

Abstract
Aberrant accumulation of amyloid β (Aβ) oligomers may underlie the cognitive failure of Alzheimer's disease (AD). All species of Aβ peptides are produced physiologically during normal brain activity. Therefore, elucidation of mechanisms that interconnect excitatory glutamatergic neurotransmission, synaptic amyloid precursor protein (APP) processing and production of its metabolite, Aβ, may reveal synapse-specific strategies for suppressing the pathological accumulation of Aβ oligomers and fibrils that characterize AD. To study synaptic APP processing, we used isolated intact nerve terminals (cortical synaptoneurosomes) from TgCRND8 mice, which express a human APP with familial AD mutations. Potassium chloride depolarization caused sustained release from synaptoneurosomes of Aβ42 as well as Aβ40, and appeared to coactivate α-, β- and γ-secretases, which are known to generate a family of released peptides, including Aβ40 and Aβ42. Stimulation of postsynaptic group I metabotropic glutamate receptor (mGluRs) with DHPG (3,5-dihydroxyphenylglycine) induced a rapid accumulation of APP C-terminal fragments (CTFs) in the synaptoneurosomes, a family of membrane-bound intermediates generated from APP metabolized by α- and β-secretases. Following stimulation with the group II mGluR agonist DCG-IV, levels of APP CTFs in the synaptoneurosomes initially increased but then returned to baseline by 10 min after stimulation. This APP CTF degradation phase was accompanied by sustained accumulation of Aβ42 in the releasate, which was blocked by the group II mGluR antagonist LY341495. These data suggest that group II mGluR may trigger synaptic activation of all three secretases and that suppression of group II mGluR signaling may be a therapeutic strategy for selectively reducing synaptic generation of Aβ42.
Introduction
Alzheimer's disease (AD) causes learning and memory dysfunction, leading to dementia, and the illness has been postulated to involve synaptic dysfunction even at preclinical stages. A body of evidence shows that intracellular and/or extracellular accumulation of soluble amyloid β (Aβ) oligomers disrupts normal neuronal plasticity in vitro and in vivo (Wang et al., 2004; Tyszkiewicz and Yan, 2005; Shankar et al., 2008; Li et al., 2009). Aging-dependent accumulation of Aβ oligomers causes neuronal and synaptic damage, eventually leading to degeneration of both (LaFerla et al., 2007).
Formation of Aβ oligomers is dependent on the relative concentrations of a family of monomeric peptides, and the process of production and release of these peptides is regulated by synaptic activity (Kamenetz et al., 2003; Cirrito et al., 2005). In turn, high levels of Aβ oligomers (largely composed of Aβ42) inhibit long-term potentiation (LTP) (Haass and Selkoe, 2007). Curiously, low picomolar levels of Aβ42 can actually enhance LTP (Puzzo et al., 2008), suggesting that precisely controlled production and release of Aβ42 from the synapse is likely to play an important role in regulation of synaptic plasticity. Signal transduction via protein phosphorylation, initiated by neurotransmitters and hormones, is known to modulate total Aβ generation (Gandy et al., 1993; Small and Gandy, 2006), although the molecular bases for translation of signals into protein processing events have remained elusive.
One of the most investigated therapeutic approaches for AD is the deployment of a variety of Aβ-lowering strategies, one of which involves reduction of Aβ production. Among these approaches is the use of memantine, a moderate-affinity blocker of the ionotropic (NMDA) class of glutamate receptors (NMDARs). Several independent studies have shown that memantine can lower amyloid burden and stabilize cognitive functions in amyloid-forming amyloid precursor protein (APP) transgenic mice (Minkeviciene et al., 2004; Scholtzova et al., 2008). Although suppression of NMDAR with memantine or MK-801 was reported to reduce Aβ production and, conversely, application of NMDAR agonist led to greater Aβ production (Alley et al., 2009; Hoe et al., 2009), another recent study has shown that activation of NMDAR stimulates the α-secretase pathway and lowers Aβ generation (Hoey et al., 2009). Further investigation using identical systems and comparing acute and chronic NMDAR activation and blockade will be required to resolve these apparently contradictory results.
