

Abstract
Background
Paroxysmal nocturnal hemoglobinuria (PNH) is associated with an increased risk of thrombosis through unknown mechanisms.
Design and Methods
We studied 23 patients with PNH, before and after five and 11 weeks of treatment with eculizumab. We examined markers of thrombin generation and reactional fibrinolysis (prothrombin fragment 1+2 (F1+2), D-dimers, and plasmin antiplasmin complexes (P-AP), and endothelial dysfunction tissue plasminogen activator (t-PA), plasminogen activator inhibitor (PAI-1), soluble thrombomodulin (sTM), intercellular adhesion molecule 1 (sICAM-1), vascular cell adhesion molecule (sVCAM-1), endothelial microparticles (EMPs), and tissue factor pathway inhibitor (TFPI).
Results
At baseline, vWF, sVCAM-1, the EMP count, and F1+2 and D-dimer levels were significantly elevated in the patients, including those with no history of clinical thrombosis. Treatment with eculizumab was associated with significant decreases in plasma markers of coagulation activation (F1+2, P=0.012, and D-dimers, P=0.01), and reactional fibrinolysis (P-AP, P=0.0002). Eculizumab treatment also significantly reduced plasma markers of endothelial cell activation (t-PA, P=0.0005, sVCAM-1, P<0.0001, and vWF, P=0.0047) and total (P=0.0008) and free (P=0.0013) TFPI plasma levels.
Conclusions
Our results suggest a new understanding of the contribution of endothelial cell activation to the pathogenesis of thrombosis in PNH. The terminal complement inhibitor, eculizumab, induced a significant and sustained decrease in the activation of both the plasma hemostatic system and the vascular endothelium, likely contributing to the protective effect of eculizumab on thrombosis in this setting.
Keywords: paroxysmal nocturnal hemoglobinuria, thrombosis, eculizumab
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a hematopoietic stem cell disorder characterized by intravascular hemolysis due to the absence of the GPI-anchored complement regulatory proteins CD55 and CD59. CD59 blocks the formation of the terminal complement complex (the membrane-attack complex) on the cell surface, thereby preventing erythrocyte lysis and in vitro platelet activation. Affected red blood cells are rendered sensitive to complement mediated lysis leading to free hemoglobin release.1 Chronically, and during severe bouts of hemolysis (paroxysms), hemoglobin can saturate biochemical systems resulting in hemoglobinuria. Excessive or persistent intravascular hemolysis in patients with PNH causes anemia, hemoglobinuria and complications related to the presence of plasma free hemoglobin, including abdominal pain, dysphagia, erectile dysfunction, possibly pulmonary hypertension and chronic kidney disease, and most importantly venous and arterial thrombosis.2
Possible mechanisms include: procoagulant microparticles released by complement-mediated platelet activation;3 chronic hypofibrinolysis through altered plasminogen activation, possibly due to a decrease in urinary plasminogen activator receptor (u-PAR) expression on leukocyte surfaces; 4,5 release of free hemoglobin by chronic hemolysis, leading to nitric oxide (NO) depletion and, subsequently, endothelial dysfunction and platelet activation.6,7 Primary or secondary prophylaxis with anticoagulants, vitamin-K antagonists (VKA) or low molecular weight heparin (LMWH), carries a high risk of complications and is insufficient to prevent thrombosis in this setting.8
Arterial thrombosis and venous thromboembolism (VTE) are potentially life-threatening complications of PNH9 and are the leading cause of death in this disease.10 VTE in critical anatomic sites (cerebral and splanchnic circulation) is the major cause of morbidity and mortality in PNH. Retrospective studies have suggested that the risk of thrombosis might correlate with the size of the PNH granulocyte clone.11 Thrombosis has been reported in patients without overt evidence of hemolysis, with smaller clones, mild anemia and no transfusions.12,13 The etiology of the increased thrombotic risk in patients with PNH is unclear.
Eculizumab, a humanized antibody that blocks cleavage of the complement component C5, thereby preventing complement-mediated RBC lysis,14 has been shown to reduce intravascular hemolysis, hemoglobinuria, and transfusion requirements,15 with an associated improvement in the quality of life of patients with PNH. Other benefits include less chronic kidney disease,16 and pulmonary hypertension.17 Eculizumab also prevents thrombosis in PNH.13
The purpose of this study was to examine the potential contributions of activation of the coagulation and/or fibrinolysis systems, and activation of the vascular endothelial cell surface, to the prothrombotic state in patients with PNH. Additionally, showing the modifications of these systems in PNH will improve understanding of the mechanisms by which eculizumab prevents clinical thrombosis.
Design and Methods
Study design
From January 2007 to August 2008, PNH patients who started to receive eculizumab for a hemolytic form of PNH were enrolled in 10 French centers. Eculizumab was given by intravenous infusion as follows: an induction phase with a dose of 600 mg every seven days for a total of 4 doses; then 900 mg seven days later; followed by a maintenance phase with a dose of 900 mg every 14±2 days, as previously described.15
Blood collection and plasma preparation
Three venous blood samples were collected atraumatically from each patient after an overnight fast. Blood was collected in 3.2% sodium citrate at baseline, once just prior to eculizumab infusion, once at week 5 just before the first dose of 900 mg, and once at week 11±2, during eculizumab maintenance treatment. Platelet-poor plasma was prepared within two hours by two centrifugation steps at 2500 g for 15 min at 15°C, then aliquoted and stored at −80°C until testing. All samples were tested by the same laboratory, and one aliquot of each plasma sample was thawed at 37°C immediately before the assay. A control group of 30 healthy blood donors was used to determine the normal range of endothelial microparticles. The procedures were conducted in accordance with the Helsinki Declaration of 1975, as revised in 2000, and all participants gave their informed consent. The study was approved by the IRB of Saint Louis Hospital, Paris, France.
