

Abstract
Objectives
To describe the clinical features of patients who presented with “idiopathic” interstitial pneumonia but who were ultimately diagnosed with anti-synthetase syndrome based on clinical features and positive anti-PL-7 or -PL-12 antibodies.
Methods
Over a 24 month period, in our interstitial lung disease (ILD) program, we evaluated 37 patients who presented with clinical features suggestive of anti-synthetase (AS) syndrome, negative anti-JO-1 antibodies, and who were assessed for other anti-tRNA synthetase (anti-tRS) antibodies. All data were abstracted from the medical record.
Results
Nine (24%) were confirmed to have non-anti-Jo-1 positive AS syndrome based on clinical features and the presence of other anti-tRS antibodies (seven with anti-PL-7 and two with anti-PL-12 antibodies). Five were women; seven were Caucasian. All nine presented with dyspnea as the initial symptom and with ILD as the first manifestation. Elevated CPK was identified in three patients (median 75, range 22–925), but only two had muscle weakness. Pulmonary physiology revealed restriction (forced vital capacity 60% of predicted) and impaired gas transfer (diffusing capacity for carbon monoxide 40% of predicted). All had similar findings on thoracic HRCT scans, with extreme basilar predominance of abnormalities and patterns suggestive of non-specific interstitial pneumonia and organizing pneumonia. Immunomodulatory therapies were used to treat the ILD—responses were variable, but some subjects clearly improved.
Conclusion
Anti-PL-7 and PL-12 antibodies may be more common among patients presenting with “idiopathic” interstitial pneumonia than formerly considered and should be checked in patients with features of AS syndrome despite a negative anti-nuclear or anti-JO-1 antibodies. Further research is needed to advance understanding of anti-PL-7 or anti PL-12 positive AS syndrome, including its prognosis, optimal approaches to therapy, and to determine how its course differs from other forms of ILD.
Keywords: antisynthetase syndrome, idiopathic interstitial pneumonia, Anti-JO-1
Introduction
The interstitial lung diseases (ILD) comprise a diverse group of disorders characterized histologically by varying degrees of inflammation and fibrosis1,2. Two major categories of causes for ILD include exposures (e.g., aerosolized organic antigens, dusts, drugs) and connective tissue disease (CTD). Many ILDs, including the idiopathic interstitial pneumonias (IIP), have no identifiable etiology.. The IIP comprise a group of conditions with similar clinical, radiologic, and physiologic findings, but different histologic patterns in surgical lung biopsy specimens 1. These histologic patterns are not specific to the IIP and may be seen, for example, in ILD related to underlying CTD. Recent data suggest that, for a given histologic pattern, CTD-related ILD has a more favorable prognosis than IIP, thus arguing for the careful evaluation of patients labeled with idiopathic ILD in an attempt to identify underlying CTD 3,4.
Recognition of CTD is particularly challenging when ILD is its first or lone manifestation or when extrathoracic features of CTD are subtle5–7. Attempts to identify underlying CTD most often include a thorough history, physical examination, and serologic assessment for the presence of autoantibodies (e.g., anti-nuclear antibodies [ANA] and rheumatoid factor [RF]). It is unclear whether these attempts are sufficient or whether additional testing is useful or necessary to identify the presence of CTD.
The association between ILD and the myositis spectrum of CTD is well-known 8,9. Patients with myositis (either polymyositis [PM] or dermatomyositis [DM]) are considered to have the anti-synthetase (AS) syndrome when they are found to have an anti-tRNA synthetase (anti-tRS) autoantibody and one or more of these clinical features in decreasing order of frequency; myositis, ILD, arthritis or arthralgias, Raynaud’s phenomenon (RP), “mechanic’s hands” (fissured, roughened skin over the tips and thenar side of the fingers), and fever10. Esophageal dysmotility is a well known manifestation of CTD, in general; and it is often seen with myositis or the AS syndrome, in particular.