Metabotropic glutamate receptor (mGluR) subtype-specific effects on Aβ production have not yet been clarified in detail. There are, however, several studies suggesting that such clarification might offer novel insights into pathogenesis and/or therapy. Application of a general (non-subtype-specific) mGluR agonist to primary neuronal cultures and brain slices has been reported to yield soluble APP secretion, indicating that one or more mGluRs are linked to α-secretase processing of APP (Lee et al., 1995; Kirazov et al., 1997). Also, one mGluR subtype, mGluR5, has been reported to mediate de novo synthesis of APP in synaptoneurosomes (Westmark and Malter, 2007). Herein, also using synaptoneurosomes, we describe a more detailed study of the potential roles that might be played by a panel of mGluR subtypes in the modulation of Aβ metabolism at the synapse.
Materials and Methods
Animals and preparation of synaptoneurosomes.
Synaptoneurosomes were prepared from the cerebral cortices of 10- to 14-d-old heterozygous pups of TgCRND8 mice overexpressing a mutant human APP 695 [“Swedish” K670N/M671L and “Indiana” V717F (Chishti et al., 2001)]. Briefly, mice were decapitated, brains were removed and dissected, and cortices were homogenized in a glass-Teflon homogenizer in homogenizing buffer [50 mm HEPES, pH 7.5, 125 mm NaCl, 100 mm sucrose, 2 mm potassium chloride and protease inhibitor mixture (Pierce)], filtered through a series of nylon mesh filters (149, 62, and 30 μm) (Small Parts) and finally through a 10 μm polypropylene filter (Gelman Sciences). Filters were washed at each step with the homogenizing buffer. The final filtrate was spun briefly (4000 × g, 1 min), and the supernatant was spun (7000 × g, 15 min) to pellet synaptoneurosomes.
Stimulation.
Synaptoneurosomes were resuspended in fresh homogenizing buffer. Before drug treatment, this suspension was stirred and incubated on ice in the presence of 1 μm tetrodotoxin (Tocris Bioscience) for 5 min then at room temperature for another 5 min. Further reactions were also conducted at room temperature. Each synaptoneurosome preparation was divided into smaller pools, which either were not stimulated (controls) or stimulated as described below. Samples were removed and instantly put on ice at 0 min (before adding stimulant), and 1, 3, 5, and 10 min after stimulation. Potassium chloride (KCl) was used at 40 mm, 3,5-dihydroxyphenylglycine (DHPG; Tocris Bioscience) at 100 μm, and 1R,2R-3-[(1S)-1-amino-2-hydroxy-2-oxoethyl-cyclopropane-1,2,dicarboxylic acid (DCG-IV; Tocris Bioscience) at 2 μm. In some experiments, preincubation with 500 nm LY341495 (Tocris Bioscience) was performed for 15 min before adding DCG-IV to selectively block group II mGluRs. Preincubation with the γ-secretase inhibitor N-[(3,5-difluorophenyl)acetyl]-l-alanyl-2-phenyl]glycine-1,1-dimethylethyl ester (DAPT; Tocris Bioscience) at 1 μm was performed for 30 min before adding KCl. Each sample was spun (20,000 × g, 10 min), and the supernatant was collected and designated as the synaptoneurosome “releasate.” The pellet (synaptoneurosomes) was lysed in 0.5% Triton X-100, 150 mm NaCl, 10 mm HEPES, pH 7.4, protease and phosphatase inhibitor mixtures (Pierce). Protein LoBind tubes (Eppendorf) were used for the reactions and sample collections.
Western blot analysis and ELISA.
For each sample, 30 μg of lysed synaptoneurosome proteins was separated in 16% Tricine gels (Invitrogen), blotted to nitrocellulose membranes, and stained with rabbit mAb369 specific for the APP/APLP2 (β-amyloid precursor-like protein) cytoplasmic tail. HRP-labeled secondary anti-rabbit antibody (Cell Signaling Technology) was detected by enhanced chemiluminescence (Pierce). To quantify and standardize protein levels, total protein was detected with Amido Black (Sigma). Chemiluminescence was measured in an LAS-4000 Intelligent Dark Box imager (Fuji Film), and relative optical densities were determined by using AlphaEaseFC software, version 4.0.1 (Alpha Innotech), normalized to total protein loaded (Aldridge et al., 2008). To quantify Aβ levels in the releasate, human Aβ1-40/ 1-42 ELISA kits (Wako) were used according to the manufacturer's instructions.
Statistics.
Student's t tests were used to compare unstimulated controls versus post-treatment differences among secreted Aβ40 and Aβ42, as well as C83 (α-) and C99 (β-) C-terminal fragments (CTFs). p < 0.05 was considered significant.
