

Abstract
The repair of DNA double-strand breaks (DSB) is tightly regulated during the cell cycle. In G1 phase, the absence of a sister chromatid means that repair of DSB occurs through non-homologous end-joining (NHEJ) or microhomology-mediated end-joining (MMEJ)1. These pathways often involve loss of DNA sequences at the break site and are therefore error-prone. In late S and G2 phases, even though DNA end-joining pathways remain functional2, there is an increase in repair of DSB by homologous recombination (HR), which is mostly error-free3,4. Consequently, the relative contribution of these different pathways to DSB repair in the cell cycle has a profound influence on the maintenance of genetic integrity. How then are DSB directed for repair by different, potentially competing, repair pathways? Here we identify a role for CtIP in this process in DT40. We establish that CtIP is not only required for repair of DSB by HR in S/G2 phase, but also for MMEJ in G1. The function of CtIP in HR, but not MMEJ, is dependent on the phosphorylation of serine residue 327 and recruitment of BRCA1. Cells expressing CtIP protein that cannot be phosphorylated at serine 327 are specifically defective in HR and exhibit decreased level of single-stranded DNA (ssDNA) after DNA damage, while MMEJ remains unaffected. Our data support a model in which phosphorylation of serine 327 of CtIP as cells enter S-phase and the recruitment of BRCA1 functions as a molecular switch to shift the balance of DSB repair from error-prone DNA end-joining to error-free homologous recombination (Supplementary Fig. 1).
Since NHEJ, MMEJ and HR pathways all operate in S and G2 phases, the repair of a DSB by a particular pathway is not determined simply by limiting the availability of specific repair factors to defined phases of the cell cycle. We reasoned that this choice is more likely established by proteins that function in the initial steps of DSB repair, involving the recognition and processing of DNA ends. Moreover, that a potential candidate for this role is CtIP, which is known to promote resection of DNA ends to the single-stranded DNA (ssDNA) tails that are essential for HR5,6.
Analysis of CtIP in mammalian cells is difficult as homozygous deletion of CtIP in mice results in early embryonic lethality7. To circumvent this problem we generated ctip null mutant cells from the avian B cell line DT40. In DT40 CtIP is present in three copies due to a chromosomal duplication of chromosome 28. We disrupted all three CtIP alleles, deleting exons 1 and 2, containing the initiation codon and 5′ untranscribed region of the gene, and replaced them with antibiotic resistance cassettes (Supplementary Fig. 2a). We confirmed the generation of ctip (-/-/-) mutant cells by Southern blot and the loss of CtIP protein expression by Western blot (Supplementary Fig. 2b,c).
In common with other DNA repair-defective mutants9,10, ctip cells exhibit reduced proliferation rate, compared with wild-type cells (Supplementary Fig. 3a). Moreover, in clonogenic survival assays they are highly sensitive to X-rays, which cause DSBs (Fig. 1a). They are also sensitive to cisplatin (CDDP), which generates interstrand DNA crosslinks that may also lead to the generation of DSB during replication (Fig. 1a). In contrast, ctip cells are not very sensitive to UV light, which causes pyrimidine dimers and 6-4 photoproducts11 (Fig. 1a). Importantly, expression of human CtIP (HsCtIP) in ctip mutant cells fully restored the resistance of these cells to X-rays, confirming that the human and avian CtIP proteins are functionally conserved (Fig. 1a, Supplementary Fig. 2d).
Figure 1. Sensitivity of ctip mutant cells to DNA damaging agents.

Clonogenic survival assays with asynchronous cell populations after exposure to X-rays (upper panel), cisplatin (CDDP, middle panel) and ultraviolet light (UV, lower panel). In this and the following figures, the data presented with error bars are the mean of three independent experiments, and the error bar indicates one standard deviation.
