to elucidate the in vivo mechanism for nitric oxide detoxification in C. jejuni.
The current work demonstrates that Cgb possesses structural and electrochemical characteristics of a protein that performs ligand chemistry rather than oxygen transport. Our data represent the first structural and potentiometric characterization of a monomeric bacterial single-domain globin and will be of importance for the understanding of how Campylobacter can survive nitrosative stress in the human and avian hosts.
Supplementary Material
Acknowledgments
The staff at the Daresbury Laboratory are acknowledged for running the synchrotron facility. We thank Dr. Linda Britton for help with the structure analysis.
This work was supported by Biotechnology and Biological Sciences Research Council Grant BB/EO10504/1 (to R. K. P.).

The on-line version of this article (available at http://www.jbc.org) contains Supplemental Tables S1 and S2 and Figs. S1–S3.
The atomic coordinates and structure factors (code 2WY4) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The residues in Cgb are named using standard globin nomenclature as follows: E7, Gln-52; B10, Tyr-28; H23, Glu-134; G5, Tyr-94; FG4, Val-89; G8, Val-97; H8, Trp-119; A11, Cys-12; H27, Tyr-138; H12, Tyr-123.
- NO
- nitric oxide
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- swMb
- sperm whale myoglobin
- Vgb
- V. stercoraria single-domain globin
- Hmp
- E. coli flavohemoglobin.
REFERENCES
- 1.Keilin D. (1953) Nature 172, 390–393 [DOI] [PubMed] [Google Scholar]
- 2.Elvers K. T., Wu G., Gilberthorpe N. J., Poole R. K., Park S. F. (2004) J. Bacteriol. 186, 5332–5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Membrillo-Hernández J., Coopamah M. D., Anjum M. F., Stevanin T. M., Kelly A., Hughes M. N., Poole R. K. (1999) J. Biol. Chem. 274, 748–754 [DOI] [PubMed] [Google Scholar]
- 4.Ermler U., Siddiqui R. A., Cramm R., Friedrich B. (1995) EMBO J. 14, 6067–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ermler U., Siddiqui R. A., Cramm R., Schröder D., Friedrich B. (1995) Proteins Struct. Funct. Genet. 21, 351–353 [DOI] [PubMed] [Google Scholar]
- 6.Ilari A., Bonamore A., Farina A., Johnson K. A., Boffi A. (2002) J. Biol. Chem. 277, 23725–23732 [DOI] [PubMed] [Google Scholar]
- 7.Vasudevan S. G., Armarego W. L., Shaw D. C., Lilley P. E., Dixon N. E., Poole R. K. (1991) Mol. Gen. Genet. 226, 49–58 [DOI] [PubMed] [Google Scholar]
- 8.Poole R. K., Hughes M. N. (2000) Mol. Microbiol. 36, 775–783 [DOI] [PubMed] [Google Scholar]
- 9.Wittenberg J. B., Bolognesi M., Wittenberg B. A., Guertin M. (2002) J. Biol. Chem. 277, 871–874 [DOI] [PubMed] [Google Scholar]
- 10.Pesce A., Couture M., Dewilde S., Guertin M., Yamauchi K., Ascenzi P., Moens L., Bolognesi M. (2000) EMBO J. 19, 2424–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu C., Egawa T., Wainwright L. M., Poole R. K., Yeh S. R. (2007) J. Biol. Chem. 282, 13627–13636 [DOI] [PubMed] [Google Scholar]
- 12.Ouellet H., Ouellet Y., Richard C., Labarre M., Wittenberg B., Wittenberg J., Guertin M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5902–5907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarricone C., Calogero S., Galizzi A., Coda A., Ascenzi P., Bolognesi M. (1997) Proteins Struct. Funct. Genet. 27, 154–156 [DOI] [PubMed] [Google Scholar]
- 14.Tarricone C., Galizzi A., Coda A., Ascenzi P., Bolognesi M. (1997) Structure 5, 497–507 [DOI] [PubMed] [Google Scholar]
- 15.Bolognesi M., Boffi A., Coletta M., Mozzarelli A., Pesce A., Tarricone C., Ascenzi P. (1999) J. Mol. Biol. 291, 637–650 [DOI] [PubMed] [Google Scholar]
- 16.Joshi M., Dikshit K. L. (1994) Biochem. Biophys. Res. Commun. 202, 535–542 [DOI] [PubMed] [Google Scholar]
- 17.Tari C., Parulekar S. J., Stark B. C., Webster D. A. (1998) Biotechnol. Bioeng. 59, 673–678 [DOI] [PubMed] [Google Scholar]
