

Abstract
Background
Additional chromosomal aberrations in Philadelphia chromosome-positive chronic myeloid leukemia are non-random and strongly associated with disease progression, but their prognostic impact and effect on treatment response is not clear. Point mutations in the BCR-ABL kinase domain are probably the most common mechanisms of imatinib resistance.
Design and Methods
We assessed the influence of additional chromosomal aberrations and BCR-ABL kinase domain mutations on the response to the second-generation tyrosine kinase inhibitor nilotinib after imatinib-failure. Standard cytogenetic analysis of metaphases was performed to detect additional chromosomal aberrations and the BCR-ABL kinase domain was sequenced to detect point mutations.
Results
Among 53 patients with a median follow-up of 16 months, of whom 38, 5 and 10 were in chronic phase, accelerated phase and blast crisis, respectively, 19 (36%) had additional chromosomal aberrations and 20 (38%) had BCR-ABL kinase domain mutations. The 2-year overall survival rate of all patients with-out additional chromosomal aberrations (89%) was higher than that of patients with such aberrations (54%) (P=0.0025). Among patients with chronic phase disease, overall survival at 2 years was 100% and 62% for patients without or with additional chromosomal aberrations, respectively (P=0.0024). BCR-ABL kinase domain mutations were associated with lower remission rates in response to nilotinib, with 9 of 20 (45%) of these patients achieving a major cytogenetic remission as compared to 26 of 33 (79%) patients without mutations (P<0.05). However, overall survival was not affected by BCR-ABL kinase domain mutations.
Conclusions
Whereas BCR-ABL kinase domain mutations may confer more specific resistance to nilotinib, which will predominantly affect response rates, the presence of additional chromosomal aberrations may reflect genetic instability and, therefore, intrinsic aggressiveness of the disease which will be less amenable to subsequent alternative treatments and thus negatively affect overall survival. Conventional cytogenetic analyses remain mandatory during follow-up of patients with chronic myeloid leukemia under tyrosine kinase inhibitor therapy.
Keywords: chronic myeloid leukemia, nilotinib, additionl chromosomal aberrations
Introduction
In chronic myeloid leukemia (CML) the pathogenic role of the oncogenic BCR-ABL tyrosine kinase, which results from a reciprocal translocation between chromosomes 9 and 22 to form the Philadelphia chromosome (Ph), sparked the development of the orally active small mole-cule kinase inhibitor, imatinib.1 Unprecedented clinical activity established imatinib as standard first-line therapy for Ph-positive CML and as a successful paradigm for targeted therapy. However, more than 30% of patients with newly diagnosed chronic phase CML discontinued imatinib within 6 years2 and acquired resistance after initial response develops in a substantial proportion of patients. Various mechanisms have been described to cause resistance, but point mutations within the kinase domain of BCR-ABL are probably the clinically most relevant and best characterized.3,4 Several types of mutations, causing varying degrees of resistance in vitro and in vivo, have been described.4
Nilotinib is a second-generation tyrosine kinase inhibitor that was designed to be both more specific and more potent than imatinib in inhibiting the wild-type ABL tyrosine kinase, as well as most kinase domain mutations conferring resistance to imatinib.5,6 Given its significant clinical activity in imatinib-resistant or -intolerant patients with Ph-positive CML, nilotinib has been approved as a second-line agent in chronic and accelerated phases of this disease.7–9 Resistance to nilotinib due to BCR-ABL kinase domain mutations has been predicted from in vitro screens,10–13 but clinical evidence on the relevance of these mutations is still incomplete.
The occurrence of additional chromosomal aberrations (ACA) in Ph-positive CML is strongly associated with disease progression and has been interpreted as both a sign of clonal evolution and chromosomal instability.14 These abnormalities are non-random, with the most common being +8, +Ph, i(17q), +19, −Y, +21, +17, and −7, which have, therefore, been dubbed major route abnormalities. Although their clinical impact is variable,14 in the pre-imatinib era abnormalities involving chromosome 17 seemed to be associated with an inferior prognosis.15 Several studies have assessed the impact of ACA on the clinical efficacy of imatinib under the assumption that these abnormalities may confer BCR-ABL-independent proliferation and decrease sensitivity to imatinib.16–21 Results have been variable, with evidence for an impact on the duration of remission,17 survival,16 risk of hematologic relapse,18 and response.21 However, clonal evolution did not necessarily impair prognosis when considered as the sole criterion for acceleration of disease19 or within separate disease stages.20
Given the clinical importance of predicting resistance to nilotinib we investigated the influence of ACA and BCR-ABL mutations on the clinical efficacy of nilotinib in patients with Ph-positive CML.