It was reported that CtIP promotes resistance to DSB-inducing agents exclusively during S and G2 phases5,12. Accordingly, ctip cells isolated by elutriation in S/G2 phase (Supplementary Fig. 4), are 5- to 6-fold more sensitive to X-ray damage (LD10 = 2.5 Gy) than are wild-type cells and approximately 2-fold more sensitive than NHEJ-defective ku70 mutant cells (Fig. 2a, upper panel). Surprisingly, ctip cells isolated in G1 phase are also sensitive to X-ray-induced DNA damage (LD10 = 2.2 Gy) (Fig. 2a, lower panel), suggesting that CtIP function is not limited to S/G2 phase, as previously proposed, but contributes to the repair of DSB throughout the cell cycle. And, since HR does not function in G1 phase, CtIP must be involved in a second pathway for repairing DSBs.
Figure 2. ctip mutant cells are sensitive to X-rays in both G1 and S/G2 phases of the cell cycle.
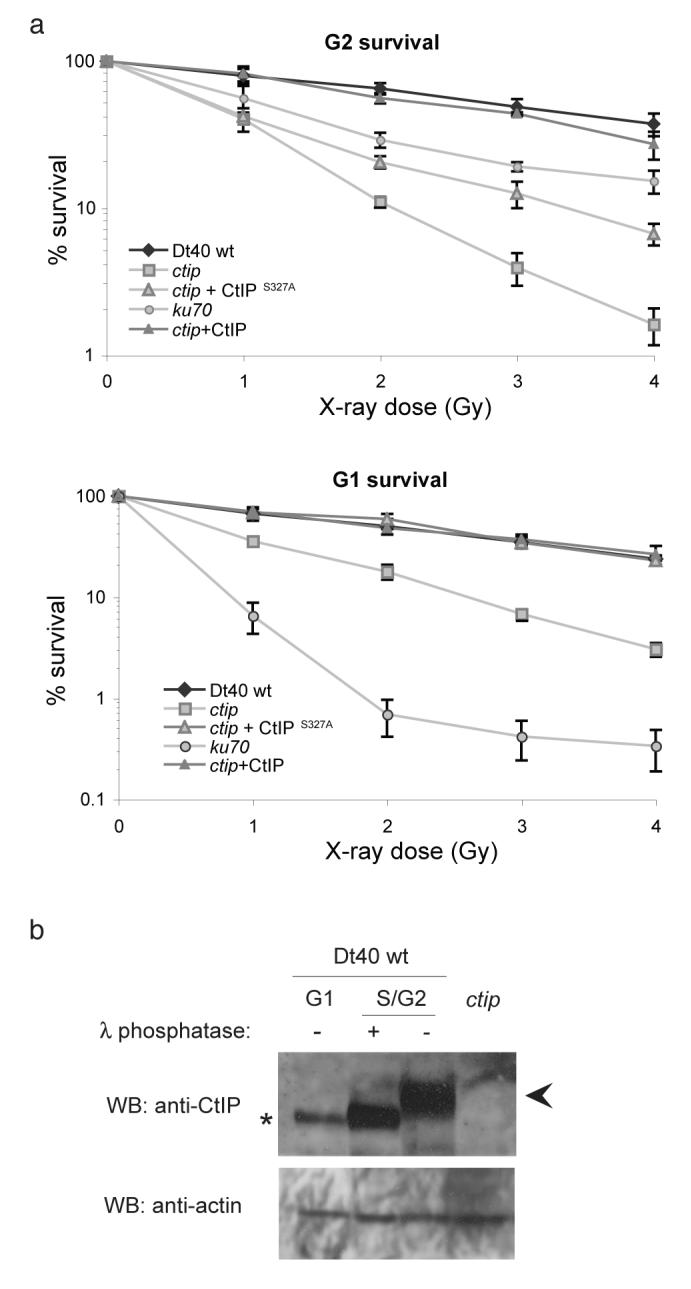
(a) Clonogenic colony survival assay after exposure of cell lines to X-rays, synchronised by elutriation in either S/G2 (upper panel) or G1 (lower panel) stages of the cell cycle. Data represent three independent experiments. (b) Western-blot showing the presence of CtIP in G1 and S/G2. Phosphorylated CtIP is indicated with an arrow. Only unphosphorylated CtIP (*) is seen in G1. Whole cell extracts were prepared from elutriated cell poulations. Where indicated, S/G2 cell extract were treated with λ phosphatase for 2 h at 30°C. Western blot with antibody against actin was used as protein loading control.