Results
Synaptoneurosome preparations provide an excellent model to study the events occurring at the synapses under a variety of physiological conditions and have been used extensively in the study of regulation of neurotransmitter release. Synaptoneurosome preparations contain a population of highly purified and resealed presynaptic processes attached to resealed postsynaptic processes (Hollingsworth et al., 1985). Both presynaptic and postsynaptic regions retain the ability to display many of the molecular effects that characterize their counterparts in the intact brain, including neurotransmitter release, receptor-mediated signal transduction, and protein synthesis (Weiler et al., 1997). Generation of Aβ is somewhat more complex than the typical application of synaptoneurosomes, since APP metabolism is a multistep process, involving transmembrane substrates and a series of transmembrane proteases, all of which are subject to tight regulation of their subcellular localization and sorting (Caporaso et al., 1994).
To study the generation of Aβ at the synapse, we measured the synaptoneurosomal-associated α- and β-CTFs of APP and, in their releasates, Aβ peptides. In our initial characterization of regulated Aβ generation, we used 40 mm KCl to cause potassium depolarization of cortical synaptoneurosomes from 10- to 14-d-old TgCRND8 mice. This depolarization induces opening of L-type Ca2+ channels, which, in turn, conduct the entry of Ca2+ into synaptoneurosomes. Levels of both C83 and C99 in the synaptoneurosomes dropped after depolarization, suggesting that KCl stimulation caused rapid activation of γ-secretase in synaptoneurosomes (Fig. 1 A). To examine whether KCl depolarization activated α-secretase and/or β-secretase as well, we preincubated synaptoneurosomes with γ-secretase inhibitor DAPT and then applied KCl. Under these conditions, in contrast with what we observed with potassium depolarization and no DAPT, we observed rapid accumulation of C83 and C99, suggesting that potassium depolarization also activates α- and β-secretases (Fig. 1 A). All three secretases were activated at apparently similar initial velocities after KCl stimulation. KCl depolarization triggered rapid accumulation of Aβ40 and Aβ42 in the releasate, as one would predict since these peptides are known to be released at the synapse (Kamenetz et al., 2003) (Fig. 1 B). In unstimulated synaptoneurosomes (controls), we did not see any changes in synaptosomal levels of C83 or C99 or in releasate levels of Aβ40 and Aβ42 during the experiments, consistent with the formulation that secretase activity was negligible or undetectable in the absence of stimulation.
Figure 1.

Synaptic APP processing following KCl depolarization. A, Cortical synaptoneurosomes from 10- to 14-d-old TgCRND8 mice were either stimulated by 40 mm KCl with no preincubation or stimulated by 40 mm KCl following a preincubation with 1 μm DAPT (γ-secretase inhibitor). After KCl stimulation with no preincubation, a rapid reduction in C99 and C83 levels was observed. DAPT pretreatment followed by KCl stimulation caused rapid accumulation of C99 (β-CTF) and C83 (α-CTF). Overall, this experiment shows that KCl depolarization coactivates α-, β-, and γ-secretases at the synapse. The representative Western blot images are from the KCl only (the upper bands in the doublets are phosphorylated C83 and C99) and the DAPT + KCl experiments. Control samples were collected from an unstimulated pool at 0 min (ctrl 0′) and 10 min (ctrl 10′). The values were normalized to the baseline (t = 0). n = 6 for each. B, Levels of Aβ40 and Aβ42 in the releasate were measured using ELISA. Both Aβ peptides were released from synaptoneurosomes after KCl depolarization. The values were normalized to the baseline (t = 0). n = 7 for Aβ40; n = 8 for Aβ42. Data are presented as means ± SEM. *p < 0.05; **p < 0.01; # p < 0.001; ## p < 0.0001.
mGluRs have been classified into three groups and eight subtypes according to (1) their respective second messenger cascades, (2) the specificity of various agonist ligands, and (3) the similarity of their sequences (Pin and Duvoisin, 1995). Group I mGluRs (mGluR1 and mGluR5) are predominantly located postsynaptically, and their activation causes phospholipase C to hydrolyze phosphoinositide phospholipids (Schoepp et al., 1999). We stimulated synaptoneurosomes with 100 μm DHPG, a specific agonist for the group I mGluR. We observed rapid accumulation of C83 and C99 in synaptoneurosomes (Fig. 2 A), consistent with the formulation that stimulation of group I mGluR activates α- and β-secretases but may not activate detectable γ-secretase activity in the postsynaptic neuron. DHPG stimulation triggered release of Aβ40 but not of Aβ42 (Fig. 2 B).