Measurement of hemostatic parameters and endothelial activation markers
Plasma levels of the following factors were quantified using enzyme linked immunosorbent assays (ELISA): tissue-type plasminogen activator (t-PA), plasminogen activator inhibitor-1 (PAI-1), total and free tissue factor pathway inhibitor (TFPI), and thrombomodulin (respectively, Asserachrom® t-PA, Asserachrom® PAI-1, Asserachrom® total TFPI, Asserachrom® free TFPI and Asserachrom® thrombomodulin; Diagnostica Stago, Asnières, France); prothrombin fragment F1+2 (F1+2) (Enzygnost F1+2 micro and Enzygnost TAT micro, Dade Behring, Marburg, Germany); plasmin-antiplasmin (P-AP) complexes (Kordia Life Sciences, Leiden, Netherland); soluble inter-cellular adhesion molecule 1 (sICAM-1), vascular cell adhesion molecule (sVCAM-1) and su-PAR (respectively, Quantikine human soluble ICAM-1, Quantikine human soluble VCAM-1, and Quantikine human soluble u-PAR; R&D Systems, Minneapolis, USA). Von Willebrand factor antigen (vWF: Ag) and D-dimers were measured with a STAR analyzer, using an immuno-turbidimetric assay (STA® Liatest®; Diagnostica Stago, Asnières, France). All assays were performed according to the manufacturers’ recommendations and using their normal ranges.
Endothelial microparticle quantification
(see Online Supplementary Data)
Microparticle-linked procoagulant activity measurement
Microparticle-linked procoagulant activity was determined with the Zymuphen MP-activity kit (Hyphen BioMed, Neuvillesur-Oise, France). This method is based on a prothrombinase assay after microparticle capture on a micro-titration plate coated with streptavidin and biotinylated annexin V. Results were expressed as nanomolar phosphatidylserine equivalent by using a calibrator prepared from a washed and lysed platelet concentrate containing a known concentration of microparticles expressed in nM phosphatidylserine equivalent (nM eq PS).
Statistical analysis
(see Online Supplementary Data)
Results
Patients’ characteristics
Twenty-three subjects with hemolytic PNH were analyzed. Fifteen patients had never had thrombotic events and did not receive anticoagulant therapy. Eight patients, 7 of whom had a history of VTE, received anticoagulant therapy for VTE prophylaxis (one fondaparinux, one danaparoid and 6 VKA). None of the characteristics listed in Table 1 differed between patients with and without anticoagulant therapy, apart from the prothrombin time, which was lower in patients on anticoagulants.
Table 1.
Patients’ characteristics at baseline.

Effect of eculizumab on intravascular hemolysis and thrombosis occurrence
Eculizumab reduced intravascular hemolysis as shown by the decrease in lactate dehydrogenase levels from 2975 U/mL [1675–4000] (median [interquartile range]) at baseline to 411 U/mL [347–514] at week 5 (P<0.0001, week 5 vs. baseline) and 447 U/mL [383–497] at week 11 (normal LDH values < 250 U/mL). Transfusion requirements were also reduced in patients on eculizumab.
No thrombosis occurred during eculizumab treatment.
Markers of thrombin generation and fibrinolysis
To assess the activation of coagulation, we studied F1+2, a direct marker of prothrombin activation to thrombin, D-dimers and P-AP complexes, as markers of reactional fibrinolysis induced by thrombin generation or by complement dysregulation. Before eculizumab therapy, F1+2 plasma levels were increased (Table 2). Eculizumab treatment resulted in a significant decrease in F1+2 plasma levels (P=0.012) at week 5, which was sustained at week 11 (Figure 1A).
Table 2.
Baseline markers values.

Figure 1.
