

Abstract
Alpha‐1‐antitrypsin (A1AT) deficiency is characterized by increased neutrophil elastase (NE) activity and oxidative stress in the lung. We hypothesized that NE exposure generates reactive oxygen species by increasing lung nonheme iron. To test this hypothesis, we measured bronchoalveolar lavage (BAL) iron and ferritin levels, using inductively coupled plasma (ICP) optical emission spectroscopy and an ELISA, respectively, in A1AT‐deficient patients and healthy subjects. To confirm the role of NE in regulating lung iron homeostasis, we administered intratracheally NE or control buffer to rats and measured BAL and lung iron and ferritin. Our results demonstrated that A1AT‐deficient patients and rats postelastase exposure have elevated levels of iron and ferritin in the BAL. To investigate the mechanism of NE‐induced increased iron levels, we exposed normal human airway epithelial cells to either NE or control vehicle in the presence or absence of ferritin, and quantified intracellular iron uptake using calcein fluorescence and ICP mass spectroscopy. We also tested whether NE degraded ferritin in vitro using ELISA and western analysis. We demonstrated in vitro that NE increased intracellular nonheme iron levels and degraded ferritin. Our results suggest that NE digests ferritin increasing the extracellular iron pool available for cellular uptake.
Keywords: alpha‐1‐antitrypsin deficiency, inflammation, iron
Introduction
Inhaled exposures (e.g., microbials, particles, and fibers) can mobilize host iron, disrupt the normal homeostasis of this metal in the lower respiratory tract, and increase its availability to participate in an oxidative stress. Subsequently, host protective mechanisms against iron‐induced oxidative injury are necessary for lung health. These focus on host re‐acquisition of its own metal with subsequent transport and storage to a catalytically less reactive state. 1 There are several pathways of iron acquisition employed by living systems. Siderophores and ferrireductases are prominent among these. 2 However, a third approach has been described with the recognition that microbes can utilize proteases in the acquisition of iron. Proteases may cleave iron‐transport and ‐storage proteins allowing use of the metal by the microbe. 3 , 4 Reflecting potential interactions between metal availability and proteases, increased iron concentrations can impact expressionand activity of collagenase, 5 elastase, 6 alkaline proteinase, 7 and metalloproteases. 8
Animal and human studies similarly suggest a participation of proteases in iron homeostasis. The intratracheal instillation of a single dose of neutrophil elastase in an animal model increased lung iron concentrations approximately 18 months later. 9 Iron homeostasis is disrupted among cystic fibrosis patients in whom airway elastase content and activity is excessive; CF patients have elevated iron and ferritin concentrations in both the sputum and bronchoalveolar lavage (BAL). 10 , 11 Finally, alpha‐1‐antitrypsin deficient patients have a propensity to develop cirrhosis which may be related to abnormal iron homeostasis; 12 , 13 this observation raises the possibility that unopposed neutrophil elastase activity alters iron homeostasis in both the lungs and systemically among A1AT‐deficient patients.
We tested the hypothesis that a disruption of the normal balance between proteases and antiproteases can be associated with an increased availability of iron in the lower respiratory tract of A1AT‐deficient patients, in the lower respiratory tract of a rat model exposed to intratracheal neutrophil elastase, and in normal human bronchial epithelial cells exposed to neutrophil elastase. We suggest that among the mechanisms of increased iron availability would be a proteolytic cleavage of iron‐transport and ‐storage proteins with subsequent increase in nonheme iron.
Methods
A1AT‐deficient patients
The characterization of A1AT‐deficient and control subjects is reported in Table 1 . Alpha‐1‐antitrypsin patients (Beaumont Hospital, Dublin, Ireland) had the diagnosis of A1AT deficiency and PiZZ phenotype confirmed by nephelometry and isoelectric focusing. The screening procedures for each subject included a history and physical examination, routine hematologic and biochemical tests, and pulmonary function tests. Fiberoptic bronchoscopy and bronchoalveolar lavage (BAL) for A1AT‐deficient patients was performed following patient consent and according to standardized guidelines as approved by the Beaumont Hospital Review Board committee.