Figure 2.
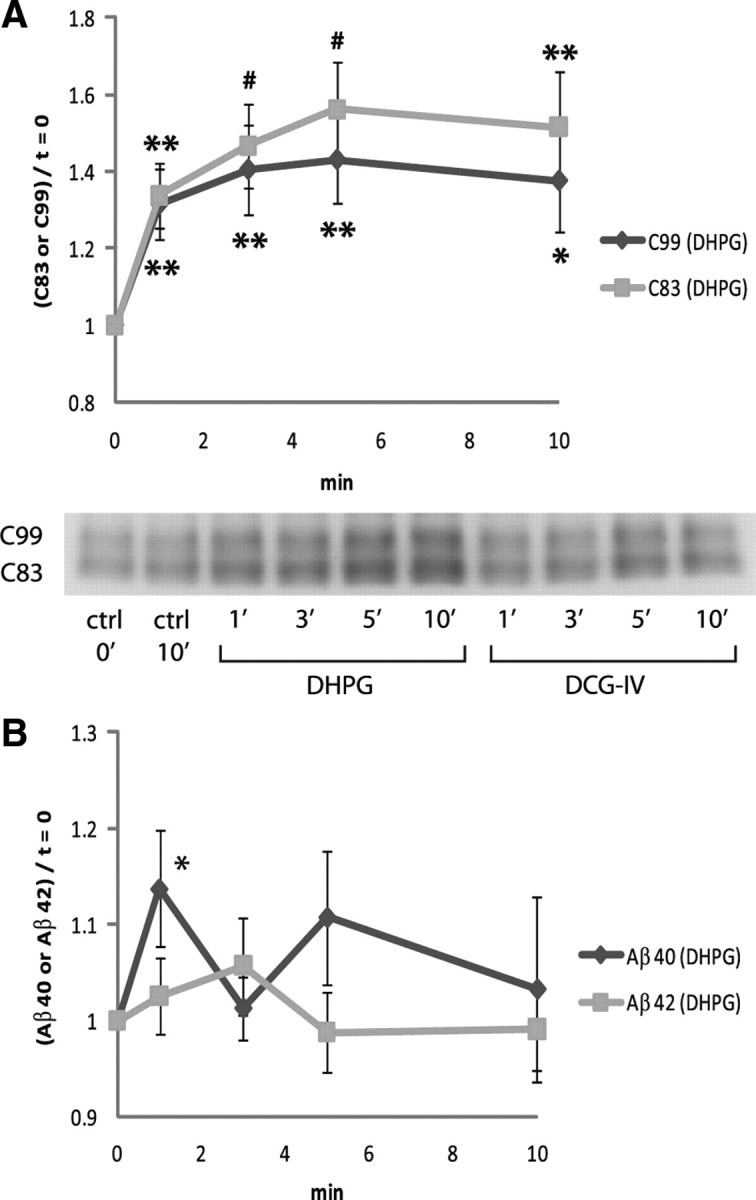
Group I mGluR-mediated APP processing in the synapse. A, Cortical synaptoneurosomes were stimulated with 100 μm DHPG (group I mGluR agonist), and synaptoneurosome-associated CTF levels were measured by Western blot. DHPG stimulation caused rapid accumulation of C83 and C99, indicating activation of α- and β-secretases. In the representative Western blot image, “DCG-IV”-labeled bands are for Figure 3 A. The values were normalized to the baseline (t = 0). n = 7 for each. B, Group I mGluR stimulation triggered release of Aβ40 but not Aβ42 from synaptoneurosomes. The values were normalized to the baseline (t = 0). n = 4 for Aβ40; n = 8 for Aβ42. Data are presented as means ± SEM. *p < 0.05; **p < 0.01; # p < 0.001.