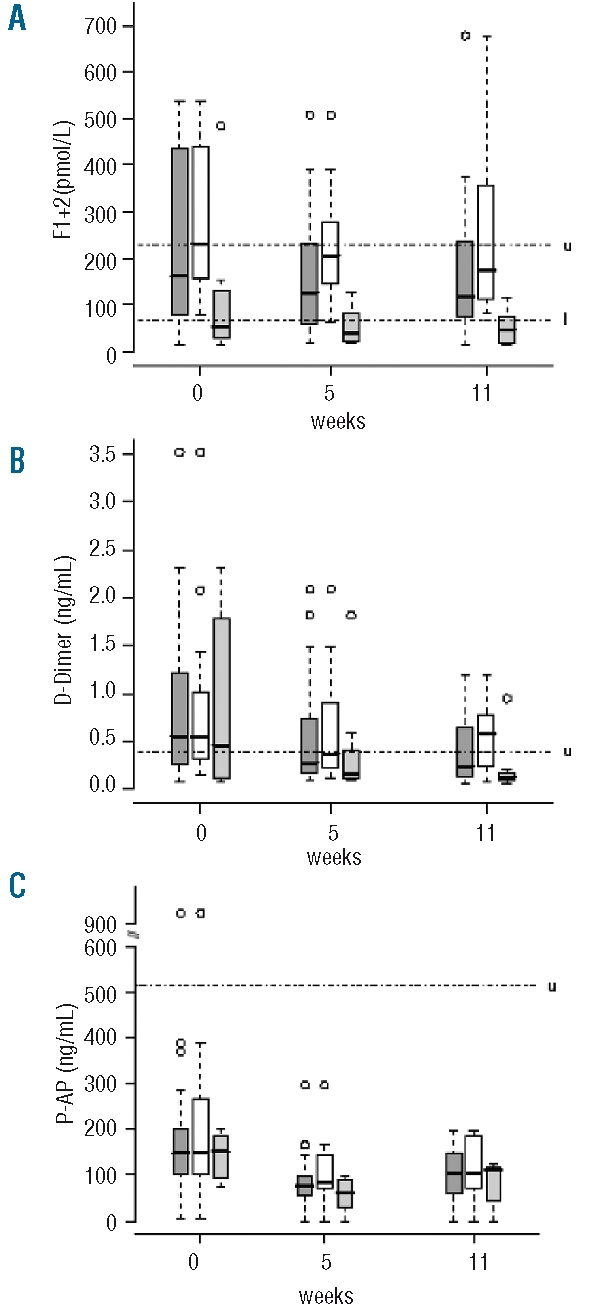
Distribution of prothrombin fragment 1+2 (A), D-dimers (B), and P-AP complex (C) plasma levels in the whole cohort ( ), and in patients without (□) and with anticoagulant treatment (
), and in patients without (□) and with anticoagulant treatment ( ) at baseline, and at week 5 and week 11 of eculizumab treatment. Boxes and whisker plots represent the median (full line) and the 25th and 75th percentiles (boxes), and the outer whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. Upper and lower limits of normal are indicated, by u and l, respectively.
) at baseline, and at week 5 and week 11 of eculizumab treatment. Boxes and whisker plots represent the median (full line) and the 25th and 75th percentiles (boxes), and the outer whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. Upper and lower limits of normal are indicated, by u and l, respectively.
Before eculizumab, F1+2 plasma levels were increased in the group without anticoagulants and normal in the group with anticoagulants (P=0.007). A decrease of F1+2 plasma levels was observed in both groups of patients during eculizumab treatment. Thus, the F1+2 plasma levels remained lower in patients on anticoagulants than in other patients at week 5 (0.04 nmol/L [0.02 to 0.07] (median [interquartile range]) vs. 0.21 nmol/L [0.15 to 0.28, P=0.0001] and week 11 (0.05 nmol/L [0.02 to 0.08] vs. 0.17 nmol/L [0.11 to 0.35], P=0.0001) (Figure 1A).
Before eculizumab, D-dimer plasma levels were elevated (Table 2), and eculizumab treatment resulted in a significant reduction in D-dimer plasma levels (P=0.01) at week 5, persisting at week 11 (Figure 1B). At baseline there was no significant difference according to the use of anticoagulant therapy (P=0.55), and the reduction in D-dimer plasma levels during eculizumab treatment was similar in the two groups (P=0.07).
Before eculizumab treatment P-AP complex plasma levels were normal, with no difference between the two groups (Table 2). Eculizumab treatment was associated with a significant decrease (P=0.0002) in plasma levels in the overall patient population at week 5, persisting at week 11 (Figure 1C).
Markers of endothelial cell activation
To evaluate the potential for endothelial cell activation and damage which could induce thrombosis, we studied several measures of endothelial function.
Before eculizumab, sVCAM-1 and vWF Ag were both above normal range, while t PA, PAI 1, sTM and sICAM 1 were normal. PAI-1 and t-PA were significantly higher in patients with anticoagulant therapy than in those without anticoagulant therapy (P=0.034 and P=0.013, respectively) (Table 2). Eculizumab treatment was associated with significant decreases at weeks 5 and 11 in plasma levels of t-PA (P=0.0005) in two groups according to anticoagulant therapy, and in sVCAM-1 (P<0.0001) and vWF Ag (P=0.0047) in the overall group of patients (Figure 2 AC); the effect on sVCAM-1 was more pronounced in the group with anticoagulant therapy (P=0.015) (Figure 2B). PAI-1, sTM and sICAM-1 plasma levels did not change during eculizumab treatment (data not shown).
Figure 2.