Design and Methods
Patients and samples
Between September 2004 and February 2009, 53 patients with Ph-positive CML and imatinib-resistance or -intolerance were treated with nilotinib in our center and are analyzed in this study. Most patients (>90 %) received nilotinib as part of still ongoing clinical trials (registered at Clinicaltrials.gov: NCT00109707 and NCT00905593). Both trials were conducted in accordance with the applicable regulatory requirements. Patients gave their written informed consent to participate in a clinical trial or to have their data analyzed retrospectively. All procedures were performed in accordance with the Helsinki Declaration and approved by the local ethics committee. Patients received nilotinib 400 mg twice daily with dose reductions, as necessary, in the case of toxicity. Patients were followed regularly at intervals of 3 to 6 months. Follow-up included clinical assessment and bone marrow evaluation with metaphase cytogenetics and determination of BCR-ABL transcript levels in bone marrow or peripheral blood samples by quantitative polymerase chain reaction (PCR) analysis. Disease stage and response were determined according to standard criteria. 5,9
Karyotype analysis and fluorescence in situ hybridization
Cytogenetic analysis of metaphases and fluorescence in situ hybridization (FISH) of bone marrow samples were performed according to standard protocols. Results are reported using the International System for Human Cytogenetic Nomenclature. Cytogenetic responses, based on the analysis of at least 20 metaphases, were defined as complete (no Ph-positive metaphases), partial (1–35% Ph-positive metaphases), minor (36–65% Ph-positive metaphases) or minimal (66–95% Ph-positive metaphases). Major cytogenetic responses included complete and partial cytogenetic responses.
BCR-ABL sequencing
In order to detect mutations in the kinase domain of BCR-ABL, RNA from either bone marrow or peripheral blood samples was reverse transcribed and amplified by nested PCR using previously published primers to generate an amplicon covering the entire BCR-ABL kinase domain.22 Sequencing was performed on an ABI PRISM 310 Genetic Analyzer 310 (Applied Biosystems, Foster City, CA, USA). At least one sample was sequenced for each patient: (i) preferentially before the start of nilotinib treatment, and (ii) in each case of disease progression or loss of response.
Statistical analysis
Descriptive statistical analyses were performed using the χ2-test or the Mann-Whitney-test when appropriate. Kaplan-Meier curves were constructed for overall survival and progression-free survival. Differences were analyzed with the log-rank test. Overall survival was calculated from the start of nilotinib therapy to death. Progression-free survival was calculated from the time of the first major cytogenetic response to loss of this response, disease progression to accelerated phase or blast crisis, discontinuation of nilotinib because of resistant disease, or death, whichever occurred first. Patients were censored at the last follow-up. A P-value less than 0.05 was considered statistically significant. All tests were two-sided.
Results
Patients’ characteristics
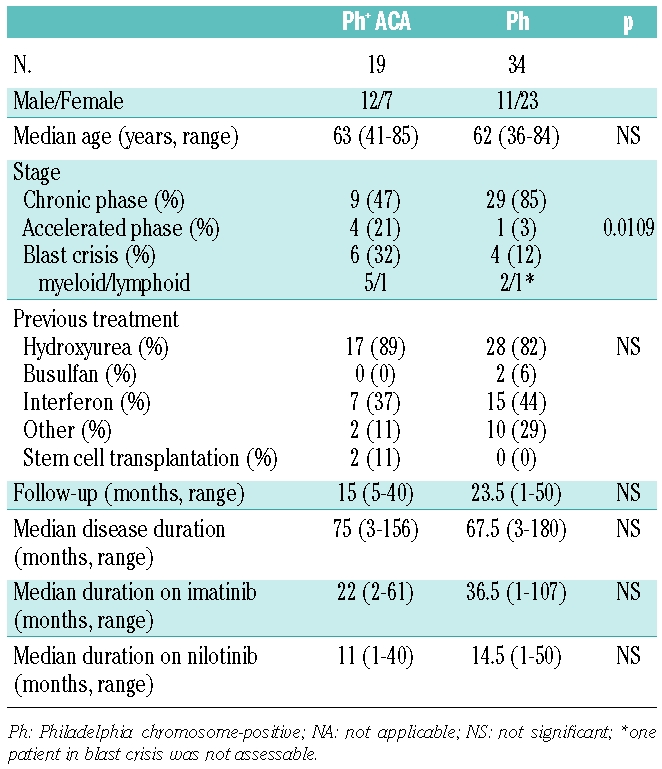
In total, 53 patients (median age, 63 years; range, 36 – 85) with a median follow-up of 16 months (range, 1 – 50) were analyzed (Table 1). Of these, 38, 5 and 10 patients were in chronic phase, accelerated phase and blast crisis, respectively. The median duration of previous treatment with imatinib was 33 months (range, 1 – 107). Prior to imatinib 45 of 53 (85%) had received hydroxyurea and 22 of 53 (42%) interferon. The median duration of nilotinib therapy was 14 months (range, 1 – 50). Thirty-five of the 53 patients (66%) had a major cytogenetic response. Twenty-two of the 53 patients (42%) discontinued nilotinib treatment, mostly (16 of 22, 73%) because of clinical resistance, e.g. loss of response or disease progression. Of these 22 patients, 11 were either in accelerated phase (n=3) or blast crisis (n=8) at baseline. Nine patients received treatment after nilotinib including dasatinib (n=6), INNO-406 (n=4), homoharringtonin (n=1), conventional chemotherapy (n=3) and/or hematopoetic stem cell transplantation (n=2).