Group II mGluRs (mGluR2 and mGluR3) are located in the presynaptic and postsynaptic elements, and presynaptic group II mGluRs are believed to act as autoreceptors that modulate glutamate release (Tamaru et al., 2001; Pinheiro and Mulle, 2008). Stimulation of group II mGluRs with a specific agonist downregulates cAMP formation while activating the MAP (mitogen-activated protein) kinase and PI-3 (phosphatidylinositol-3) kinase pathways (Phillips et al., 1998; Ferraguti et al., 1999). We used 2 μm DCG-IV to stimulate group II mGluR on synaptoneurosomes. Levels of C99 increased transiently but then fell, consistent with the formulation that group II mGluR stimulation successively activates first β-secretase and then γ-secretase (Fig. 3 A). However, DCG-IV stimulation was associated with a sustained increase in C83, indicating increased action of α-secretase on APP but no increase in γ-secretase activity on C83 (Fig. 3 A). This differential action of γ-secretase may be a function of the subcellular localization of C83 (generated at the plasma membrane) vs C99 (generated at the trans-Golgi network and in endosomes) (Skovronsky et al., 2000). Interestingly, DCG-IV induced sustained release of Aβ42 but only transient release of Aβ40 (Fig. 3 B). To confirm that group II mGluR signaling is preferentially linked to synaptic Aβ42 production and release, we preincubated synaptoneurosomes with 500 nm LY341495, a group II mGluR antagonist, before stimulating those receptors with DCG-IV. Inhibition of group II mGluR completely abolished DCG-IV-induced Aβ42 release from synaptoneurosomes (Fig. 3 C).
Figure 3.
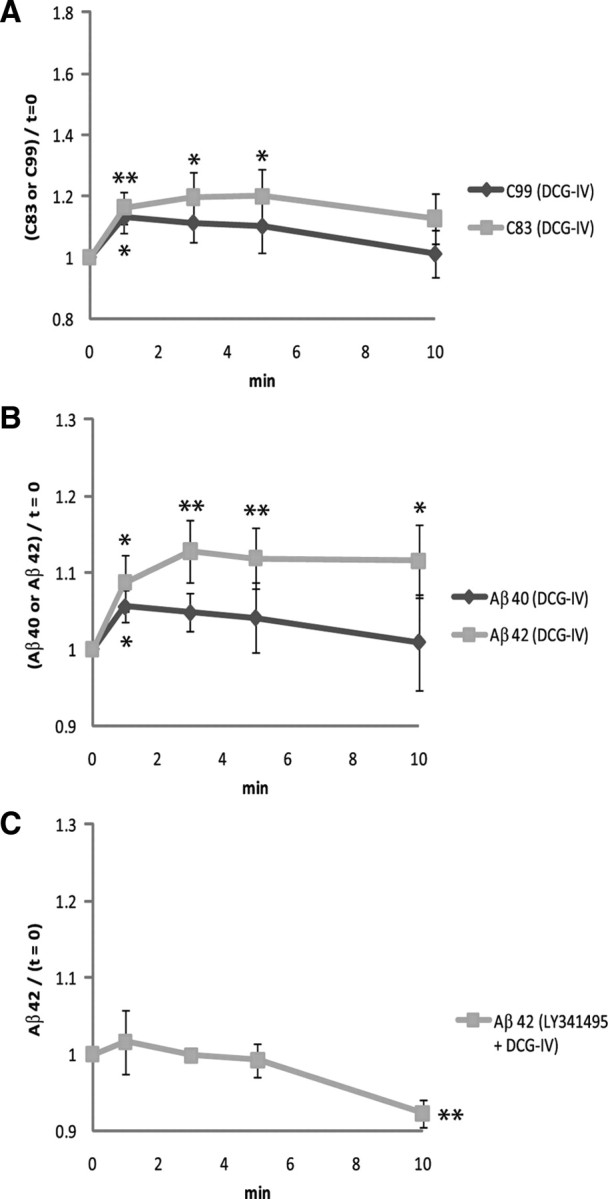
Group II mGluR stimulation is preferentially linked to synaptic Aβ42 production and release. A, Group II mGluRs on cortical synaptoneurosomes were stimulated with 2 μm DCG-IV, and then levels of synaptosomal C83 and C99 were measured by Western blots. Rapid accumulation of C83 and C99 at t = 1′ in response to DCG-IV shows that both α- and β-secretase pathways were activated. Differential γ-secretase cleavage of C99 vs C83 probably represented the differential subcellular localization of each CTF. The representative Western blot picture is shown in Figure 2 A. The values were normalized to the baseline (t = 0). n = 7 for each. B, DCG-IV triggered sustained accumulation of Aβ42 in the releasate but only transient release of Aβ40. The values were normalized to the baseline (t = 0). n = 5 for Aβ40; n = 8 for Aβ42. C, Preincubation with 500 nm LY341495 (group II mGluR antagonist) suppressed DCG-IV-induced Aβ42 release from synaptoneurosomes (compare with the Aβ42 graph in B). The values were normalized to the baseline (t = 0). n = 4. Data are presented as means ± SEM. *p < 0.05; **p < 0.01.