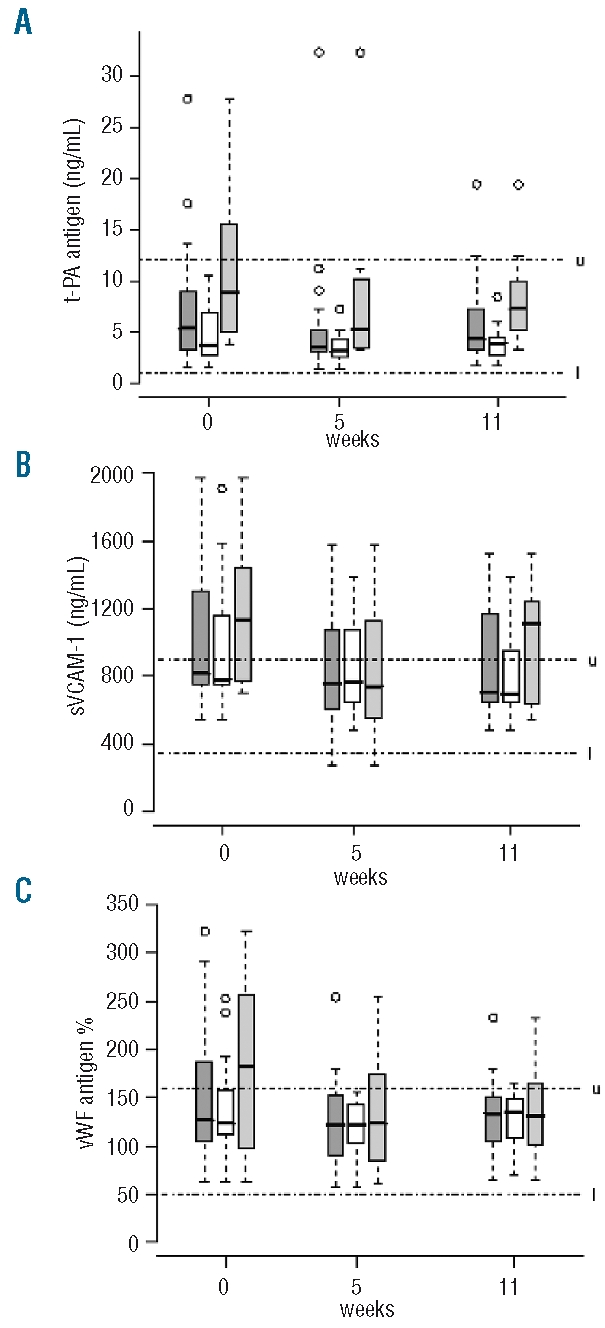
Distribution of t-PA (A), sVCAM-1 (B), and vWF Ag (C) plasma levels in the whole cohort (), and in patients without (□) and with anticoagulant therapy () at baseline, and at week 5 and week 11 of eculizumab treatment. Boxes and whisker plots represent the median (full line) and the 25th and 75th percentiles (boxes), and the outer whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. Upper and lower limits of normal are indicated, by u and l, respectively.
Plasma levels of su-PAR were elevated in both groups before eculizumab treatment (Table 2). There was a difference in Su-PAR kinetics between the groups (P=0.0004), with a significant decrease at week 5 in the anticoagulant group only (P=0.002), persisting at week 11; but su-PAR plasma levels remained elevated in both groups (data not shown). Thus, endothelial cells were strongly activated and showed a potentially prothrombotic phenotype (elevated sVCAM-1 and vWF) in these patients with PNH. Eculizumab treatment significantly reduced the plasma levels of parameters associated with endothelial cell activation or damage (t–PA, sVCAM-1 and vWF).
TFPI
TFPI, the inhibitor of coagulation cascade initiation, is synthesized and released by endothelial cells.
Before treatment, total TFPI plasma levels were normal and free TFPI plasma levels were at the upper limit of normal (Table 2). Eculizumab treatment was associated with significant decreases in both total TFPI (P=0.0008) and free TFPI (P=0.0013) plasma levels at week 5, persisting at week 11, with no difference according to anticoagulant therapy (Figure 3 A and B).
Figure 3.
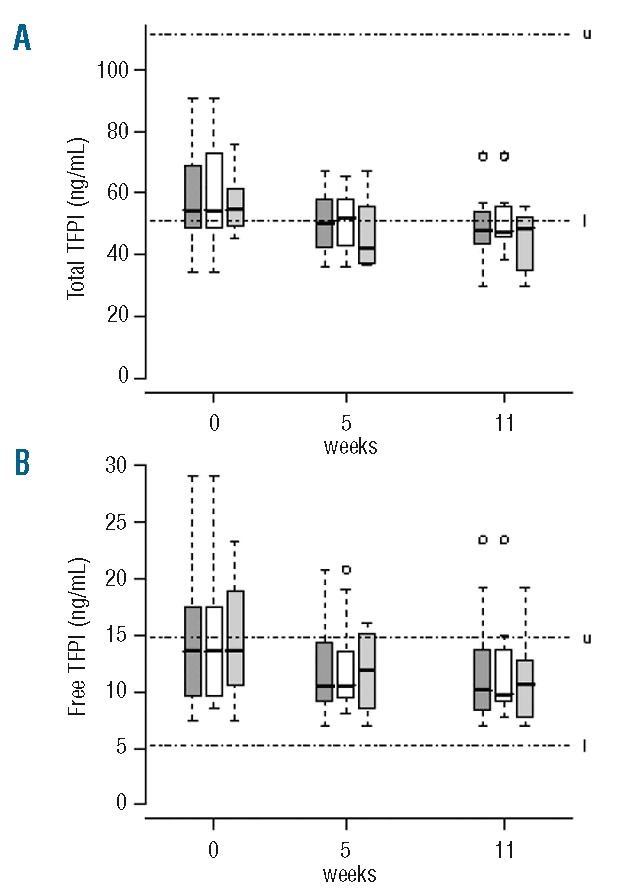
Distribution of total TFPI (A) and free TFPI (B) antigen plasma levels in the whole cohort (), and in patients without (□) and with anticoagulant therapy () at baseline, and at week 5 and week 11 of eculizumab treatment. Boxes and whisker plots represent the median (full line) and the 25th and 75th percentiles (boxes), and the outer whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the box. Upper and lower limits of normal are indicated, by u and l, respectively.