The anti-tRS autoantibodies target aminoacyl-transfer RNA synthetases that catalyze the binding of specific amino acids to their cognate tRNA during protein synthesis. The most commonly identified and readily commercially tested anti-tRS antibody is anti-JO-1 (anti-histidyl-tRNA synthetase)11. Others include anti-PL-7 (anti-threonyl), anti-PL-12 (anti-alanyl), anti-OJ (anti-isoleucyl), anti-EJ (anti-glycyl), anti-KS (anti-asparaginyl), anti-ZO (anti-phenylalanyl), and an anti-tyrosyl tRS antibody12. Anti-JO-1 is found in about 30%, anti-PL-7 or anti-PL-12 in 3–4%, and the other anti-tRS antibodies in < 2% of patients with myositis13. Numerous studies have elucidated the link between anti-JO-1 antibodies and ILD14,15; however, there are few data on the characteristics of myositis patients with other anti-tRS antibodies.
We conducted this study in an attempt to achieve three specific goals: First, to add to the limited literature of and raise awareness for what we believe to be an under-recognized cause of fibrotic ILD—non-anti-Jo-1 AS syndrome. Second, we aimed to highlight the chest HRCT findings of ILD associated with the AS syndrome. Finally, we wished to emphasize the challenges physicians face in treating ILD in the AS syndrome. The objective of this study was to describe the clinical features of a series of patients who presented with “idiopathic” interstitial pneumonia but were diagnosed with AS syndrome based on clinical features and positive anti-PL-7 or anti-PL-12 antibodies.
Methods
Over a 24-month period, we identified 37 patients evaluated at our center for “idiopathic” interstitial pneumonia who had clinical features suggestive of AS syndrome, negative anti-JO-1 antibodies, and were assessed for other anti-tRS antibodies. We abstracted from medical records data on demographics, clinical presentation, pulmonary physiology, chest radiology, histology, and therapy. No patient presented with a disease or exposure linked with ILD. As part of their comprehensive ILD evaluation, all patients underwent standardized comprehensive clinical testing that included serologic testing, thoracic high-resolution computed tomography (HRCT), barium contrast esophagography, and pulmonary function testing. Most patients were seen by both ILD and rheumatology specialists. The decision to test for anti-tRS antibodies other than anti-Jo-1 was a clinical one made by the consulting physicians. This testing was conducted by ordering the “Myositis Antibody Panel” through Oklahoma Medical Research Foundation (OMRF) Clinical Immunology Laboratory in Oklahoma City, OK. This was a HIPAA-compliant protocol, approved by our institutional review board. Data are presented as counts or medians with interquartile ranges.
Results
All patients presented with ILD of unknown etiology and were anti-JO-1 antibody negative. Nine (24%) of thirty-seven patients were confirmed to have AS syndrome based on clinical features and confirmatory anti-tRS antibody positivity. Seven had anti-PL-7 antibodies and two had anti-PL-12 antibodies. No other anti-tRS antibody was identified. Clinical characteristics of these nine patients are presented in Table 1. Six patients had esophageal dysmotility, four had Raynauds phenomenon with abnormal wide-field nailfold capillaroscopy, four had inflammatory arthritis, three had “mechanics hands, and two had Gottrons papules. Elevated CPK was identified in three patients (median 75, range 22–925), but only two had muscle weakness. The other twenty-eight patients were considered to have either an undifferentiated CTD or remained unclassifiable/idiopathic and were not further analyzed.
Table 1.