Table 1.
Clinical profiles of human subjects.
| A1AT‐deficient | Normal controls patients | |
|---|---|---|
| Age (years) | 46 ± 2.8 | 24.2 ± 3.8 |
| Gender (% males) | 80 | 60 |
| Race (% Caucasians) | 100 | 100 |
| FEV1 (% predicted) | 43.2 ± 3.7 | 102 ± 4.4 |
Data are presented as mean ± SEM. A1 AT‐deficient patients are all PiZZ phenotype, confirmed by nephelometry and isoelectric focusing.
Healthy subjects
Healthy nonsmoking volunteers (control subjects) underwent fiberoptic bronchoscopy with BAL at the National Health and Environmental Effects Research Laboratory, U.S. Environmental Protection Agency (EPA) Research Triangle Park, NC, USA; the protocol and consent form were approved by the University of North Carolina School of Medicine Committee on the Protection of the Rights of Human Subjects. After adequate sedation and analgesia, the fiberoptic bronchoscope was wedged into a segment of the lingula or the right middle lobe and lavaged with five sequential 50 mL aliquots of sterile saline which were infused quickly with no dwell time between infusion and aspiration. BAL fluid from both lobes was combined and the percentage of recovered volume measured. Cells were separated from the BAL fluid at 2,000 rpm, 15 minutes. BAL was separated and stored at −70°C.
Bronchoalveolar lavage measures of iron and ferritin
Lavage (0.5 mL) was added to 0.5 mL 6 N HCl/20% trichoroacetic acid. This was hydrolyzed at 70°C for 18 hours, and centrifuged at 20,000 ×g for 10 minutes. Metal concentrations were determined in the supernatants using inductively coupled plasma optical emission spectroscopy (ICPOES; Model Optima 4300D, Perkin Elmer, Norwalk, CT, USA) operated at a wavelength of 238.204 nm. 14 Ferritin concentrations in BAL supernatant were quantitated by enzyme immunoassay according to the manufacturer's instructions (Microgenics, Concord, CA, USA). 14
In vivo rat experiments with neutrophil elastase intratracheal instillation
The Environmental Protection Agency's Institutional Animal Care and Use Committee reviewed and approved all procedures on animals. After anesthesia with 2% to 5% halothane (Aldrich Chemicals, Milwaukee, WI, USA), 60‐day‐old (250 g) male Sprague–Dawley rats (total n= 96) were intratracheally instilled with either 0.5 mL buffer or 50 μg human neutrophil elastase in 0.5 mL buffer. At 7, 14, and 28 days after exposure, rats were again anesthetized with halothane and euthanized. Specimens acquired included tracheal lavage with saline (35 mL/kg of body weight; n= 8 /exposure/time point) and inflation‐fixed lung (10% formalin; n= 4/exposure/time point).
BAL protein and lactate dehydrogenase (LDH) were evaluated as measures of NE‐induced lung injury. Lavage protein was determined using the Pierce Coomassie Plus Protein Assay Reagent (Pierce, Rockford, IL, USA). Bovine serum albumin served as the standard. Lavage LDH concentration was measured using a commercially prepared kit (Sigma, St. Louis, MO, USA). Both assays were modified for automated measurement (Cobas Fara II centrifugal analyzer).