The fact that CtIP is required for repair of X-ray-induced DSBs in G1 and S/G2 phases raised the possibility that it functions commonly in both HR and DNA end-joining. To address this we made use of several GFP-reporter assays that measure the repair of a restriction enzyme-induced DSB by different repair pathways (Supplementary Fig. 5).
For HR we measured the repair of a defined I-SceI-induced genomic DSB in a defective GFP reporter gene (Sce-GFP13) (Supplementary Fig. 5). In line with previous studies7 we observed a 10-fold defect in HR for ctip mutant cells compared to wild-type cells (Fig. 3a). A second form of homology directed DNA repair is single-strand annealing (SSA). This occurs when a DSB is generated between two directly repeated sequences and is achieved by resection of DNA ends to produce homologous ssDNA tails that can anneal to promote joining14. Again we found that ctip mutant cells are 10-fold defective in SSA compared with wild-type cells (Fig. 3b). We conclude that CtIP plays an important role in DSB repair by HR and this accounts for the sensitivity of ctip cells to X-ray damage in S and G2 phases.
Figure 3. ctip mutant cells are defective for homologous recombination and microhomology-mediated end joining (MMEJ).
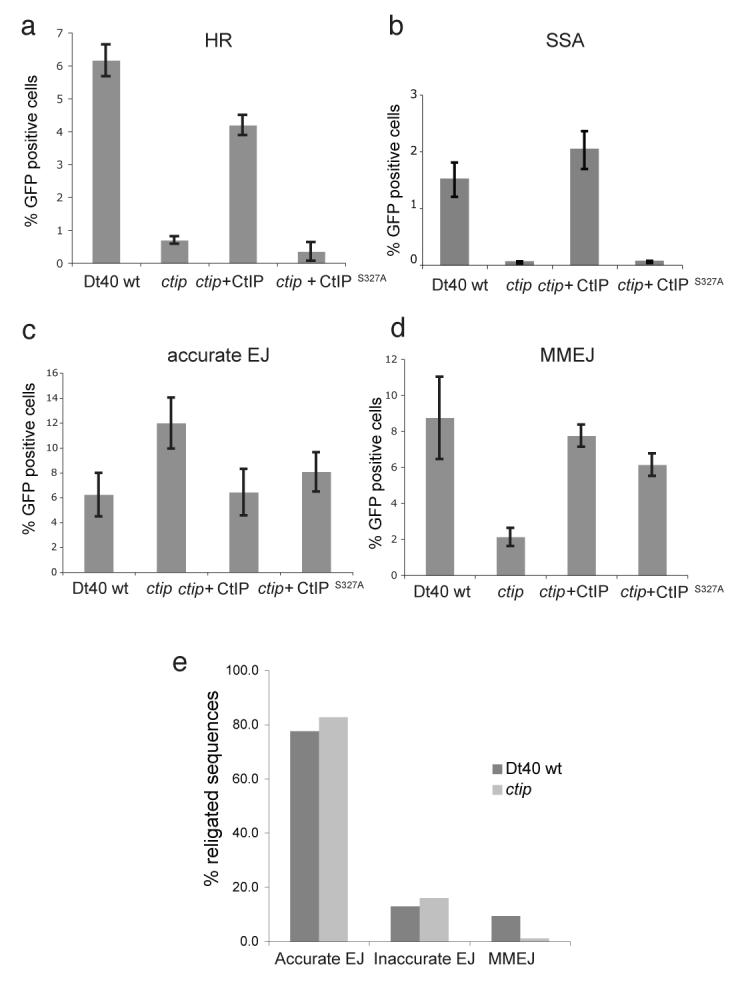
Repair is indicated by percentage of cells expressing GFP as described in Supplementary Fig. 5. Repair by (a) homologous recombination (HR); (b) single-strand annealing (SSA); (c) accurate non-homologous end-joining (accurate EJ); (d) microhomology-mediated end joining (MMEJ); (e) analysis of DNA sequences at repaired break sites in wild-type and ctip mutant cells. Individual sequences (shown in Supplementary Fig. 4) were classified according to the nature of their joints into “accurate EJ”, “inaccurate EJ” and ”MMEJ”.