Baseline Characteristics of Study Subjects
| Subjects N = 9 |
|
|---|---|
| Age in years | 60.1 (41.1–67.3) |
| Female:Male | 5:4 |
| Ever cigarette smoker | 4 |
| Ethnicity | |
| PL-7 | White 5 |
| Asian 1 | |
| Latino 1 | |
| PL-12 | White 1 |
| Latino 1 | |
| Clinical features | |
| Dyspnea | 9 |
| Raynaud’s phenomenon | 4 |
| Arthritis | 4 |
| Esophageal disease | 6 |
| Laboratory | |
| Elevated CPK | 3 |
| Autoantibodies | |
| ANA | |
| SSA | 0 |
| PL-7 | 2 |
| PL-12 | 7 |
| 2 | |
| Pulmonary physiology | |
| FVC% | 60 (50–65) |
| DLCO% | 40 (37–46) |
| HRCT pattern | |
| fNSIP | 9 |
| Surgical biopsy | 5 |
| mNSIP + OP | 2 |
| fNSIP + OP | 1 |
| fNSIP (upper) + UIP (lower) | 1 |
| ALI + OP | 1 |
Data are counts, median (interquartile range); CPK=creatine phosphokinase; ANA=anti-nuclear antibody; FVC%=percent predicted forced vital capacity; DLCO%=percent predicted diffusing capacity of the lung for carbon monoxide; mNSIP= mixed cellular and fibrotic non-specific interstitial pneumonia; OP=organizing pneumonia; fNSIP= fibrotic NSIP; UIP=usual interstitial pneumonia; ALI=acute lung injury
Dyspnea was the initial symptom, and ILD was the presenting manifestation, in all nine subjects. Dyspnea was present for a median 17 months prior to the diagnosis of AS syndrome. In the two patients with muscle weakness and elevated CPK, dyspnea pre-dated muscle symptoms by two and five months respectively. Over a median 12.6 months of follow-up, none of the six subjects with normal CPK levels at presentation developed CPK enzyme elevation.
Therapeutic choices and responses to them are presented for each subject in Table 2. All subjects with AS are living. Representative slices from HRCT scans are displayed in Figure 1; they show the characteristic findings of AS-related ILD, including extreme basilar ground glass and reticular opacities, areas of consolidation, and traction bronchiectasis—a constellation of findings that suggests a mixed histologic pattern of nonspecific interstitial pneumonia (NSIP) and organizing pneumonia.
Table 2.
Response to Therapy in Each Subject
| Subject | Gender | Synthetase Ab | Muscle involvement | Pre-Tx FVC(%)/DLCO(%) | Rx | Duration | On Rx FVC(%)/DLCO(%) |
|---|---|---|---|---|---|---|---|
| 1 | M | PL-7 | Yes | 2.71(50)/15.6(40) | P, CYC | 6mos | 3.23(60)/13.9(36) |
| 2 | M | PL-7 | Yes | 3.22(74)/10.32(37) | P, MMF | - | - |
| 3 | M | PL-7 | No | 3.5(65)/30.1(78) | None | 10mos | 3.73(70)/30.9(80) |
| 4 | F | PL-7 | No | 2.47(76)/10.5(39) | MMF | 21mos | 2.55(74)/11.1 (43) |
| 5 | M | PL-7 | Yes | 2.99(63)/13/66 (40) | P, MMF | 16mos | 3.38 (72)/14.5 (43) |
| 6 | F | PL-7 | No | 1.6(61)/13 (68) | P, MMF | 4mos | 1.7(62)/13.5 (69) |
| 7 | F | PL-7 | No | 1.65(58)/10.4(46) | None | - | - |
| 8 | F | PL-12 | No | 1.66(45)/10.1(35) | P, AZA | 6mos | 1.4(38)/8.11(28) |
| 9 | F | PL-12 | No | 0.9(23)/unable | P, CYC | 2mos | 1.0(25)/3.52(12) |
P=prednisone; CYC=cyclophosphamide; MMF=mycophenolate mofetil; AZA=azathioprine
Figure 1. HRCT Findings in AS Syndrome.