To quantify the inflammatory cell influx in the lung following animal exposures, a modified Wright's stain (Diff‐Quick stain; American Scientific Products, McGaw Park, IL, USA) was used and cell differentials were expressed as the percentage of total cells recovered. After hydrolysis in an equivalent volume of 6 N HCl/20% trichloroacetic acid, BAL nonheme iron concentrations were quantified using ICPOES, Model Optima 4300D, (Perkin Elmer) operated at a wavelength of 238.204 nm. Lavage ferritin concentrations were measured using an enzyme immunoassay (Microgenics Corporation). Nonheme iron in resected lung tissue was measured after adding 10.0 mL 3 N HCl/10% trichloroacetic acid/gram tissue, heating to 70°C for 18 hours, and centrifuging at 20,000 g for 10 minutes. Metal concentrations were determined in the supernatants using ICPOES. Lavage cells (at 1.0 × 106/1.0 mL) were cytocentrifuged (0.2 mL) onto slides and stained for iron using Perl's Prussian Blue for iron. Lungs were inflation‐fixed with 10% formalin for 24 hours, parafin embedded, and 4 μm sections prepared for histology and immunohistochemistry. Iron was detected using Perl's Prussian blue stain. Immunohistochemical staining for ferritin was performed following blocking of endogenous peroxidase activity with 30% hydrogen peroxide in 30 mL methanol for 8 minutes. After treatment with Cyto Q Background Buster (Innovex Biosciences, Richmond, CA, USA) for 10 minutes at room temperature, slides were incubated with the primary antibody (rabbit antihuman L ferritin antibody; 1:100 dilution in 1% BSA in PBS) (Dako, Carpenteria, CA, USA) at 37°C, 45 minutes. Slides were then incubated with biotinylated antirabbit IgG antibody from Stat Q Staining System (Innovex Biosciences) for 10 minutes at room temperature, washed in PBS, and labeled with peroxidase enzyme label from Stat Q Staining System (Innovex Biosciences). Slides were developed with 3,3’ diaminobenzidine tetrahydrochloride for 3 minutes at room temperature and counterstained with hematoxylin. 15
Normal human bronchial epithelial cell culture
Primary normal human bronchial epithelial (NHBE) cells were harvested from human tracheobronchial tissues of donors obtained from the Lung Transplant Program and the Department of Pathology, Duke University Medical Center. The protocol was approved by the Institutional Review Board for Clinical Investigations, Duke University Medical Center. After initial harvest and expansion, cells were cultured submerged on 12‐well plastic plates in a small airway basal medium (SABM; Clonetics/Lonza, Walkersville, MD, USA): Dulbecco's modified Eagles Medium (DMEM) [1:1 ratio; Invitrogen, Carlsbad, CA] supplemented with 12 factors: insulin (4 μg/mL; Sigma), holotransferrin (5 μg/mL; Sigma), EGF (0.5 ng/mL; BD Biosciences, San Jose, CA, USA), dexamethasone (0.1 μM; Sigma), cholera toxin (20 ng/mL; List Biological Laboratories, Inc., Campbell, CA, USA), bovine hypothalamic extract (1:500; Pel‐Freeze Biologicals, Rogers, AR), nystatin (20 U/mL; Sigma), gentamicin (50 μg/mL; Invitrogen), amphotericin B (250 ng/mL; Invitrogen), HEPES (1.5mM; Invitrogen), T3 (6.5 ng/mL; Sigma), and BSA (0.5 mg/mL; Sigma). NHBE cells were grown to confluence for all studies.
Assessment of intracellular iron using calcein‐acetoxymethyl ester
The assay for intracellular iron concentration is based on the fluorescent and metal chelating properties of calcein‐acetoxymethyl ester (Calcein‐AM; Invitrogen). Calcein‐AM is a nonfluorescent lipophilic ester that penetrates the cell membrane and is cleaved by cytosolic esterases resulting in intracellular capture of a fluorescent alcohol with the capacity to chelate catalytically active iron. Upon iron chelation, the calcein green fluorescence is quenched, and this property can be used to quantitate the intracellular labile iron pool. 16 Fluorescence decreases as the amount of nonheme iron increases. Cells were loaded with calcein‐AM (0.5 μM, 1 hour) in the media described above. Following loading, the media was removed and cells were treated in one of five conditions in an iron‐free media (RPMI; Invitrogen): control, neutrophil elastase alone (NE, 100 nM; 875 U/mg protein, Elastin Products, Co., Owensville, MO, USA), ferric ammonium citrate (FAC, positive control, 200 μM; Sigma), ferritin alone (500 ng/mL; horse spleen ferritin, Sigma, Cat # F4503), and ferritin plus NE. Cells were incubated for 4 hours at 37°C. After incubation, media was removed and cells were rinsed twice with PBS. Then 500 μL of PBS (Invitrogen) were added to each well and the plate was analyzed for fluorescence in a fluorescent plate reader (Safire II, TECAN USA, Research Triangle Park, NC, USA). The data are expressed as a percentage decrease in absorbance compared to control‐treated cells.