DNA end-joining is achieved either through NHEJ, whereby broken DNA ends are directly rejoined, or by MMEJ in which DNA ends are resected locally to reveal short regions of complementary DNA (4 to 6 nucleotides) which stabilise broken ends for ligation1. We tested ctip cells for both types of end-joining and found no defect in accurate NHEJ (Fig. 3c). In fact we observed a slight increase in NHEJ activity in the ctip mutant compared with wild-type cells. On the other hand, for MMEJ we observed a 4- to 5-fold defect in the ctip mutant compared with wild-type cells (Fig. 3d).
In a second assay we transfected cells with linearised plasmid, recovered the repaired plasmids after 24 hours and examined the DNA sequences surrounding the joints (Supplementary Fig. 6). Both wild-type and ctip mutant cells repaired the majority of breaks through a combination of accurate and inaccurate NHEJ (Fig 3e). Where NHEJ was inaccurate, the spectrum of deletions at the break site was similar in ctip and wild-type cells. Nevertheless, in the ctip mutant cells we again detected a reduction in MMEJ (Fig. 3e). Together, with observations in 293 cells that siRNA-mediated knockdown of CtIP alters the balance of DSB repair15, these data establish a role for CtIP in MMEJ and provide an explanation for the defects in DSB repair observed in ctip cells during G1 phase.
A role for CtIP in DSB repair during G1 was unexpected. Previous studies suggested that CtIP is present at very low levels outside of the S and G2 phases12,16. Nevertheless, CtIP was present in extracts from DT40 cells in G1 albeit at reduced levels compared to cells in S/G2 (Fig. 2b). Furthermore, whereas in G1 phase CtIP is largely unmodified, the majority of CtIP in S/G2 is phosphorylated (Fig. 2b).
Previously Yu and Chen demonstrated that CtIP is phosphorylated on serine residue 327 as cells enter S phase, which mediates its interaction with the tumour suppressor BRCA1 that is required for the transient G2/M checkpoint12,17. Therefore, we next considered whether phosphorylation of serine 327 might also regulate the function of CtIP in DSB repair. To investigate this we expressed a mutant form of HsCtIP, in which serine 327 was substituted by alanine (HsCtIPS327A), in ctip cells and examined its sensitivity to X-rays. While expression of HsCtIPS327A improved the survival of ctip cells to X-rays, complementation was only partial (Fig.1a), suggesting that mutation of serine 327 results in loss of some but not all the repair functions of CtIP.
The picture became clearer when we looked at the survival to X-ray damage in different phases of the cell cycle. We found that expression of either HsCtIP or HsCtIPS327A in ctip cells fully restored resistance to X-rays in G1 phase and restored MMEJ to wild-type levels in a plasmid assay (Fig 3d), suggesting that phosphorylation on serine 327 is not required for the repair of DSB by MMEJ (Fig 2a). However, expression of HsCtIPS327A in ctip cells did not restore HR, suggesting that phosphorylation of serine 327 is important for this function (Fig. 3a,b). Accordingly, the sensitivity ctip cells to X-rays in S/G2, was only partially restored by HsCtIPS327A (Fig. 2a), which can be accounted for by restoration of MMEJ, but not HR, in these cells.