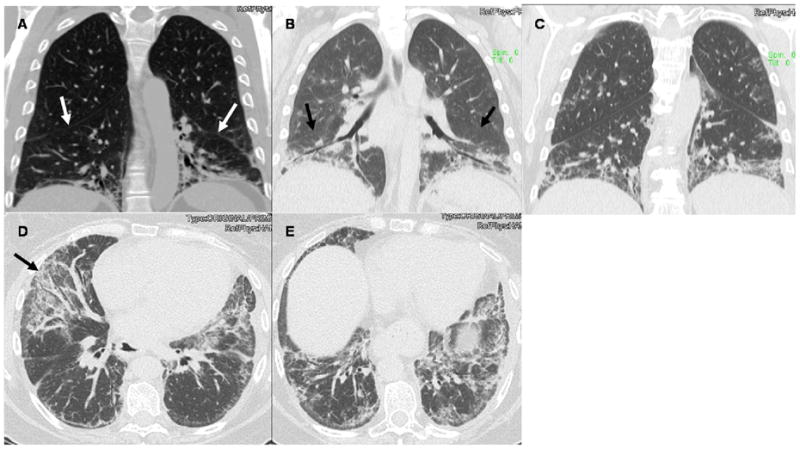
Panels A–C display coronal slices from three patients with AS syndrome. Note the extreme basilar predominance of findings, the extensive traction bronchiectasis in Panel B, and how the opacities in all three panels hug or “pancake” the diaphragms. White arrows in Panel A and black arrows in Panel B point to the major fissures being pulled caudally from the extensive lower lobe volume loss. Similar findings are seen in Panel C. Panels D and E are transverse slices from the same patient as Panel C. The black arrow in Panel D points to the extreme caudal aspect of the middle lobe affected by extensive ground glass opacities and fraction bronchiectasis. Note the patulous esophagus. Similar findings in the lung parenchyma are seen in Panel E.
Discussion
Over the last 24 months, among 37 patients labeled with idiopathic ILD, we identified identified nine with anti-PL-7 or anti-PL-12. positive AS syndrome. In each subject, ILD was the initial manifestation that prompted clinical evaluation. Most of the patients were evaluated by both pulmonologists and rheumatologists as part of their comprehensive ILD evaluation. In general, the response of the ILD to potent immunomodulatory therapy - glucocorticoids with or without a cytotoxic agent - was variable.
To date, at least seven anti-tRS antibodies have been identified; anti-JO-1 is the most common, and it is the most widely recognized myositis-specific antibody 16. Anti-tRS antibodies appear to be specific for the AS syndrome; they appear to be mutually exclusive (patients with AS syndrome have only one AS antibody); and they do not cross-react among aminoacyl-tRNA synthetase enzymes—that is, each enzyme is specific for only one amino acid16. It remains to be determined precisely how AS antibodies are generated and whether they are pathogenic (i.e. responsible for tissue injury) in patients with AS syndrome. Some studies have suggested viral infection might induce the production of anti-tRS antibodies17,18.
Interstitial lung disease is a common manifestation of the AS syndrome and strongly associated with the presence of anti-tRS antibodies. Love and colleagues examined a large National Institutes of Health myositis database and noted the presence of ILD in nearly 90% of subjects with anti-tRS antibodies14. Among the 212 subjects in that study, 48 (23%) had anti-tRS antibodies most had anti-JO-1, five had anti-EJ, four had anti-PL-7, two had anti-OJ, and one had anti-PL-12 antibodies. Sixty-two percent of subjects with anti-tRS antibodies were Caucasian, 23% were black, 54% met diagnostic criteria for DM, and 40% met criteria for PM. Among the subgroup with anti-tRS antibodies, findings at presentation included fever in 87%, RP in 62%, myalgias in 84%, arthritis in 94%, “mechanic’s hands” in 71%, and positive ANA in only 44%. The five-year survival rate for subjects with anti-tRS antibodies was nearly 75%.