Preparation of 57Fe‐loaded ferritin
Horse spleen apoferritin (Sigma) was loaded with 57FeCl2 as previously described. 17 Briefly, an apo‐ferritin solution (0.25 μM, 2.5 mL) was prepared in 0.1 M Mops buffer pH 7.4, 0.05 M NaCl, and iron ions were added from a 0.010 M 57FeCl2 stock solution to a final concentration of 50 μM (approximately 200 Fe molecules/ferritin molecule). The iron loading into ferritin was monitored spectrophotometrically at 350 nm. This process was repeated 9 times to achieve a theoretical loading of 2000 Fe molecules/ferritin molecule. After the loading of iron, unbound iron was removed using Amicon Ultra centrifugal filter devices with a 30,000 molecular weight cutoff. 57Fe‐loaded ferritin was diluted in 0.1 M MOPS solution and protein concentrations were determined using the BioRad assay (Bio‐Rad, Hercules, CA, USA).
57Fe‐loaded ferritin experiment
Cells were treated in one of four conditions in an iron free media (RPMI): control, NE (100 nM) alone, 57Fe‐ loaded ferritin (100 ng/mL), and 57Fe‐ loaded ferritin plus NE. Cells were incubated for 4 hours at 37°C, media was removed, and cells were collected using 10% Tricarboxylic acid/HCl. 57Fe concentration was quantified on a Perkin Elmer Elan 6000 inductively coupled plasma mass spectrometer (ICPMS) with a Scott cross‐flow nebulizer to minimize oxide formation. The settings were: power = 700 W, nebulizer flow = 1.3 L/min, lens = 3 V (static). Co was added at a level of 50 ppb as an internal standard using an online internal standard addition kit (Perkin Elmer, N0690673).
Ferritin degradation assay
Degradation of ferritin by neutrophil elastase was evaluated by ELISA and western analysis. For the ELISA, 50 ng/mL ferritin (type V from human spleen; Sigma, Cat # F6879, Lot 078K1463)/mL Dulbecco's PBS (Invitrogen) was incubated for 1 hour at 37°C with neutrophil elastase (0 to 250 nM). At the end of incubation, the sample was frozen at −80°C. After thawing, ferritin in the sample was measured using an ELISA (Alpha Diagnostics International, San Antonio, TX, USA). This ELISA utilizes two different mouse monoclonal antibodies to capture and detect intact human ferritin but the specific epitopes of each antibody are unknown. For the western analysis, horse spleen ferritin (Sigma, Cat # F4503, Lot 098K7009) was vortexed vigorously for 30 seconds prior to dilution to 1 μg/mL in Dulbecco's PBS (+Ca/+Mg). Neutrophil elastase (1 μM) or control vehicle was added to 20 μL of this diluted ferritin and incubated for 1 hour, at 37°C with shaking. SDS‐PAGE loading buffer was added to the incubation reactions and mixed but not boiled, and all samples were separated on a 4% to 15% Tris‐HCl SDS‐ polyacrylamide gel (Bio‐Rad). After transfer to nitrocellulose (Bio‐Rad), membranes were blocked with 5% milk in TBS‐Tween 20 (0.01%), 1 hour, room temperature. Ferritin was detected by incubating overnight with rabbit antihorse spleen ferritin antibody (1:1000 dilution; Sigma, Cat # F6136, Lot 019K4787) in 5% BSA in TBS‐Tween 20. After washing, membranes were incubated with peroxidase‐conjugated goat antirabbit IgG used as the secondary antibody (1:2000 dilution; Cell Signaling, Danvers, MA, USA) and developed with ECL‐Plus according to the manufacturer's instructions (GE Healthcare Biosciences Corp., Piscataway, NJ, USA). Autoradiographs were scanned and densitometric analysis was performed with ImageQuant TL software (GE Healthcare Bio‐sciences Corp.).