How might phosphorylation of serine 327 on CtIP increase the contribution of HR during S phase? It is known that CtIP promotes the resection of DNA ends5 and that this is an important step in both HR and MMEJ. However, since MMEJ requires small local regions of microhomology it is likely that the resection required for this pathway is less extensive than for strand exchange in HR. We reasoned, therefore, that phosphorylation of CtIP might upregulate the generation of ssDNA.
To test this we took advantage of the fact that BrdU incorporated into the genome is detectable by anti-BrdU antibody only in regions of ssDNA18. We cultured cells in BrdU, treated them with X-rays and stained for BrdU at intervals up to 2 hours (Fig. 4a). Approximately 25% of unirradiated cells exhibit 10 or more BrdU foci. After exposure to X-rays (8 Gy) we observed a time-dependent increase in BrdU staining in wild-type cells until, after 2 hours, approximately 50% contained 10 or more BrdU foci. Over the same time period only 30% of ctip mutant cells stained with BrdU, suggesting that while these mutant cells are not completely defective in the generation of ssDNA after exposure to X-rays, it occurs more slowly. Moreover, expression of HsCtIP in the ctip fully rescued this delay. On the other hand, expression of HsCtIPS327A cells did not complement this defect in the ctip mutant, suggesting that DNA damage-dependent increase in ssDNA is linked to the phosphorylation of serine 327. The delayed generation of ssDNA is not linked to reduced growth rate as cells expressing HsCtIPS327A exhibit reduced BrdU foci but proliferate normally (Supplementary Fig. 7).
Figure 4. Phosphorylation of S327 is required for generation of ssDNA in DT40 cells.

(a) Cells were labelled with 20 μM BrdU and treated with 8 Gy X-rays (where indicated). ssDNA was detected over time by staining with antibody against BrdU and analysed using confocal microscopy. Staining was performed under DNA denaturing (2N HCl) or native conditions as indicated. (b) Kinetics of ssDNA generation from (a). Cells containing 10 or more foci were scored as positive. (c) HR assay, performed as in Fig. 3 with indicated cell lines. Shows that expression of “constitutively activated” CtIPPM restores HR in ctip mutant cells but not in brca1 cells.
Our data place CtIP at the ‘crossroads’ between DNA end-joining and HR pathways for the repair of DSB, with phosphorylation of serine 327 acting as a cell-cycle dependent switch that regulates CtIP-dependent DNA end resection. Phosphorylation of serine 327 is known to control the interaction of CtIP with BRCA1 in DT4012 (Supplementary Fig. 8a). Moreover, like serine 327 of CtIP, BRCA1 is required for repair of DSB by HR but not MMEJ (Fig. 4c and Supplementary Fig. 8b), suggesting that the recruitment of BRCA1 to CtIP may be a determining factor in this switch.
Serine 327 lies within a weak CDK consensus site (SP/TP) in CtIP which, combined with the presence of a cyclin interaction motif (ZRXL)19, makes it a likely substrate for cyclin-dependent kinases. This residue is specific to CtIP in higher eukaryotes and is not present in Sae2, the CtIP—like protein of the budding yeast, S. cerevisiae. In yeast, HR and DNA end resection are promoted by CDK-dependent phosphorylation of Sae2 on serine 26720,21. Interestingly, this site is conserved at threonine residue 847 of vertebrate CtIP21, which we show here performs a similar function to S267 of Sae2 (Supplementary Figs 9 and 10). In CtIP, substitution of T847 to alanine causes defects in the repair of DSB by HR in S phase, but does not affect MMEJ. Accordingly, cells expressing CtIPT847A exhibit increased mutagenic repair of DSB (Supplementary Fig. 10d). Moreover, as in yeast, the requirement for phosphorylation of T847 can be circumvented by the use of a phosphomimic mutation where T847 in CtIP is replaced by aspartate or glutamate (CTIPPM). This mutant exhibits normal resection and restoration of HR without phosphorylation at T847 (Supplementary Figs 10 and 11).