Microparticles
To further assess the activation and possible damage of endothelial cells, we measured endothelial microparticles release into the circulation and total microparticles shed from all cells exposing phosphatidylserine, which conferred a procoagulant activity.
Before eculizumab, microparticles procoagulant activity, absolute plasma numbers of total EMPs and ICAM-1 (CD54)-expressing EMPs were elevated, while the percentage of ICAM-1+ EMPs was normal (Table 2). No difference in these four parameters was found between the groups before eculizumab treatment (Table 2), and eculizumab had no effect on these markers in either group (data not shown).
Discussion
The pathogenesis of thrombotic complications in PNH is unclear. In particular, no deficiencies in antithrombin, protein C or protein S have been described in patients with PNH.18
It was recently suggested that the most frequent symptoms of PNH are consistent with smooth muscle perturbation through the cell-free plasma hemoglobin and NO scavenging. 2 The lack of complement regulatory proteins CD55 and CD59 on the surface of blood cells, including platelets, that make these cells more sensitive to complement-mediated activation or cytolysis, may contribute to the thrombotic tendency in PNH.1 Additionally, patients with PNH may experience NO-depletion and plasma heme-induced platelet activation, a source of oxidative stress resulting in endothelial cell dysfunction.
The granulocyte clone size correlates with the risk of thrombosis in PNH.11,12 In our study, all the patients had large clones (> 57%; 20 patients > 70%) and were thus at a high thrombotic risk. As reported by Kakkar et al, patients with thrombosis and treated with anticoagulants have a reduction in their F1+2 plasma levels.19 This could explain why, in our study, the patients without anticoagulant therapy had higher F1+2 plasma levels than the 8 patients with anticoagulant therapy (7 with previous thrombosis, one on primary prophylaxis). In our study, before eculizumab treatment, the elevated F1+2 and D-dimer plasma levels in PNH patients (compared to the reference values) confirm the activation of both the coagulation and fibrinolytic system,20 as observed in a previous study of a small number of PNH patients.21 This prothrombotic activity indicates that PNH patients, and particularly those not treated with anticoagulants, have significant activation of coagulation despite the absence of overt thrombosis. However, we found that plasma levels of P-AP (a marker of plasmin generation) were normal, suggesting that fibrinolytic system activation was impaired. The increase in su-PAR, also observed in previous studies,22,23 might inhibit reactional fibrinolysis, as suggested by Grünewald et al.21 These latter authors also reported a negative correlation between clone size and activation of the fibrinolytic system. Our results suggest that patients have coagulation activation in the absence of active thrombosis.
We found normal plasma levels of t-PA, a marker released only by endothelial cells; PAI-1, an acute phase reactant synthesized by endothelial cells, monocytes and hepatocytes; sTM, a marker of endothelium activation due to a cleavage by neutrophil elastase increased by inflammation or infection; and sICAM-1. We found elevated plasma levels of vWF, an acute phase reactant that can be released by endothelial cells, as well as of sVCAM-1 and EMPs; this could not be explained by a systemic inflammation, as C-reactive protein serum levels were normal. This absence of systemic inflammation could explain why EMPs were mainly ICAM-1-negative (indicating continuous endothelial stimulation and/or injury), while EMP ICAM-1-positive EMPs reflect the inflammatory status of endothelial cells.24 This lack of generalized inflammation is also supported by the contrast between increased vWF levels and normal sTM levels; indeed, in the absence of inflammation, as in atrial fibrillation, sTM is normal while vWF is elevated.25 Setty et al.26 have shown that heme induces tissue factor expression on endothelial cells in culture, independently of the effect of inflammatory cytokines (IL-1α or TNF-α). Thus, our data point to endothelial cell activation in patients with PNH, in the absence of systemic inflammation.
Interestingly, eculizumab significantly reduced the plasma levels of F1+2, D-dimers and P-AP complexes during the induction phase (week 5) and during chronic treatment (week 11), and even at week 47 in the 9 patients tested (data not shown). These results indicate a decrease in coagulation activation and in reactional fibrinolysis during eculizumab therapy. Patients receiving anticoagulant treatment after a thrombotic event had even lower F1+2 plasma levels probably related to the anticoagulant treatment itself.
Recently, Weitz et al. reported, in an abstract form, that eculizumab reduced D-dimer and thrombin-antithrombin complex levels, independently of hemolysis suppression based on LDH assay. This reduction correlated with a decrease in IL-6, an inflammatory maker.27 However, this effect on IL-6 could also be related to a decrease in IL-6 production by endothelial cells due to decreased thrombin generation. 28 In our study, LDH levels normalized in all patients after the eculizumab induction phase. The lack of correlation, observed by Weitz et al.27 between decreased LDH and thrombin generation suggests that the potential reduction in clinical thrombosis by eculizumab is not only due to a decrease in hemolysis leading to the hypothesis of the path-ogenic role of the endothelium in PNH-related thromboembolism.