Table 1.
Characteristics of patients with or without additional chromosomal aberrations (ACA).
Identification of additional cytogenetic aberrations prior to nilotinib therapy
Cytogenetic analysis on bone marrow metaphases identified ACA in 19 patients (36%), with 44 single abnormalities detected. About half of the ACA (21 of 44, 48%) consisted of the previously described major route aberrations, 14 including +8, +Ph, i(17q), +19, −Y, +21, +17, and −7 (Table 2). Five of the 44 abnormalities involved chromosome 17. The occurrence of ACA was significantly associated with disease stage and was detected in 24%, 80% and 60% of the patients in chronic phase, accelerated phase and blast crisis, respectively (Table 1). There were no significant differences with respect to age, disease duration or duration of imatinib therapy between patients with or with-out ACA. During therapy with nilotinib three patients developed new ACA. After 3 months, one patient in blast crisis with +Ph at baseline developed del(7)(q21), followed by an additional del(6)(q2¿1) 1 month later. Of two other patients in chronic phase without ACA at baseline, one developed der(17)t(17;¿)(p13;¿)del(17)(p13)t(17;¿) (q25;¿) with progression to blast crisis after 6 months and the other +Ph after 7 months. Two patients had chromosomal aberrations in Ph-negative clones at baseline. One patient with +8 is in complete molecular remission at the last follow-up and one patient with two different clones, with +12, −18, +mar and +1, −5, +12, died after 17 months.
Table 2.
Additional chromosomal aberrations detected after imatinib and before nilotinib treatment.

Frequency and spectrum of BCR-ABL kinase domain mutations prior to and during nilotinib therapy
Overall, sequencing identified BCR-ABL kinase domain mutations in 20 of 53 patients (38%, Tables 3 and 4). Most of these patients (14 of 20, 70%) had a single mutation. However, five patients presented with two different mutations (either in a single clone or in two different clones) and one patient had three mutations. Nine of 38 patients (26%) who were assessed at baseline had mutations prior to nilotinib therapy. Of these, four had completely different mutations at later time points with loss of the mutation present at baseline mutation, three acquired mutations in addition to the baseline mutation, one patient (with H396R) lost his mutation and one patient (with E255K) had the same mutation at the end of nilotinib therapy. Six of 53 patients (11%) had more than one mutation at baseline or during follow-up with the following genotypes: M351T/D276G/F359I, Y253H/F359V, Y253F/E255K, G250E/T315I (twice), G250E/F359I, G250E/M351T. One patient had two different combinations at baseline and during follow-up. BCR-ABL point mutations tended to occur more often in patients with ACA (53%) than in patients without ACA (29%). Overall, ten patients had ACA with mutations, nine patients had ACA without mutations, ten patients had mutations without ACA, and 24 patients had neither ACA nor mutations. Mutations predicted to confer resistance to nilotinib with an IC50 of greater than 150 nM23 (Y253H, E255K/V, T315I and F359V) were found in 15 cases. Six had sensitive mutations (IC50 <150 nM) and ten cases had mutations with unknown sensitivity to nilotinib (Table 4).
Table 3.
Relative frequency of BCR-ABL kinase domain mutations detected before and after treatment with nilotinib.
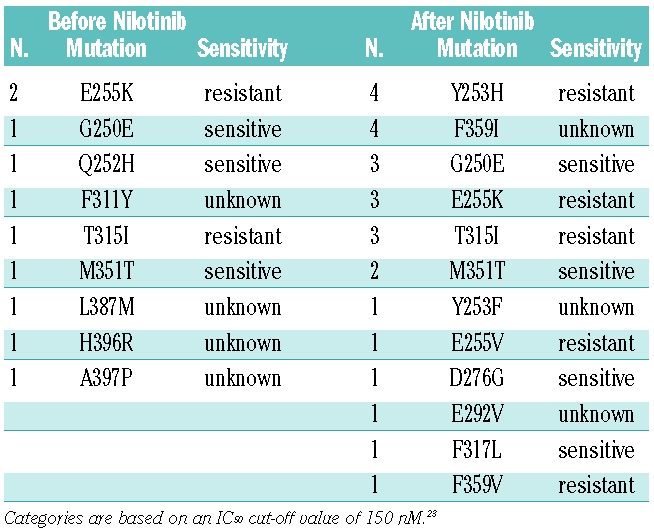
Table 4.