While in yeast the switch to accurate DSB repair in S phase is controlled by phosphorylation of a single CDK site in Sae2, our data demonstrate that phosphorylation of CtIP at two independent CDK sites (S327 and T847) is required in vertebrates. The particular importance of S327 and the requirement of BRCA1 for this switch were established in two ways. Firstly, while expression of CtIPPM restores HR to ctip mutant cells without the requirement for phosphorylation at residue 847, we found that the CtIPS327A,PM mutant, in which phosphorylation at S327 is not possible, is defective in HR (Supplementary Fig. 10e). Secondly, we show that a CtIPPM mutant does not restore HR to brca1 mutant cells, confirming that recruitment of BRCA1 by CtIP is required for efficient HR function independently from the activation at T847 (Fig 4c).
Together these data establish a pivotal role for CtIP and BRCA1 in a switch that has profound consequences for the maintenance of genetic integrity in DNA-damaged cells by facilitating a shift from predominantly error-prone repair of DSB by DNA end-joining in G1 to the accurate repair afforded by HR in S and G2 phases.
Methods Summary
DT40 chicken cells were propagated in standard RPMI supplemented media11 at 37 °C, 6% CO2. Transfections were carried out by electroporation as previously described11. For the generation of CtIP knockout cells genomic DNA sequences were amplified with LA-TaKara and targeting vectors assembled (neomycin, histidinol and blasticidin). For each transfection, 30 μg of the selected vector were linearized using NotI (NEB) and transfected into DT40 cells. Clones were obtained after 2-4 weeks of growth under selective media. Genomic DNA from individual clones was prepared with PURAGENE DNA Purification Kit (Gentra). Twenty μl of each were digested with PacI and XmnI (NEB) at 37 °C overnight, and used in Southern blotting to detect targeted integration. Membranes were hybridized with a 32P GgCtIP probe (Supplementary Fig. 2b). Primers used for generating targeting vectors are listed in Supplementary Methods. Expression vectors for complementation studies were generated by insertion of CtIP cDNA into pCR2.1-TOPO and subsequent subcloning into pcDNA3.1/zeo(+). Primers used for cloning CtIP are listed in Supplementary Methods. Clonogenic survival assays were performed as previously described11. Cells were isolated at specific stages of the cell cycle by elutriation. Briefly, 109 DT40 cells were collected by centrifugation, resuspended in 20 ml RPMI medium and drawn into a standard (4 ml) chamber at 4000 rpm in a Beckman JE-5.0 centrifugal elutriation rotor. Cells were maintained at a constant flow rate (38 ml/min) and were elutriated by decreasing the rotor speed at intervals of 250 rpm. For each elutriation interval, a 150 ml fraction was collected and cells spun down, resuspended in 3 ml cold PBS and counted. An aliquot of 1×105 cells was incubated for 10 minutes with 1 μl of the cell-permeable DNA dye Draq5 (Biostatus Ltd) and the cell cycle profile determined by FACS analysis on a FACScan cytometer (Becton Dickinson).
Supplementary Material
Acknowledgements
The authors would like to thank Dr Maria Jasin (Sloane-Kettering) for kind gifts of DR-GFP and pHPRT-SSA-GFP, Dr Shunichi Takeda for gift of ku70 DT40 and Dr Javier Di Noia (IRCM, Montreal) for DT40 DTDR-17. We would also like to thank our colleagues Drs Cristina Rada and Julian Sale (MRC, Cambridge) for helpful comments and suggestions during the preparation of this manuscript. M.H.Y. is a Milstein Student of the Darwin Trust, Edinburgh, Scotland.