The fact that t-PA was significantly reduced by eculizumab treatment while sICAM-1 and sTM plasma levels were unaffected does not rule out a possible effect of eculizumab treatment on endothelial cell activation; indeed sICAM-1 is ubiquitous and sTM levels are not modified in the absence of inflammation.25
Total and free TFPI were significantly reduced by eculizumab treatment; however, the levels of free TFPI, the only active form in anticoagulation, remained over the 5th percentile and are thus not associated with an increased risk of thrombosis.29 Low molecular weight heparin is known to enhance TFPI plasma levels,30 but only one of our patients received this treatment.
Su-PAR levels were substantially above the upper limit but eculizumab reduced plasma su-PAR levels only in patients with concomitant anticoagulant treatment. In this group F1+2 levels and, thus, thrombin generation were lower (see above). Thus, platelet activation due to thrombin must be lower; and, as reported recently by Sloand et al., part of su-PAR in PNH patients is released from activated platelets.31 Moreover, the decrease in hemolysis, reduces ADP secretion and NO-depletion, and thus platelet activation.
Among all the tested markers, D-dimers, which decreased during eculizumab treatment and can be routinely measured, could be used in future to perform a prospective, multi-center study to detect PNH patients with high thrombotic risk. Unfortunately, apart from vWF antigen assay, specific markers of endothelial activation are not routinely tested.
Importantly, terminal complement inhibition with eculizumab significantly reduced plasma levels of endothelial activation markers during the induction phase. This decrease was sustained in the maintenance phase. These effects may be due partly to an eculizumab-mediated improvement of hemolysis, which had a positive impact on hemostasis via reduction in NO-depletion;32 they could also be due to the decrease in complement-mediated pathological activation of endothelial cells. Indeed, several findings support pathological endothelial cell activation and/or activation of a pathological PNH phenotype endothelial cell. (i) VWF and sVCAM-1 plasma levels were elevated in patients with PNH, and were significantly reduced during eculizumab treatment. Previous studies of cultured normal endothelial cells showed that complement proteins C5b-C9 induce vWF secretion,33,34 increase VCAM-1 expression,35 and trigger the release of procoagulant microparticles.36 The membrane attack complex also induces endothelial cells to produce and secrete IL-1, which can stimulate endothelial cells, in an autocrine or paracrine fashion, to generate the activated phenotype.37 (ii) TFPI, the main source of which is the endothelium, involves a GPI mediated membrane anchor-age in localizing to the surface of endothelial cells.38 Indeed, part of TFPI is directly secreted by endothelial cells. Most of the TFPI tightly, but reversibly, binds to a GPI-anchored co-receptor in the endoplasmic reticulum/Golgi apparatus. The co-receptor then acts as a molecular chaperone for TFPI by trafficking it to the surface of normal endothelial cells, or to lysosomes in cells lacking GPI-anchored proteins.39 This could explain why plasma levels of total TFPI were reduced in PNH patients, while free TFPI levels were normal. (iii) The identification of a new thrombophilic disease in which an autosomal recessive inherited GPI deficiency is not associated with apparent hemolysis supports the contribution of a non-hemolytic mechanism to thrombosis in a context of GPI deficiency.40 (iv) We found that ECFCs isolated from peripheral blood of a PNH patient and a healthy control showed that the patient had a two and three populations of ECFCs based on CD55 and CD59 expression, respectively, possibly corresponding to the presence of a pathological endothelial cell population (see Online Supplementary Appendix). This defect of GPI-anchored protein expression on ECFCs suggests that endothelial cells could be affected by a PIG-A mutation. In this case, eculizumab might prevent clinical thrombosis not only by inhibiting hemolysis and subsequent NO-depletion, but also by protecting pathological endothelial cells from complement activation.8 To our knowledge there are no reported attempts to identify endothelial cells with the PNH phenotype, probably due to the large amount of blood necessary for such assay. Further investigations with more patients are obviously necessary to confirm this hypothesis.
In conclusion, we demonstrate an endothelial dysfunction in PNH, which could be due to NO-depletion via hemolysis and/or platelet dysfunction; however, our results suggest a possible contribution of endothelial cell activation and dysfunction to the increased risk of thrombosis in PNH. Treatment with the terminal complement inhibitor eculizumab led to a significant and sustained decrease in activation of both the plasmatic hemostatic system and of the endothelium. This biological effect may help to explain the observed clinical benefit of eculizumab to reduce thrombosis in patients with PNH.8 Our results thus suggest a new role of endothelial cells in the occurrence of VTE in this setting. Interestingly, while the endothelial cell has never been studied directly in PNH patients, it may not be coincidental that we found a decrease in endothelial cell activation during treatment with eculizumab and a possible direct impairment of endothelial cells in GPI-anchored proteins. A prospective study with more patients is underway to confirm this apparent key role of endothelial cells in PNH-associated thrombosis.
Acknowledgments
the authors thank Yann Burnel and Emeline Levionnois for their excellent assistance with the laboratory work.
Appendix
The authors thank all the participating centers belonging to the French Society of Haematology: CHU Bicêtre, Kremlin Bicêtre (O. Lambotte, T. Boutekedjiret); CHU de Brest (C. Berthou, G. Guillerm, M.T. Blouch); CHU de Limoges (D. Bordessoule, T. Mohamed, N. Gachard, M Donnard, A. Julia, M.P. Chaury); CHU de Nice (E. Rosenthal, F. Sanderson, A. Rosenthal-Allieri); CHU de Rouen (F. Jardin, J.Y. Borg, P. Chamouni); CHU Hôtel Dieu, Paris (A. Vekhoff); CHU Necker-Enfants Malades, Paris (B. Deau-Fischer, C. Brouzes); Centre Hospitalier de Mulhouse (B. Drenoud); Hôpital Gilles de Corbeil, Corbeil-Essonnes (C. Salanoubat, I. Lemaire).