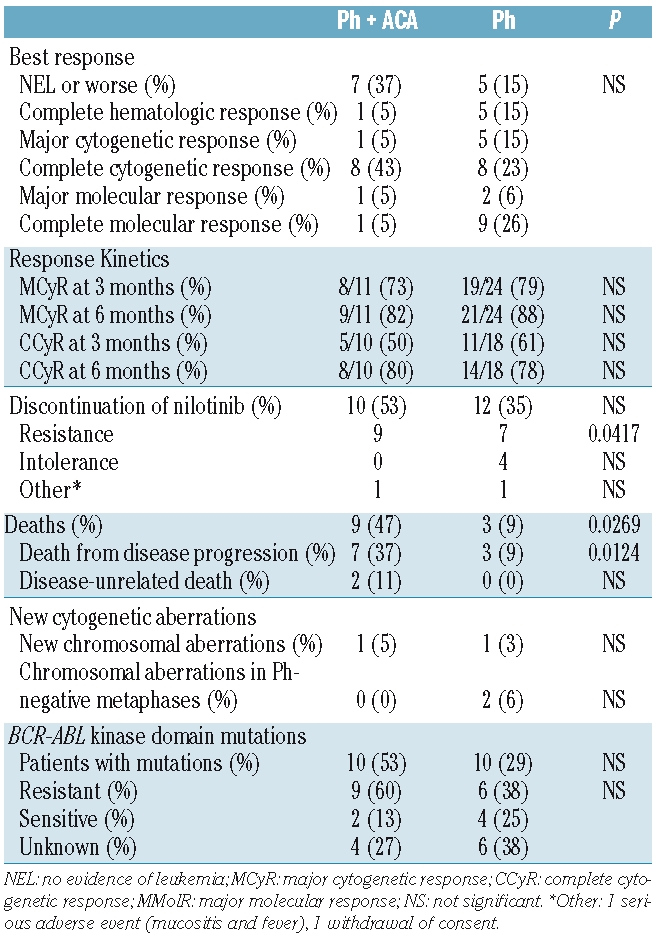
Response to nilotinib and spectrum of BCR-ABL kinase domain mutations in patients with or without additional chromosomal aberrations.
Clinical response to nilotinib in patients with additional chromosome aberrations and BCR-ABL point mutations
Overall, the rate of major cytogenetic response in chronic phase was 74%, and the rate of any response in accelerated phase or blast crisis was 73%. There was no statistically significant difference with respect to response between patients with and without ACA (Table 4). However, more patients with ACA discontinued nilotinib (53% versus 35%), mostly due to resistant disease (47% versus 21%, P<0.05). Three of the five patients with abnormalities of chromosome 17 had resistant disease (60%). Patients with ACA more frequently tended to have BCR-ABL mutations (53% versus 29%) and more resistant mutations (60% versus 38%, Table 4). Patients with mutations were less likely to respond to nilotinib, with 9 of 20 (45%) having a major cytogenetic response as compared to 26 of 33 (79%) patients without kinase domain mutations (P<0.05). None of four patients with T315I had a cytogenetic response. In terms of response kinetics, there was no difference between patients with our without ACA (Table 4) or patients with or without BCR-ABL kinase domain mutations (data not shown).
Additional chromosomal aberrations negatively affect overall survival independently of BCR-ABL kinase domain mutations
The overall survival and progression-free survival of the whole cohort at 2 years was 76% and 58%, respectively. Patients with ACA had significantly worse overall survival (54%) than patients without ACA (89%, P=0.0025, Figure 1A). The progression-free survival rates at 2 years were 37% and 69%, respectively (P=NS). Since the presence of ACA was significantly associated with disease stage, we assessed the influence of ACA for each disease stage. Overall survival at 2 years among patients in chronic phase was 62% and 100% for patients with or without ACA, respectively (P=0.0024) (Figure 1B). In multivariate analysis, only stage was significantly associated with survival, while ACA was not statistically significant (P=0.09, data not shown). Survival was not different for patients with accelerated phase or blast crisis (data not shown). Three patients with ACA (16%) and six patients without ACA (18%) received treatment after nilotinib. Both patients who underwent hematopoietic stem cell transplantation had ACA. One died of disease progression, and one is in complete molecular remission 18 months after transplantation. Abnormalities involving chromosome 17 and duplication of the Philadelphia chromosome did not affect survival. The presence of BCR-ABL kinase domain mutations did not affect survival when analyzed independently or in the context of ACA (Figure 1C). However, when stratified for presumed sensitivity to nilotinib, there was a trend towards an elevated survival with higher sensitivity to nilotinib (data not shown).