APPENDIX
Full Methods
Primers
GgCtIP knock out
For the construction of the targeting vectors the following primers were used:
Left arm :
5′SalI-CtIP L arm: 5′-gtcgacCAGAATCCCTCAGGTCCTGGAATG-3′
3′BglII-CtIP L arm: 5′-agatctATTTGTGGTCTGCTGGCTGTTGGG-3′
Right arm:
5′BglII-CtIP R arm: 5′-agatctTGACCATTTTGGTCCAGTTAAATC-3′
3′NotI-CtIP R arm: 5′-gcggccgcGCACTCTATTGGAGGTATTGCC-3′
For the screening of clones by Southern blot, a probe was designed with the following primers:
Probe forward: 5′-CCTGATGAATGTCTACTGTGGTGCATTCTT-3′
Probe reverse: 5′-CGCCTTTAATAAGTTAGATCATCAGTATGA -3′
pCMV/cyto/GFP* primers
EJfwd: 5_-CTAGGGATAACAGGGTAATCGGCTAG-3_
EJrev: 5_-CCATTACCCTGTTATCCCTAGCTAG-3_
CtIP cloning primers
5′BamHI CtIP (pcDNA3.1): 5′-ggatccaccATGAACATCTTGGGAAGCAGCTGTG-3′
3′NotI CtIP (pcDNA3.1): 5′-gcggccgcCTATGTCTTCTGCTCCTTGCCT-3′
CtIP mutagenesis CtIPS327A
Forward primer: 5′-CCTACTCGAGTGTCAGCTCCTGTATTTGGAG-3′
Reverse primer: 5′-CTCCAAATACAGGAGCTGACACTCGAGTAGG-3′
Inmunoprecipitation and Western Blotting
Whole cell extracts were prepared from 15×106 chicken DT40 cells lysed in NET-N buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% NP-40) supplemented with protease and phosphatase inhibitors (1 mM NaF, 1 mM Na3VO4 and 10 mM β—glycerophosphate). Nuclear extracts were prepared as previously described.
For immunoprecipitation analysis, nuclear or whole cell extract were incubated with 30-10μl of the indicated antibodies and 40 μl of protein A/G-sepharose beads (Amersham). Beads were collected by centrifugation, washed 5 times with 1 ml NET-N buffer and boiled for 5 minutes in 50 μl of 2X SDS loading buffer (0.1 mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 0.1% β-mercaptoethanol, 0.004% bromophenol blue). Twenty μl of each sample were resolved by SDS-PAGE. Proteins were identified by standard Western blot using the following primary antibodies: CtIP (T-16) sc-5970 (Santa Cruz Inc.) 1:500 dilution, GgBRCA1 (non commercial) 1:1000 dilution. Proteins were visualized using ECL-plus (Amersham).
Cell cycle analysis
Cells were pulsed with 20 μM BrdU (Sigma) for 20 min, washed them twice with PBS and fixed them overnight in 70% ethanol at -20 °C. Cells were stained against BrdU and propidium iodide and cell cycle analysis performed as described10.
HR and SSA assays
For the HR assay, DT40 cells stably expressing a single copy of DR-GFP (M. Jasin, Sloan-Kettering) were a gift from J. Di Noia (Université de Montréal). For the SSA assay, the vector pHPRT-SSA-GFP (M. Jasin, Sloan-Kettering) was stably transfected into DT40 or ctip mutant cells. The clones obtained after puromicin selection were screened for single integrants by Southern blotting. To perform each of the experiments, cells were transfected with either pCASce13 or the control plasmid pDsRed1-N1 (Clontech) using the Amaxa Nucleofection system. They were then suspended in 5 ml of medium, incubated at 37 °C for 24 h and washed in 500 μl of PBS. Eight μg ml-1 propidium iodide was added to the samples transfected with pCASce. Cells expressing GFP were quantified using a FACScan cytometer (Becton Dickinson). Data are corrected for transfection efficiency as measured by the percentage of cells that express DsRed.