Targoff and Arnett reported clinical features of eight patients with anti-PL-12 antibodies16. Five were black, two were white, and one was Latino. Seven were female; six had elevated CPK; three had arthritis; and three had RP. Significant ILD was identified in seven. In a study from Japan, Yamasaki and colleagues identified 36 subjects who were evaluated at their center from 1991–2002 and found to have anti-tRS antibodies—six with anti-PL-7, eight with anti-JO-1, and two with anti-PL-12 antibodies19. All six subjects with anti-PL-7 and all eight subjects with anti-JO-1 antibodies had ILD. Interesgingly, La Corte and colleagues studied 21 subjects with AS and found those with SSA antibodies had more extensive ILD by HRCT.20 This was not true in our subjects, but only two had SSA antibodies.
Data on therapy for anti-PL-7 or anti-PL-12 associated ILD are few, and systematic evaluations are lacking. Glucocorticoids have been the mainstay of therapy. Immunomodulatory agents used in combination with glucocorticoids have included azathioprine, calcineurin antagonists, and cyclophosphamide—effects on symptoms and pulmonary physiology have been variable21–24. In a few of our nine subjects, potent anti-inflammatory/immunomodulatory therapy led to physiological improvement—mirroring the variable response to therapy reported in previously published studies. As with ILD from other causes, in which the clinical course is variable and inexorable progression is not necessarily the rule, whether patients whose physiology remained stable on therapy might have declined if untreated is uncertain.
As in these prior series, the subjects in the current study had various features of the AS syndrome; the one constant finding was fibrotic ILD that was moderate to very severe in the majority. It is noteworthy to highlight the unique HRCT features in our cohort. The extreme basilar predominance of findings, including reticular and groundglass opacities and traction bronchiectasis with or without consolidation, that hug or “pancake” the diaphragms, and the lower lobe volume loss, with the major fissures being dragged caudally are, in our experience, fairly specific for AS-related ILD. Other investigators have examined HRCT scan findings in patients with AS: the most common radiologic features in a study of 17 subjects with anti-Jo-1 antibodies and ILD by Karadimitrakis and colleagues25 were ground glass opacities (41%), reticular opacities (35%), and honeycombing (35%). Although the authors commented that the ground glass opacities were lower zone predominant and that the radiologic features suggested an underlying histologic pattern of NSIP, they did not mention the diaphragmatic “hugging” we observed in our subjects.
When confronted with an “idiopathic” interstitial pneumonia, there is no standard approach – or uniform set of serologies to obtain – to reliably exclude underlying CTD. ILD may be the forme fruste presentation of CTD, and in our experience, effective cross-specialty interactions among rheumatologists and pulmonologists is needed to optimize the evaluation of such patients. None of our subjects had a positive ANA; thus, practitioners should not be dissuaded from considering CTD (including the AS syndrome) as a potential etiology for ILD simply because the ANA is negative. In our cohort, subtle clinical features such as detecting RP, “mechanic’s hands”, and esophageal dysmotility were relevant and aided in the overall clinical assessment. Clinicians involved in the evaluation of patients with ILD need to thoroughly probe for subtle extra-thoracic features and have a low-threshold to involve rheumatologic expertise in the evaluation process5–7. The assessment for anti-tRS antibodies allowed accurate CTD identification in 24% (9/37) of our patients. The other twenty-eight subjects were considered to have either undifferentiated CTD based on clinical features along with non-specific autoantibody positivity (such as ANA and Anti-SSA) or remain unclassified/idiopathic in the absence of any identifiable autoantibodies. Interestingly, in a study of 21 patients with AS, La Corte and co-investigators20 observed that the presence of anti-SSA antibodies was associated with more severe ILD as defined by HRCT scan criteria. However, the presence of anti-SSA appeared not to adversely affect survival.