Footnotes
Funding: this work was supported by an unrestricted grant from France HPN, a non profit association.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
DH designed and performed research, analyzed data, and wrote the paper; RPL and CAR designed and performed research; GS and AMF designed research and contributed to the writing of the paper; JFS contributed to the critical revision of the article; JM, and IGF, performed research and validated the data; AD, performed research; RP performed statistical analysis.
DH, RPL, JFS, AMF and GS report receiving lecture and consulting fees from Alexion Pharmaceutical. No other potential conflicts of interests relevant to this article were reported.
References
- 1.Parker CJ. The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2007;35(4):523–33. doi: 10.1016/j.exphem.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 2.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. Jama. 2005;293(13):1653–62. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 3.Hugel B, Socie G, Vu T, Toti F, Gluckman E, Freyssinet JM, et al. Elevated levels of circulating procoagulant microparticles in patients with paroxysmal nocturnal hemoglobinuria and aplastic anemia. Blood. 1999;93(10):3451–6. [PubMed] [Google Scholar]
- 4.Ploug M, Plesner T, Ronne E, Ellis V, Hoyer-Hansen G, Hansen NE, et al. The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1992;79(6):1447–55. [PubMed] [Google Scholar]
- 5.Plesner T, Behrendt N, Ploug M. Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR. Stem Cells. 1997;15(6):398–408. doi: 10.1002/stem.150398. [DOI] [PubMed] [Google Scholar]
- 6.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–9. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 7.Schafer A, Wiesmann F, Neubauer S, Eigenthaler M, Bauersachs J, Channon KM. Rapid regulation of platelet activation in vivo by nitric oxide. Circulation. 2004;109(15):1819–22. doi: 10.1161/01.CIR.0000126837.88743.DD. [DOI] [PubMed] [Google Scholar]
- 8.Hillmen P, Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–8. doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]
- 9.Ziakas PD, Poulou LS, Rokas GI, Bartzoudis D, Voulgarelis M. Thrombosis in paroxysmal nocturnal hemoglobinuria: sites, risks, outcome. An overview. J Thromb Haemost. 2007;5(3):642–5. doi: 10.1111/j.1538-7836.2007.02379.x. [DOI] [PubMed] [Google Scholar]
- 10.Peffault de Latour R, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008;112(8):3099–106. doi: 10.1182/blood-2008-01-133918. [DOI] [PubMed] [Google Scholar]
- 11.Moyo VM, Mukhina GL, Garrett ES, Brodsky RA. Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol. 2004;126(1):133–8. doi: 10.1111/j.1365-2141.2004.04992.x. [DOI] [PubMed] [Google Scholar]
- 12.Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2003;102(10):3587–91. doi: 10.1182/blood-2003-01-0009. [DOI] [PubMed] [Google Scholar]
- 13.Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840–7. doi: 10.1182/blood-2007-06-094136. [DOI] [PubMed] [Google Scholar]
- 14.Hillmen P, Hall C, Marsh JC, Elebute M, Bombara MP, Petro BE, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–9. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 15.Hillmen P, Young NS, Schubert J, Brodsky RA, Socie G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–43. doi: 10.1056/NEJMoa061648. [DOI] [PubMed] [Google Scholar]
- 16.Hillmen P, Elebute M, Kelly R, Urbano-Ispizua A, Rother RP, Fu CL, et al. High Incidence of Progression to Chronic Renal Insufficiency in Patients with Paroxysmal Nocturnal Hemoglobinuria (PNH) Blood. 2007;110(11) Abstract 3678. [Google Scholar]
- 17.Hill A, Rother RP, Wang X, Sapsford RJ, Collinson PO, Gaze DC, et al. Eculizumab reduces pulmonary hypertension through inhibition of hemolysis-associated nitric oxide consumption in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;112(11) Abstract 486. [Google Scholar]
- 18.Griscelli-Bennaceur A, Gluckman E, Scrobohaci ML, Jonveaux P, Vu T, Bazarbachi A, et al. Aplastic anemia and paroxysmal nocturnal hemoglobinuria: search for a pathogenetic link. Blood. 1995;85(5):1354–63. [PubMed] [Google Scholar]
- 19.Kakkar VV, Hoppenstead DA, Fareed J, Kadziola Z, Scully M, Nakow R, Breddin HK. Randomized trial of different regimens of heparins and in vivo thrombin generation in acute deep vein thrombosis. Blood. 2002;99(6):1965–70. doi: 10.1182/blood.v99.6.1965. [DOI] [PubMed] [Google Scholar]
- 20.Liebman HA, Feinstein DI. Thrombosis in patients with paroxysmal noctural hemoglobinuria is associated with markedly elevated plasma levels of leukocyte-derived tissue factor. Thromb Res. 2003;111(4–5):235–8. doi: 10.1016/j.thromres.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 21.Grunewald M, Siegemund A, Grunewald A, Schmid A, Koksch M, Schopflin C, et al. Plasmatic coagulation and fibrinolytic system alterations in PNH: relation to clone size. Blood Coagul Fibrinolysis. 2003;14(7):685–95. doi: 10.1097/00001721-200310000-00011. [DOI] [PubMed] [Google Scholar]