MMEJ and EJ assays
For the MMEJ assay, the vector pCMV/cyto/myc/GFP (Invitrogen) was modified by insertion of a pre-annealed oligo (EJfwd and EJrev) carrying an I-SceI restriction site into the unique BmtI site present in the GFP coding sequence, to generate pCMV/cyto/myc/GFP*. The vector was digested with I-SceI at 37 °C overnight, analyzed for complete digestion and ethanol precipitated. Ten μg of either uncut pCMV/cyto/myc/GFP (transfection control) or I-SceI-digested pCMV/cyto/myc/GFP* were used for transient transfection of DT40 cells with the Amaxa Nucleofection system and analysed as described above. The same protocol was used for the EJ assay but transfections were carried out with HincII-digested pCMV/cyto/myc/GFP or its uncut version. For the analysis of joined DSBs, cells were transfected with a I-SceI linearized vector pCMV/cyto/myc/GFP* as described. Plasmid DNA was extracted and used to tranform DH5 alpha bacteria. Resistant colonies were grown in 96-well agar plates and sequence analysis performed using a pCMV-F primer (GATC biotech).
DT40 Immunofluorescence
DT40 cells were cultured for one and a half cell cycles in the presence of bromodeoxyuridine (BrdU, 20 μM, Invitrogen). After this period, cells were grown in 6-well-plates at 3×106 cells per well and exposed to X-rays as described above. Glass coverslips coated with poly-L-lysine (Sigma) were added to each sample. At different times after irradiation, cells were washed with PBS, fixed in 4% paraformaldehyde for 5 min and washed with PBS a second time. Cells subject to denaturing conditions were treated for 30 min with 2 ml of 2 N HCl/0.5% triton X-100 and neutralized with 2 ml Na tetraborate, after which they were blocked in PBSTS/1% BSA alongside cells subject to native conditions. Samples were next incubated with mouse polyclonal antibody against BrdU (BD Pharmigen) for 2 h. Coverslips were washed 2X with PBSTS/1% BSA, incubated with an antibody against mouse Ig FITC-conjugated (BD Pharmigen) for 1 h, washed twice in PBS and finally mounted on slides with mounting medium containing 4,6-diamidino-2-phenylindole (DAPI, Vectashield). Samples were analysed using a Nikon confocal microscope.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature. A figure summarising the main result of this paper is also included as SI.
REFERENCES
- 1.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 2003;23:8820–8. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim JS, et al. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J. Cell Biol. 2005;170:341–7. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takata M, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998;23:247–51. doi: 10.1016/s0968-0004(98)01232-8. [DOI] [PubMed] [Google Scholar]
- 7.Chen PL, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol. Cell. Biol. 2005;25:3535–42. doi: 10.1128/MCB.25.9.3535-3542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonoda E, Morrison C, Yamashita YM, Takata M, Takeda S. Reverse genetic studies of homologous DNA recombination using the chicken B-lymphocyte line, DT40. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:111–7. doi: 10.1098/rstb.2000.0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tauchi H, Matsuura S, Kobayashi J, Sakamoto S, Komatsu K. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene. 2002;21:8967–80. doi: 10.1038/sj.onc.1206136. [DOI] [PubMed] [Google Scholar]
- 10.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J. 2003;22:1654–64. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat. Genet. 2005;37:953–7. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 12.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell. Biol. 2004;24:9478–86. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–8. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 2004;24:9305–16. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998;273:25388–92. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg RA, et al. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlegel BP, Jodelka FM, Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006;66:5181–9. doi: 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- 19.Endicott JA, Noble ME, Tucker JA. Cyclin-dependent kinases: inhibition and substrate recognition. Curr. Opin. Struct. Biol. 1999;9:738–44. doi: 10.1016/s0959-440x(99)00038-x. [DOI] [PubMed] [Google Scholar]
- 20.Ira G, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–7. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–92. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.