Our study has limitations. The small sample size did not allow us to conduct formal statistical analyses. We were surprised by the lack of improvement in DLCO among subjects whose FVC improved on therapy; one possible and likely explanation for this is the intrinsic variability of the DLCO maneuver combined with the small sample size. Like all data generated from patients evaluated at a specialized referral center, data from this study are inherently biased, which detracts from the external validity and potential generalizability of results. In addition, the lack of a systematic decision-making regarding whether to test for anti-tRS antibodies and how to approach therapy is a pitfall of this retrospective study. However, the purpose of this study was not to decipher the best approach to ILD therapy in patients with anti-PL-7 or anti-PL-12 antibodies; rather, it was conducted in an attempt to achieve three specific goals: First, to add to the limited literature of non-anti-Jo-1 AS syndrome. The lack of, up-to-recently, commercially available testing for non-anti-Jo-1 AS antibodies meant that many patients with non-anti-Jo-1 AS syndrome were likely given a clinical summary diagnosis of idiopathic ILD. Second, we wanted to highlight the chest HRCT findings of ILD associated with the AS syndrome that include extreme basilar predominance of findings, including reticular and groundglass opacities and traction bronchiectasis with or without consolidation, that hug or “pancake” the diaphragms, and the lower lobe volume loss, with the major fissures being dragged caudally. Finally, we wished to emphasize the challenges physicians face in treating ILD in the AS syndrome. We send our samples for non-Jo-1 anti-synthetase antibody testing to the Myositis Testing Laboratory at the Oklahoma Medical Research Foundation (OMRF). Specimen handling and mailing instruction are found on their website at http://www.omrf.org/OMRF/Core/MyositisLab.asp. We are not certain whether specimens can be sent from overseas to OMRF or whether there are centers outside the U.S. performing similar testing that is commercially available. Despite meeting our goals for this study, many questions remain—for example, what is the best immunomodulatory agent to use for AS syndrome-associated ILD? Does anti-tRS antibody type inform decision-making with respect to which therapeutic agent to institute? Which AS syndrome patients should be treated? For how long? Does the ILD in patients with AS syndrome “behave” similar to other CTD-related ILD? Similar to idiopathic disease? We can only hope to begin to answer some of these questions if we are able to accurately identify and prospectively study more patients with this condition.
In conclusion, we have described the clinical characteristics of nine patients who presented with an “idiopathic” interstitial pneumonia and were ultimately diagnosed with anti-PL-7 or anti-PL-12 antibody positive AS syndrome. Each subject was ANA and anti-JO-1 negative, and each presented with a HRCT scans suggestive of NSIP with or without overlapping organizing pneumonia. Effective cross-specialty collaboration among pulmonologists and rheumatologists led to the detection subtle extra-thoracic features of the AS syndrome. Among patients with “idiopathic” NSIP, consideration should be given to AS syndrome and a full complement of anti-tRS antibodies should be checked.
Abbreviations
- ANA
anti-nuclear antibody
- AS
anti-synthetase syndrome
- DM
dermatomyositis
- HRCT
high-resolution computed tomography
- ILD
interstitial lung disease
- PM
polymyositis
- RP
Raynaud’s phenomenon
Footnotes
Disclosures: None of the authors has any real or potential conflict of interest with the data presented in this manuscript.