Figure 1.

The presence of additional chromosomal aberrations (ACA) negatively affects overall survival. (A) Overall survival of the entire population. (B) Overall survival for patients in chronic phase (CP) only. (C) Overall survival of the entire population stratified for the presence of ACA and BCR-ABL kinase domain mutations.
Discussion
In patients with Ph-positive CML the prediction of resistance to targeted therapy with tyrosine kinase inhibitors may help timely identification of those who would benefit from alternative therapies. Based on clinical response to imatinib, criteria have been established to define failure or suboptimal response in early chronic phase CML.24 Achievement of a cytogenetic response at 3–6 months under treatment with a second generation tyrosine kinase inhibitor was highly predictive for a major cytogenetic response at 12 months and was associated with increased progression-free and overall survival.25 However, these criteria are time-dependent and require observation for several months.
In our series of patients receiving nilotinib after imatinib had failed, we found that the presence of ACA prior to nilotinib treatment predicted worse overall survival. Clinical results with imatinib have been conflicting with variable impact on the duration of remission, survival, risk of hematologic relapse or response.16–21 One reason for the differences between these studies and our findings may be that our study population included patients in whom imatinib had failed and who, therefore, had advanced disease, in which differences may become more apparent. The correlation of ACA with advanced disease stage may have contributed to the worse outcome of patients with ACA. But even among patients in chronic phase, ACA were associated with decreased overall survival which may be attributable to the relatively long disease duration of our chronic phase study cohort. The presence of ACA had no impact on overall survival among patients in accelerated phase or blast crisis (data not shown), perhaps due to the low number of patients in these groups or a true lesser biological importance. The progression-free survival rate was lower among patients with ACA, but the difference was not statistically significant. One possible explanation for this is the higher number of patients in the ACA group who had died without having any response. It is unknown when ACA occurred during imatinib therapy. Indeed, the duration of ACA could be an important factor. However, at present there is no evidence to favor a clinical decision, such as switching treatment or increasing the dose of imatinib, based on the detection of ACA.
ACA were identified in 36% of patients, of which about half were so-called major route aberrations that are found in more than 5% of cases with ACA.14 Several patterns of karyotypic evolution have been correlated with previous treatment. Although limited by the comparatively small number of patients and the heterogeneous treatment before imatinib, there was no increase of unusual aberrations with the possible exception of trisomy 14 which appeared slightly more common than described in the literature. Abnormalities involving chromosome 17 or a duplication of the Philadelphia chromosome were found in five and six cases, respectively, but did not seem to confer a worse prognosis. Chromosomal aberrations in Ph-negative clones have previously been observed after treatment with imatinib,26 dasatinib,27 and nilotinib28 with undetermined significance.
Several mechanisms of resistance to imatinib have been described. However, mutations within the kinase domain of BCR-ABL are probably the clinically most important.4 Although nilotinib has been described to be effective against most mutations conferring resistance to imatinib,23 screening assays were performed to analyze potential resistance mechanisms against this second-generation tyrosine kinase inhibitor.10,11,13 Several nilotinib-resistant mutations that are only partly overlapping with imatinib and dasatinib were identified with the T315I mutation being completely resistant to all currently approved tyrosine kinase inhibitors. In vitro and in vivo, there is evidence that the Y253H, E255K/V and F359C/V mutations are relatively resistant to nilotinib23,29–32 and may negatively affect the response to nilotinib.33 Alternative mechanisms of resistance to nilotinib include the relative insensitivity of CML progenitors,34,35 survival factors,36,37 src kinase overexpression, 38 and the protective bone marrow environment.39