Results
Alpha‐1‐antitrypsin deficient patients have increased concentrations of iron and ferritin in bronchoalveolar lavage
A1AT‐deficient patients have increased oxidative stress that has been attributed to both inflammation and loss of alpha‐1‐antitrypsin activity 18 We hypothesized that this oxidative stress may be due to increased levels of catalytically active iron in the lung. Using ICP optical emission spectroscopy to quantitate iron, and an ELISA for ferritin, we observed that A1AT‐deficient patients have increased concentrations of both iron and ferritin in the BAL compared to normal control subjects ( Figure 1 ).
Figure 1.
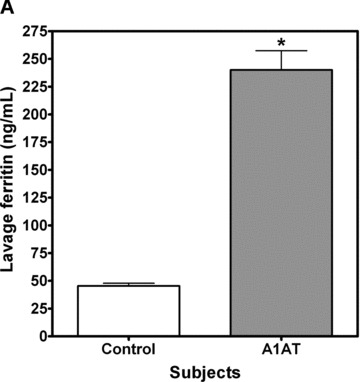
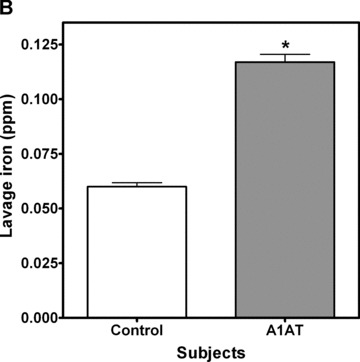
Iron and ferritin quantitation in bronchoalveolar lavage. Patients with A1AT deficiency (n= 6) and healthy control subjects (n= 6) had bronchoalveolar lavage to evaluate for iron and ferritin levels. Iron was measured by ICPOES and ferritin was determined by enzyme immunoassay. Results demonstrate that BAL ferritin (A) and BAL iron (B) were significantly increased in A1AT‐deficient subjects; (Mean ± SEM, *A1AT‐deficient patients significantly different from control subjects, p < 0.05).
Rats exposed to NE have increased concentrations of iron and ferritin in the lower respiratory tract
To determine whether NE exposure directly increased airway nonheme iron, we exposed Sprague–Dawley rats to intratracheal neutrophil elastase and evaluated whether, over 7 to 28 days following exposure, there were changes in BAL nonheme iron and ferritin. Relative to instillation of buffer only, lavage concentrations of nonheme iron were increased 7 days following elastase ( Figure 2B ). Concentrations of BAL nonheme iron remained elevated throughout the duration of study. Similarly, ferritin concentrations in the lavage were increased ( Figure 2A ). Lung nonheme iron concentrations almost doubled after elastase exposure ( Figure 2C ); levels remained elevated at 28 days after instillation. Lavage concentrations of both protein and LDH were significantly elevated at 7 days after exposure to elastase ( Figure 2D and E ). These results reflect minor injury that resolved by 14 and 28 days postinstillation.
Figure 2.



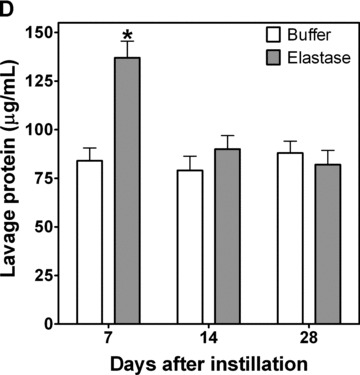
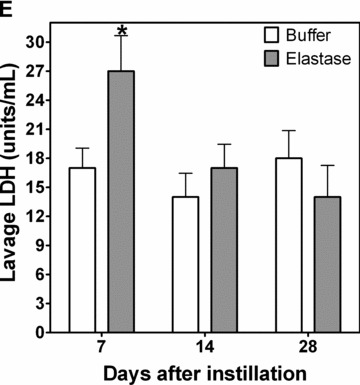
BAL ferritin and iron and lung iron in rats treated with NE. Rats were treated with a single intratracheal instillation of control vehicle or NE. At 7, 14, and 28 days postexposure, BAL was performed for ferritin quantitation (A) by enzyme immunoassay, iron quantitation (B) by ICPOES, protein quantitation (D) and LDH analysis (E). Lungs were harvested, hydrolyzed, and supernatant was prepared for iron content by ICPOES (C). BAL analysis for protein quantitation (D) and LDH analysis (E) were performed to evaluate for lung injury postelastase. Graphs summarize mean ± SEM; n= 8 rats per treatment condition and time point. *NE‐exposed rats had greater ferritin and iron levels than control‐treated rats, p < 0.05.