References
- 1.Joint Statement of the American Thoracic Society and European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 2.Collard HR, King TE., Jr Demystifying idiopathic interstitial pneumonia. Arch Intern Med. 2003;163:17–29. doi: 10.1001/archinte.163.1.17. [DOI] [PubMed] [Google Scholar]
- 3.Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165:1581–6. doi: 10.1164/rccm.2106012. [DOI] [PubMed] [Google Scholar]
- 4.Park JH, Kim DS, Park IN, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. 2007;175:705–11. doi: 10.1164/rccm.200607-912OC. [DOI] [PubMed] [Google Scholar]
- 5.Fischer A, Meehan RT, Feghali-Bostwick CA, West SG, Brown KK. Unique characteristics of systemic sclerosis sine scleroderma-associated interstitial lung disease. Chest. 2006;130:976–81. doi: 10.1378/chest.130.4.976. [DOI] [PubMed] [Google Scholar]
- 6.Strange C, Highland KB. Interstitial lung disease in the patient who has connective tissue disease. Clin Chest Med. 2004;25:549–59. vii. doi: 10.1016/j.ccm.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 7.Tzelepis GE, Toya SP, Moutsopoulos HM. Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur Respir J. 2008;31:11–20. doi: 10.1183/09031936.00060107. [DOI] [PubMed] [Google Scholar]
- 8.Frazier AR, Miller RD. Interstitial pneumonitis in association with polymyositis and dermatomyositis. Chest. 1974;65:403–7. doi: 10.1378/chest.65.4.403. [DOI] [PubMed] [Google Scholar]
- 9.Salmeron G, Greenberg SD, Lidsky MD. Polymyositis and diffuse interstitial lung disease. A review of the pulmonary histopathologic findings. Arch Intern Med. 1981;141:1005–10. [PubMed] [Google Scholar]
- 10.Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med. 1990;77:1019–38. doi: 10.1093/qjmed/77.1.1019. [DOI] [PubMed] [Google Scholar]
- 11.Nishikai M, Reichlin M. Heterogeneity of precipitating antibodies in polymyositis and dermatomyositis. Characterization of the Jo-1 antibody system. Arthritis Rheum. 1980;23:881–8. doi: 10.1002/art.1780230802. [DOI] [PubMed] [Google Scholar]
- 12.Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 2007;46:1005–8. doi: 10.1093/rheumatology/kem045. [DOI] [PubMed] [Google Scholar]
- 13.Hirakata M, Katsuki Y, Sato S. [Immunologic tests: Anti-PL 7 antibodies, anti-PL-12 antibodies, and other anti-aminoacyl tRNA synthetase antibodies] Nippon Rinsho. 2005;63 (Suppl 7):508–11. [PubMed] [Google Scholar]
- 14.Love LA, Leff RL, Fraser DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991;70:360–74. doi: 10.1097/00005792-199111000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Plotz PH, Rider LG, Targoff IN, Raben N, O’Hanlon TP, Miller FW. NIH conference. Myositis: immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med. 1995;122:715–24. doi: 10.7326/0003-4819-122-9-199505010-00010. [DOI] [PubMed] [Google Scholar]
- 16.Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol. 2000;12:475–81. doi: 10.1097/00002281-200011000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Bowles NE, Dubowitz V, Sewry CA, Archard LC. Dermatomyositis, polymyositis, and Coxsackie-B-virus infection. Lancet. 1987;1:1004–7. doi: 10.1016/s0140-6736(87)92271-9. [DOI] [PubMed] [Google Scholar]
- 18.Imbert-Masseau A, Hamidou M, Agard C, et al. Antisynthetase syndrome. Three cases and a review of the literature. Ann Med Interne (Paris) 2003;154:483–8. [PubMed] [Google Scholar]
- 19.Yamasaki Y, Yamada H, Nozaki T, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum. 2006;54:2004–9. doi: 10.1002/art.21883. [DOI] [PubMed] [Google Scholar]
- 20.La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity. 2006;39:249–53. doi: 10.1080/08916930600623791. [DOI] [PubMed] [Google Scholar]
- 21.Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum. 1996;26:459–67. doi: 10.1016/s0049-0172(96)80026-6. [DOI] [PubMed] [Google Scholar]
- 22.Oddis CV, Sciurba FC, Elmagd KA, Starzl TE. Tacrolimus in refractory polymyositis with interstitial lung disease. Lancet. 1999;353:1762–3. doi: 10.1016/S0140-6736(99)01927-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum. 2005;52:2439–46. doi: 10.1002/art.21240. [DOI] [PubMed] [Google Scholar]
- 24.Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007;46:124–30. doi: 10.1093/rheumatology/kel112. [DOI] [PubMed] [Google Scholar]
- 25.Karadimitrakis S, Plastiras SC, Zormpala A, et al. Chest CT findings in patients with inflammatory myopathy and Jo1 antibodies. Eur J Radiol. 2008;66:27–30. doi: 10.1016/j.ejrad.2007.05.017. [DOI] [PubMed] [Google Scholar]