BCR-ABL kinase domain mutations were frequent in our population, which is in agreement with previous results8 and the generally advanced stage after imatinib-failure. Patients with mutations had significantly less response to nilotinib than patients with wild-type BCR-ABL, but this did not translate into worse overall survival. In addition, the presence of BCR-ABL kinase domain mutations did not enable further stratification of patients with ACA. The correlation of single point mutations with clinical outcome is confounded by various issues. Heterogeneous definitions of resistance, e.g. cut-off values for IC50, the assays that these IC50 values are based upon, and the lack of information for a number of mutations must all be taken into account. In addition, correlating the IC50 with Cmax and/or Cmin as in vivo parameters may better reflect the physiological significance of individual mutations. Here, defining resistance with an IC50 cut-off of 150 nM, we found evidence for selection of resistant mutations with nilotinib in vivo. Overall, there were more mutations after nilotinib therapy than before, with a relative increase of resistant mutations. Most (7 of 8) patients with wild-type BCR-ABL at baseline and a point mutation during nilotinib therapy had a resistant mutation, e.g. Y253H, E255K/V or T315I. A recent analysis did not find a worse outcome for patients with T315I after failure of treatment with imatinib or a second generation tyrosine kinase inhibitor.40 However, none of four patients with T315I had a cytogenetic response and three had completely resistant disease and died in blast crisis. One patient retrospectively found to harbor T315I and G250E had lost T315I, while additionally acquiring F359I more than 3 years later, indicating that T315I is not invariably selected during progression. 22 The timing of screening for BCR-ABL kinase domain mutations triggered by rising transcript levels has been addressed in several reports.41–43 However, there are currently no consensus guidelines similar to those for defining failure and suboptimal response in early chronic phase.24 A practical approach to monitoring patients with CML has recently been proposed.44
Despite the relatively small sample size and retrospective nature of this single center analysis, the presence of ACA significantly affected overall survival and to a lesser degree response to nilotinib. However, BCR-ABL kinase domain mutations conferred resistance to nilotinib, as evidenced by fewer cytogenetic remissions, but did not affect survival. One possible explanation for this apparent discrepancy is a direct and rather specific resistance to nilotinib through BCR-ABL point mutations, whereas ACA are evidence for a generally more aggressive disease that may or may not have additional mutations. Obviously, patients in whom nilotinib had failed received subsequent treatment with alternative substances with different resistance profiles, which could circumvent resistance to nilotinib but would address the intrinsic aggressiveness of the disease to a lesser degree.
In conclusion, ACA were frequently detected and were significantly associated with worse overall survival in patients with Ph-positive CML after imatinib treatment had failed. This indicates that conventional cytogenetics on metaphases remains mandatory at diagnosis and during follow-up and should not be replaced by techniques that analyze only BCR-ABL. The decreased efficacy of nilotinib in the context of ACA may reflect intrinsic aggressiveness of the disease that should, therefore, be monitored more closely. Point mutations of the BCR-ABL kinase domain were common and associated with resistant disease. However, differential sensitivities to nilotinib and varying clinical courses with the same mutation hamper clinical decision-making based solely on mutational analysis, with the notable exception of T315I.
Acknowledgments
the authors would like to thank B. Pawlaczyk-Peter, P. Grille and K. Dietze for expert technical assistance and C. Haferlach for critical review of the manuscript.
Footnotes
Authorship and Disclosures
TDK and PlC wrote the paper and analyzed data; ST and CB performed the cytogenetic analysis; PD performed the sequencing; MS, GK, HN and BD analyzed data; all authors approved the paper.
PlC received honoraria from Novartis. No other potential conflicts of interests relevant to this article were reported.
References
- 1.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112(13):4808–17. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 2.Hochhaus A, O'Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23(6):1054–61. doi: 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- 3.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8(11):1018–29. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 4.O'Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242–9. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 5.Kim TD, Dörken B, le Coutre P. Nilotinib for the treatment of chronic myeloid leukemia. Expert Rev Hematol. 2008;1(1):29–39. doi: 10.1586/17474086.1.1.29. [DOI] [PubMed] [Google Scholar]