Discussion
Increasing evidence indicates that Aβ peptide is generated in response to synaptic activity (Buxbaum et al., 1993; Fazeli et al., 1994; Kamenetz et al., 2003; Cirrito et al., 2005). Since most excitatory synaptic transmission is mediated by glutamate receptors, we reasoned that a detailed understanding of the regulation of brain Aβ metabolism was incomplete without an elucidation of which glutamate receptor subtype(s) regulates Aβ release from the nerve terminals. In the present study, we show that activation of postsynaptically concentrated group I mGluR leads to Aβ40 release, and that group II mGluR stimulation triggers production and sustained release of Aβ42 peptide as well as transient release of Aβ40. This differential effect on Aβ speciation was not anticipated but may well be important to pathogenesis and therapy, since one action of Aβ40 appears to be maintenance of Aβ42 solubility (Kim et al., 2007). Simply increasing Aβ40 was reported to decrease Aβ deposition by 60–90% in vivo (Giuffrida et al., 2009). Therefore, physiologically relevant, subtle changes in the Aβ42/ 40 ratio are not only possible but likely.
Generation of Aβ at the nerve terminal is a complex event involving both endocytosis and release (Cirrito et al., 2008). The cell-free reconstitution of such events is frequently inefficient. For example, cell-free reconstitution of ricin endocytosis typically demonstrates a maximum fold increase of 20%, with an occasional fold increase of 30% (Bilge et al., 1995). Therefore, the effects that we observe on modulation of Aβ release (10–20% changes) and modulation of APP CTF metabolism (10–60% changes) in synaptoneurosomes are well within the expected ranges for fold effects observed in other examples of cell-free reconstitution of complex cell biological events.
The existing literature on the possible roles of mGluR subtypes in the pathogenesis of Alzheimer's dementia is fairly limited. One of the group II mGluR subtypes, mGluR2, was reported to be overexpressed in the hippocampus of Alzheimer's disease patients compared with age-matched control cases (Lee et al., 2004). Interestingly, neurofibrillary tangles (phosphorylated tau) were colocalized in brain regions enriched in neurons that overexpress mGluR2; excessive mGluR2 stimulation leads to dysregulated activation of ERK, which then directly phosphorylates tau (Lee et al., 2009). Alternatively, group I mGluR-linked phospholipase C activity is downregulated in the cerebral cortex of Alzheimer's patients (Albasanz et al., 2005). Taken in the context of our data, one possible scenario for AD pathogenesis would involve upregulated group II mGluR signaling causing increased synaptic Aβ42 generation and/or downregulated group I mGluR signaling causing decreased synaptic Aβ40 generation.
APP has been shown to be rapidly translated in the synapse in response to activation of mGluR5, one of group I mGluRs (Westmark and Malter, 2007). We confirmed that APP mRNA is abundant in FACS-sorted purified synaptoneurosomes, including the transfected mutant human APP mRNA and endogenous murine mRNA (data not shown). APP in the postsynaptic density was reported to control NMDAR function by regulating its trafficking and subunit composition (Hoe et al., 2009). Interestingly, DHPG triggered rapid production and accumulation of CTFs (C83, C99) without subsequent significant activation of γ-secretase in the cortical synaptoneurosomes, suggesting physiological roles of an APP-dependent pathway in cortical synaptic plasticity. It remains to be determined whether DHPG-induced full-length CTFs have their own as-yet-unidentified functions, or whether they are always eventually processed by γ-secretase to generate bioactive metabolites (i.e., AICD, p3, Aβ). A recent study showed that γ-secretase inhibition in vivo for 4 d caused reduction in spine density and suggested that accumulation of C83 and/or C99 might be toxic to dendritic spine maturation (Bittner et al., 2009).