- 18.Kaur R., Pathania R., Sharma V., Mande S. C., Dikshit K. L. (2002) Appl. Environ. Microbiol. 68, 152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaur R., Ahuja S., Anand A., Singh B., Stark B. C., Webster D. A., Dikshit K. L. (2008) FEBS Lett. 582, 3494–3500 [DOI] [PubMed] [Google Scholar]
- 20.Iovine N. M., Pursnani S., Voldman A., Wasserman G., Blaser M. J., Weinrauch Y. (2008) Infect. Immun. 76, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elvers K. T., Turner S. M., Wainwright L. M., Marsden G., Hinds J., Cole J. A., Poole R. K., Penn C. W., Park S. F. (2005) Mol. Microbiol. 57, 735–750 [DOI] [PubMed] [Google Scholar]
- 22.Wainwright L. M., Elvers K. T., Park S. F., Poole R. K. (2005) Microbiology 151, 4079–4091 [DOI] [PubMed] [Google Scholar]
- 23.Monk C. E., Pearson B. M., Mulholland F., Smith H. K., Poole R. K. (2008) J. Biol. Chem. 283, 28413–28425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu C., Mukai M., Lin Y., Wu G., Poole R. K., Yeh S. R. (2007) J. Biol. Chem. 282, 25917–25928 [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 26.Matthews B. W. (1968) J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project 4 (1994) Acta Crystallogr. D 50, 760–76315299374 [Google Scholar]
- 28.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Acta Crystallogr. Sect. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 29.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 30.Brünger A. T. (1992) Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 31.Lamzin V. S., Wilson K. S. (1993) Acta Crystallogr. D Biol. Crystallogr. 49, 129–147 [DOI] [PubMed] [Google Scholar]
- 32.Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 33.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 34.Egawa T., Yeh S. R. (2005) J. Inorg. Biochem. 99, 72–96 [DOI] [PubMed] [Google Scholar]
- 35.Bernhardt P. V., Chen K. I., Sharpe P. C. (2006) J. Biol. Inorg. Chem. 11, 930–936 [DOI] [PubMed] [Google Scholar]
- 36.Arcovito A., Benfatto M., Cianci M., Hasnain S. S., Nienhaus K., Nienhaus G. U., Savino C., Strange R. W., Vallone B., Della Longa S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 6211–6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamada H., Makino R., Yamazaki I. (1975) Arch. Biochem. Biophys. 169, 344–353 [DOI] [PubMed] [Google Scholar]
- 38.Finzel B. C., Poulos T. L., Kraut J. (1984) J. Biol. Chem. 259, 13027–13036 [PubMed] [Google Scholar]
- 39.Lee S. Y., Stark B. C., Webster D. A. (2004) Biochem. Biophys. Res. Commun. 316, 1101–1106 [DOI] [PubMed] [Google Scholar]
- 40.Jakob W., Webster D. A., Kroneck P. M. (1992) Arch. Biochem. Biophys. 292, 29–33 [DOI] [PubMed] [Google Scholar]
- 41.Verma S., Patel S., Kaur R., Chung Y. T., Duk B. T., Dikshit K. L., Stark B. C., Webster D. A. (2005) Biochem. Biophys. Res. Commun. 326, 290–297 [DOI] [PubMed] [Google Scholar]
- 42.Rohlfs R. J., Mathews A. J., Carver T. E., Olson J. S., Springer B. A., Egeberg K. D., Sligar S. G. (1990) J. Biol. Chem. 265, 3168–3176 [PubMed] [Google Scholar]
- 43.Mukai M., Mills C. E., Poole R. K., Yeh S. R. (2001) J. Biol. Chem. 276, 7272–7277 [DOI] [PubMed] [Google Scholar]
- 44.Dou Y., Olson J. S., Wilkinson A. J., Ikeda-Saito M. (1996) Biochemistry 35, 7107–7113 [DOI] [PubMed] [Google Scholar]
- 45.Cooper C. E., Ioannidis N., D'mello R., Poole R. K. (1994) Biochem. Soc. Trans. 22, 709–713 [DOI] [PubMed] [Google Scholar]
- 46.Rayner B. S., Stocker R., Lay P. A., Witting P. K. (2004) Biochem. J. 381, 365–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poulos T. L. (1996) J. Biol. Inorg. Chem. 1, 356–359 [Google Scholar]
- 48.Hausladen A., Gow A., Stamler J. S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 10108–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.