- 6.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–51. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 8.Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110(10):3540–6. doi: 10.1182/blood-2007-03-080689. [DOI] [PubMed] [Google Scholar]
- 9.le Coutre P, Ottmann OG, Giles F, Kim DW, Cortes J, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–9. doi: 10.1182/blood-2007-04-083196. [DOI] [PubMed] [Google Scholar]
- 10.Bradeen HA, Eide CA, O'Hare T, Johnson KJ, Willis SG, Lee FY, et al. Comparison of imatinib mesylate, dasatinib (BMS- 354825), and nilotinib (AMN107) in an N-ethyl- N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108(7):2332–8. doi: 10.1182/blood-2006-02-004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray A, Cowan-Jacob SW, Manley PW, Mestan J, Griffin JD. Identification of BCR-ABL point mutations conferring resistance to the Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study. Blood. 2007;109(11):5011–5. doi: 10.1182/blood-2006-01-015347. [DOI] [PubMed] [Google Scholar]
- 12.Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol. 2009;27(3):469–71. doi: 10.1200/JCO.2008.19.8853. [DOI] [PubMed] [Google Scholar]
- 13.von Bubnoff N, Manley PW, Mestan J, Sanger J, Peschel C, Duyster J. Bcr-Abl resistance screening predicts a limited spectrum of point mutations to be associated with clinical resistance to the Abl kinase inhibitor nilotinib (AMN107) Blood. 2006;108(4):1328–33. doi: 10.1182/blood-2005-12-010132. [DOI] [PubMed] [Google Scholar]
- 14.Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76–94. doi: 10.1159/000046636. [DOI] [PubMed] [Google Scholar]
- 15.Majlis A, Smith TL, Talpaz M, O'Brien S, Rios MB, Kantarjian HM. Significance of cytogenetic clonal evolution in chronic myelogenous leukemia. J Clin Oncol. 1996;14(1):196–203. doi: 10.1200/JCO.1996.14.1.196. [DOI] [PubMed] [Google Scholar]
- 16.Cortes JE, Talpaz M, Giles F, O'Brien S, Rios MB, Shan J, et al. Prognostic significance of cytogenetic clonal evolution in patients with chronic myelogenous leukemia on imatinib mesylate therapy. Blood. 2003;101(10):3794–800. doi: 10.1182/blood-2002-09-2790. [DOI] [PubMed] [Google Scholar]
- 17.Mohamed AN, Pemberton P, Zonder J, Schiffer CA. The effect of imatinib mesylate on patients with Philadelphia chromosome- positive chronic myeloid leukemia with secondary chromosomal aberrations. Clin Cancer Res. 2003;9(4):1333–7. [PubMed] [Google Scholar]
- 18.O'Dwyer ME, Mauro MJ, Blasdel C, Farnsworth M, Kurilik G, Hsieh YC, et al. Clonal evolution and lack of cytogenetic response are adverse prognostic factors for hematologic relapse of chronic phase CML patients treated with imatinib mesylate. Blood. 2004;103(2):451–5. doi: 10.1182/blood-2003-02-0371. [DOI] [PubMed] [Google Scholar]
- 19.O'Dwyer ME, Mauro MJ, Kurilik G, Mori M, Balleisen S, Olson S, et al. The impact of clonal evolution on response to imatinib mesylate (STI571) in accelerated phase CML. Blood. 2002;100(5):1628–33. doi: 10.1182/blood-2002-03-0777. [DOI] [PubMed] [Google Scholar]
- 20.Schoch C, Haferlach T, Kern W, Schnittger S, Berger U, Hehlmann R, et al. Occurrence of additional chromosome aberrations in chronic myeloid leukemia patients treated with imatinib mesylate. Leukemia. 2003;17(2):461–3. doi: 10.1038/sj.leu.2402813. [DOI] [PubMed] [Google Scholar]
- 21.Palandri F, Testoni N, Luatti S, Marzocchi G, Baldazzi C, Stacchini M, et al. Influence of additional cytogenetic abnormalities on the response and survival in late chronic phase chronic myeloid leukemia patients treated with imatinib: long-term results. Leuk Lymphoma. 2009;50(1):114–8. doi: 10.1080/10428190802492415. [DOI] [PubMed] [Google Scholar]
- 22.Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, et al. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106(6):2128–37. doi: 10.1182/blood-2005-03-1036. [DOI] [PubMed] [Google Scholar]
- 23.Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer. 2006;94(12):1765–9. doi: 10.1038/sj.bjc.6603170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108(6):1809–20. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 25.Tam CS, Kantarjian H, Garcia-Manero G, Borthakur G, O'Brien S, Ravandi F, et al. Failure to achieve a major cytogenetic response by 12 months defines inadequate response in patients receiving nilotinib or dasatinib as second or subsequent line therapy for chronic myeloid leukemia. Blood. 2008;112(3):516–8. doi: 10.1182/blood-2008-02-141580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bumm T, Muller C, Al-Ali HK, Krohn K, Shepherd P, Schmidt E, et al. Emergence of clonal cytogenetic abnormalities in Ph- cells in some CML patients in cytogenetic remission to imatinib but restoration of polyclonal hematopoiesis in the majority. Blood. 2003;101(5):1941–9. doi: 10.1182/blood-2002-07-2053. [DOI] [PubMed] [Google Scholar]