We propose that suppressing group II mGluR signaling might be a viable prophylactic or therapeutic strategy for AD via the following mechanisms. (1) Group II mGluR inhibitors enhance hippocampus-dependent cognitive function (Higgins et al., 2004) and have antidepressant-like effects in rodents (Chaki et al., 2004; Kawashima et al., 2005); AD patients exhibit early hippocampus-dependent episodic memory decline (Souchay et al., 2002; Starr et al., 2005), and nearly half of all AD patients are depressed (Levy et al., 1996; Mega et al., 1996). (2) Chromogranin A, which is overexpressed in AD brain, induces the potentially neurotoxic activation of microglia, while inhibition of microglial group II mGluRs can block chromogranin A-induced microglial activation and is beneficial for neuronal survival (Taylor et al., 2002). (3) Neurofibrillary tangles composed of hyperphosphorylated tau might be attenuated by a group II mGluR antagonist since one possible cause of tau hyperphosphorylation in the AD hippocampus involves overactivated group II mGluRs (Lee et al., 2004, 2009). (4) Group II mGluR antagonist increased cell proliferation in the adult mouse hippocampus (Yoshimizu and Chaki, 2004), and increasing neurogenesis has been proposed to be a possible therapeutic strategy for AD (Jin et al., 2004). Based on our data, we speculate that group II mGluR inhibition might decrease synaptic production and release of Aβ42 while increasing Aβ40 due to the elevated postsynaptic activity (Cirrito et al., 2008). Further explorations of mGluR-mediated pathogenesis of AD may reveal novel targets for the prophylaxis and/or therapy of this disease.
Footnotes
This work was supported by National Institutes of Health Grant AG10491, Cure Alzheimer's Fund, Canadian Institutes of Health Research, the Alzheimer Society of Ontario, the Alberta Prion Research Institute, the Canada Foundation for Innovation, Howard Hughes Medical Institute, and The Wellcome Trust. We thank Loren Khan-Vaughan and Justine Bonet for assistance with mouse colony management.
References
- Albasanz JL, Dalfó E, Ferrer I, Martín M. Impaired metabotropic glutamate receptor/phospholipase C signaling pathway in the cerebral cortex in Alzheimer's disease and dementia with Lewy bodies correlates with stage of Alzheimer's-disease-related changes. Neurobiol Dis. 2005;20:685–693. doi: 10.1016/j.nbd.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J Neurosci Methods. 2008;172:250–254. doi: 10.1016/j.jneumeth.2008.05.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, Banerjee PK, Lahiri DK. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J Neurosci Res. 2010;88:143–154. doi: 10.1002/jnr.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilge A, Warner CV, Press OW. Translocation of ricin A-chain into proteoliposomes reconstituted from Golgi and endoplasmic reticulum. J Biol Chem. 1995;270:23720–23725. doi: 10.1074/jbc.270.40.23720. [DOI] [PubMed] [Google Scholar]
- Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J. γ-Secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci. 2009;29:10405–10409. doi: 10.1523/JNEUROSCI.2288-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Koo EH, Greengard P. Protein phosphorylation inhibits production of Alzheimer amyloid beta/A4 peptide. Proc Natl Acad Sci U S A. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P. Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer β/A4 amyloid precursor protein. J Neurosci. 1994;14:3122–3138. doi: 10.1523/JNEUROSCI.14-05-03122.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, Yoshimizu T, Yasuhara A, Sakagami K, Okuyama S, Nakanishi S, Nakazato A. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46:457–467. doi: 10.1016/j.neuropharm.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazeli MS, Breen K, Errington ML, Bliss TV. Increase in extracellular NCAM and amyloid precursor protein following induction of long-term potentiation in the dentate gyrus of anaesthetized rats. Neurosci Lett. 1994;169:77–80. doi: 10.1016/0304-3940(94)90360-3. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Baldani-Guerra B, Corsi M, Nakanishi S, Corti C. Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur J Neurosci. 1999;11:2073–2082. doi: 10.1046/j.1460-9568.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- Gandy SE, Caporaso GL, Buxbaum JD, de Cruz Silva O, Iverfeldt K, Nordstedt C, Suzuki T, Czernik AJ, Nairn AC, Greengard P. Protein phosphorylation regulates relative utilization of processing pathways for Alzheimer beta/A4 amyloid precursor protein. Ann N Y Acad Sci. 1993;695:117–121. doi: 10.1111/j.1749-6632.1993.tb23038.x. [DOI] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. β-Amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Ballard TM, Kew JN, Richards JG, Kemp JA, Adam G, Woltering T, Nakanishi S, Mutel V. Pharmacological manipulation of mGlu2 receptors influences cognitive performance in the rodent. Neuropharmacology. 