- 22.Ploug M, Eriksen J, Plesner T, Hansen NE, Dano K. A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria. Eur J Biochem. 1992;208(2):397–404. doi: 10.1111/j.1432-1033.1992.tb17200.x. [DOI] [PubMed] [Google Scholar]
- 23.Ronne E, Pappot H, Grondahl-Hansen J, Hoyer-Hansen G, Plesner T, Hansen NE, et al. The receptor for urokinase plasminogen activator is present in plasma from healthy donors and elevated in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1995;89(3):576–81. doi: 10.1111/j.1365-2141.1995.tb08366.x. [DOI] [PubMed] [Google Scholar]
- 24.Simak J, Holada K, Risitano AM, Zivny JH, Young NS, Vostal JG. Elevated circulating endothelial membrane microparticles in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2004;125(6):804–13. doi: 10.1111/j.1365-2141.2004.04974.x. [DOI] [PubMed] [Google Scholar]
- 25.Freestone B, Chong AY, Nuttall S, Blann AD, Lip GY. Soluble E-selectin, von Willebrand factor, soluble thrombomodulin, and total body nitrate/nitrite product as indices of endothelial damage/dysfunction in paroxysmal, persistent, and permanent atrial fibrillation. Chest. 2007;132(4):1253–8. doi: 10.1378/chest.07-1185. [DOI] [PubMed] [Google Scholar]
- 26.Setty BN, Betal SG, Zhang J, Stuart MJ. Heme induces endothelial tissue factor expression: potential role in hemostatic activation in patients with hemolytic anemia. J Thromb Haemost. 2008;6(12):2202–9. doi: 10.1111/j.1538-7836.2008.03177.x. [DOI] [PubMed] [Google Scholar]
- 27.Weitz IC, Ghods M, Rochanda L, Pravazi P, Zwicker J, Furie B, et al. Eculizumab therapy results in rapid and sustained decreases in markers of thrombin generation and inflammation in patients with PNH. Blood. 2008;112(11) doi: 10.1016/j.thromres.2012.04.001. Abstract 156. [DOI] [PubMed] [Google Scholar]
- 28.Marin V, Montero-Julian FA, Gres S, Boulay V, Bongrand P, Farnarier C, et al. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol. 2001;167(6):3435–42. doi: 10.4049/jimmunol.167.6.3435. [DOI] [PubMed] [Google Scholar]
- 29.Dahm A, Van Hylckama Vlieg A, Bendz B, Rosendaal F, Bertina RM, Sandset PM. Low levels of tissue factor pathway inhibitor (TFPI) increase the risk of venous thrombosis. Blood. 2003;101(11):4387–92. doi: 10.1182/blood-2002-10-3188. [DOI] [PubMed] [Google Scholar]
- 30.Sarig G, Blumenfeld Z, Leiba R, Lanir N, Brenner B. Modulation of systemic hemostatic parameters by enoxaparin during gestation in women with thrombophilia and pregnancy loss. Thromb Haemost. 2005;94(5):980–5. doi: 10.1160/TH05-03-0212. [DOI] [PubMed] [Google Scholar]
- 31.Sloand EM, Pfannes L, Scheinberg P, More K, Wu CO, Horne M, Young NS. Increased soluble urokinase plasminogen activator receptor (suPAR) is associated with thrombosis and inhibition of plasmin generation in paroxysmal nocturnal hemoglobinuria (PNH) patients. Exp Hematol. 2008;36(12):1616–24. doi: 10.1016/j.exphem.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ataga KI. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica. 2009;94(11):1481–4. doi: 10.3324/haematol.2009.013672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem. 1989;264(15):9053–60. [PubMed] [Google Scholar]
- 34.Hamilton KK, Ji Z, Rollins S, Stewart BH, Sims PJ. Regulatory control of the terminal complement proteins at the surface of human endothelial cells: neutralization of a C5b-9 inhibitor by antibody to CD59. Blood. 1990;76(12):2572–7. [PubMed] [Google Scholar]
- 35.Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med. 1997;185(9):1619–27. doi: 10.1084/jem.185.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamilton KK, Hattori R, Esmon CT, Sims PJ. Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J Biol Chem. 1990;265(7):3809–14. [PubMed] [Google Scholar]
- 37.Brunn GJ, Saadi S, Platt JL. Differential regulation of endothelial cell activation by complement and interleukin 1alpha. Circ Res. 2006;98(6):793–800. doi: 10.1161/01.RES.0000216071.87981.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Piro O, Lu L, Broze GJ., Jr Glycosyl phosphatidylinositol anchorage of tissue factor pathway inhibitor. Circulation. 2003;108(5):623–7. doi: 10.1161/01.CIR.0000078642.45127.7B. [DOI] [PubMed] [Google Scholar]
- 39.Maroney SA, Cunningham AC, Ferrel J, Hu R, Haberichter S, Mansbach CM, et al. A GPI-anchored co-receptor for tissue factor pathway inhibitor controls its intracellular trafficking and cell surface expression. J Thromb Haemost. 2006;4(5):1114–24. doi: 10.1111/j.1538-7836.2006.01873.x. [DOI] [PubMed] [Google Scholar]
- 40.Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, et al. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med. 2006;12(7):846–51. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]