- 27.De Melo VA, Milojkovic D, Khorashad JS, Marin D, Goldman JM, Apperley JF, et al. Philadelphia-negative clonal hematopoiesis is a significant feature of dasatinib therapy for chronic myeloid leukemia. Blood. 2007;110(8):3086–7. doi: 10.1182/blood-2007-05-092437. [DOI] [PubMed] [Google Scholar]
- 28.Baldazzi C, Luatti S, Marzocchi G, Stacchini M, Gamberini C, Castagnetti F, et al. Emergence of clonal chromosomal abnormalities in Philadelphia negative hematopoiesis in chronic myeloid leukemia patients treated with nilotinib after failure of imatinib therapy. Leuk Res. 2009;33(12):e218–20. doi: 10.1016/j.leukres.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 29.Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O'Brien S, et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110(12):4005–11. doi: 10.1182/blood-2007-03-080838. [DOI] [PubMed] [Google Scholar]
- 30.Hochhaus A, Erben P, Branford S, Radich J, Kim DW, Martinelli G, et al. Hematologic and cytogenetic response dynamics to nilotinib (AMN107) depend on the type of BCR-ABL mutations in patients with chronic myelogeneous leukemia (CML) after imatinib failure. ASH Annual Meeting Abstracts; 2006 November 16; 2006. p. 749. [Google Scholar]
- 31.Hughes T, Saglio G, Martinelli G, Kim D-W, Soverini S, Mueller M, et al. Responses and disease progression in CML-CP patients treated with nilotinib after imatinib failure appear to be affected by the BCR-ABL mutation status and types. ASH Annual Meeting Abstracts; 2007 November 16; 2007. p. 320. [Google Scholar]
- 32.Soverini S, Gnani A, Colarossi S, Castagnetti F, Palandri F, Giannoulia P, et al. Philadelphia chromosome-positive leukemia patients who harbor imatinibresistant mutations have a higher likelihood of developing additional mutations associated with resistance to novel tyrosine kinase inhibitors. ASH Annual Meeting Abstracts; 2007 November 16; 2007. p. 322. [Google Scholar]
- 33.Hughes T, Saglio G, Branford S, Soverini S, Kim DW, Muller MC, et al. Impact of baseline BCR-ABL mutations on response to nilotinib in patients with chronic myeloid leukemia in chronic phase. J Clin Oncol. 2009;27(25):4204–10. doi: 10.1200/JCO.2009.21.8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9):4016–9. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 35.Konig H, Holtz M, Modi H, Manley P, Holyoake TL, Forman SJ, et al. Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia. 2008;22(4):748–55. doi: 10.1038/sj.leu.2405086. [DOI] [PubMed] [Google Scholar]
- 36.Belloc F, Airiau K, Jeanneteau M, Garcia M, Guerin E, Lippert E, et al. The stem cell factor-c-KIT pathway must be inhibited to enable apoptosis induced by BCR-ABL inhibitors in chronic myelogenous leukemia cells. Leukemia. 2009;23(4):679–85. doi: 10.1038/leu.2008.364. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Cai D, Brendel C, Barett C, Erben P, Manley PW, et al. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 2007;109(5):2147–55. doi: 10.1182/blood-2006-08-040022. [DOI] [PubMed] [Google Scholar]
- 38.Mahon FX, Hayette S, Lagarde V, Belloc F, Turcq B, Nicolini F, et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008;68(23):9809–16. doi: 10.1158/0008-5472.CAN-08-1008. [DOI] [PubMed] [Google Scholar]
- 39.Dillmann F, Veldwijk MR, Laufs S, Sperandio M, Calandra G, Wenz F, et al. Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL(+) cell to imatinib and nilotinib. Leuk Lymphoma. 2009;50(10):1676–86. doi: 10.1080/10428190903150847. [DOI] [PubMed] [Google Scholar]
- 40.Jabbour E, Kantarjian H, Jones D, Breeden M, Garcia-Manero G, O'Brien S, et al. Characteristics and outcomes of patients with chronic myeloid leukemia and T315I mutation following failure of imatinib mesylate therapy. Blood. 2008;112(1):53–5. doi: 10.1182/blood-2007-11-123950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Branford S, Rudzki Z, Parkinson I, Grigg A, Taylor K, Seymour JF, et al. Real-time quantitative PCR analysis can be used as a primary screen to identify patients with CML treated with imatinib who have BCR-ABL kinase domain mutations. Blood. 2004;104(9):2926–32. doi: 10.1182/blood-2004-03-1134. [DOI] [PubMed] [Google Scholar]
- 42.Press RD, Willis SG, Laudadio J, Mauro MJ, Deininger MW. Determining the rise in BCR-ABL RNA that optimally predicts a kinase domain mutation in patients with chronic myeloid leukemia on imatinib. Blood. 2009;114(13):2598–605. doi: 10.1182/blood-2008-08-173674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Knight K, Lucas C, Clark RE. The role of serial BCR-ABL transcript monitoring in predicting the emergence of BCR-ABL kinase mutations in imatinib-treated patients with chronic myeloid leukemia. Haematologica. 2006;91(2):235–9. [PubMed] [Google Scholar]
- 44.Radich JP. How I monitor residual disease in chronic myeloid leukemia. Blood. 2009;114(16):3376–81. doi: 10.1182/blood-2009-02-163485. [DOI] [PMC free article] [PubMed] [Google Scholar]