2004;46:907–917. doi: 10.1016/j.neuropharm.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DT, Rebeck GW. The effects of amyloid precursor protein on postsynaptic composition and activity. J Biol Chem. 2009;284:8495–8506. doi: 10.1074/jbc.M900141200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates α-secretase amyloid precursor protein processing and inhibits amyloid-β production. J Neurosci. 2009;29:4442–4460. doi: 10.1523/JNEUROSCI.6017-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR. Biochemical characterization of a filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci. 1985;5:2240–2253. doi: 10.1523/JNEUROSCI.05-08-02240.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Karasawa J, Shimazaki T, Chaki S, Okuyama S, Yasuhara A, Nakazato A. Neuropharmacological profiles of antagonists of group II metabotropic glutamate receptors. Neurosci Lett. 2005;378:131–134. doi: 10.1016/j.neulet.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Aβ40 inhibits amyloid deposition in vivo . J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirazov L, Löffler T, Schliebs R, Bigl V. Glutamate-stimulated secretion of amyloid precursor protein from cortical rat brain slices. Neurochem Int. 1997;30:557–563. doi: 10.1016/s0197-0186(96)00119-2. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lee HG, Ogawa O, Zhu X, O'Neill MJ, Petersen RB, Castellani RJ, Ghanbari H, Perry G, Smith MA. Aberrant expression of metabotropic glutamate receptor 2 in the vulnerable neurons of Alzheimer's disease. Acta Neuropathol. 2004;107:365–371. doi: 10.1007/s00401-004-0820-8. [DOI] [PubMed] [Google Scholar]
- Lee HG, Zhu X, Casadesus G, Pallàs M, Camins A, O'Neill MJ, Nakanishi S, Perry G, Smith MA. The effect of mGluR2 activation on signal transduction pathways and neuronal cell survival. Brain Res. 2009;1249:244–250. doi: 10.1016/j.brainres.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RK, Wurtman RJ, Cox AJ, Nitsch RM. Amyloid precursor protein processing is stimulated by metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 1995;92:8083–8087. doi: 10.1073/pnas.92.17.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ML, Cummings JL, Fiorello T, Gornbein J. The spectrum of behavioral changes in Alzheimer's disease. Neurology. 1996;46:130–135. doi: 10.1212/wnl.46.1.130. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mega MS, Miller BL, Cummings JL, Fairbanks LA, Craig A. Alzheimer disease and frontotemporal dementias: behavioral distinctions. Arch Neurol. 1996;53:687–690. doi: 10.1001/archneur.1996.00550070129021. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Banerjee P, Tanila H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2004;311:677–682. doi: 10.1124/jpet.104.071027. [DOI] [PubMed] [Google Scholar]
- Phillips T, Barnes A, Scott S, Emson P, Rees S. Human metabotropic glutamate receptor 2 couples to the MAP kinase cascade in chinese hamster ovary cells. Neuroreport. 1998;9:2335–2339. doi: 10.1097/00001756-199807130-00034. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Pinheiro PS, Mulle C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat Rev Neurosci. 2008;9:423–436. doi: 10.1038/nrn2379. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, Banerjee P, Wisniewski T. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer's-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res. 2008;86:2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souchay C, Isingrini M, Gil R. Alzheimer's disease and feeling-of-knowing in episodic memory. Neuropsychologia. 2002;40:2386–2396. doi: 10.1016/s0028-3932(02)00075-1. [DOI] [PubMed] [Google Scholar]
- Starr JM, Loeffler B, Abousleiman Y, Simonotto E, Marshall I, Goddard N, Wardlaw JM. Episodic and semantic memory tasks activate different brain regions in Alzheimer disease. Neurology. 2005;65:266–269. doi: 10.1212/01.wnl.0000168907.44632.55. [DOI] [PubMed] [Google Scholar]
- Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/s0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Cuzner ML, Pocock JM. Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer's disease. J Neurochem. 2002;82:1179–1191. doi: 10.1046/j.1471-4159.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- Tyszkiewicz JP, Yan Z. beta-Amyloid peptides impair PKC-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J Neurophysiol. 2005;93:3102–3111. doi: 10.1152/jn.00939.2004. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimizu T, Chaki S. Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem Biophys Res Commun. 2004;315:493–496. doi: 10.1016/j.bbrc.2004.01.073. [DOI] [PubMed] [Google Scholar]