Iron‐laden macrophages were detected in NE‐treated rats starting at 7 days and these persisted at 28 days post‐NE instillation ( Figure 3A–D ); no sideromacrophages were observed in those animals exposed to buffer. Lung section immunohistochemistry similarly revealed increased ferritin staining in macrophages after elastase exposure ( Figure 3E and F ). There was no evidence of pulmonary hemorrhage/alveolar red blood cells at time points > 7 days (data not shown).
Figure 3.
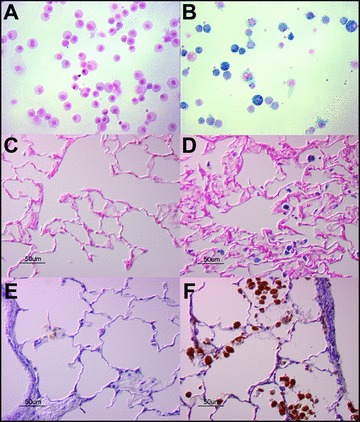
Immunohistochemistry for iron and ferritin in rat lung sections following NE or control vehicle intratracheal instillation. Sprague‐Dawley rats were exposed to NE (50 μg; B, D, F) or to control vehicles (A, C, E) via oral aspiration. Twenty‐eight days following exposure, rats were sacrificed and were lavaged (A, B) or lungs were harvested and fixed for histology and immunohistochemistry (C–F). BAL cell pellets (A, B) were stained with Perl's Prussian blue stain for iron. Lung sections were stained for iron (C, D) and for ferritin (E, F). Micrographs are representative of n= 4 animals. NE increased both iron staining and ferritin staining.
Importantly, BAL cell dif erentials revealed an inflammatory response to NE at 7 days. Neutrophils were significantly increased at this time point (21.6 ± 9.2% and 1.2 ± 0.8 of total cell count after NE and buffer, respectively). By 14 days, this inflammatory influx had resolved. These results were consistent with the findings from A1AT‐deficient patients suggesting that unopposed airway NE activity could induce increased airway iron and ferritin.
NHBE exposed to NE have increased intracellular nonheme iron
We have previously shown that NE increases reactive oxygen species (ROS) in A549 lung cancer cells and exposure to desferrioxamine, an iron chelator, blocked this ROS production. 19 To evaluate whether NE increased cell iron levels, we loaded cells with the fluorescent probe, calcein‐AM, which is modified by cytosolic esterases resulting in an intracellular fluorescent probe. Calcein chelates nonheme iron resulting in quenching of fluorescence and has been used to quantify the labile iron pool in the cytosol. 16 NHBE cells exposed to NE had increased nonheme iron concentrations as detected by decreased calcein fluorescence ( Figure 4 ). A positive control, ferric ammonium citrate, also caused significant quenching of calcein fluorescence consistent with increased intracellular nonheme iron. The response to NE required the presence of both the protease and the iron‐containing protein, ferritin, in the media; NE alone in iron‐free, protein‐free medium had no effect on intracellular nonheme iron content. Therefore, we hypothesized that NE degrades iron‐containing proteins in the extracellular milieu resulting in the release of iron and its uptake into the cell. To test this hypothesis, we exposed NHBE to 57Fe‐loaded ferritin in the presence or absence of NE ( Figure 5 ). Although in the presence of 57Fe‐loaded ferritin alone there was a small increase in intracellular 57Fe, the increase in intracellular 57Fe was significantly greater in the presence of NE.
Figure 4.
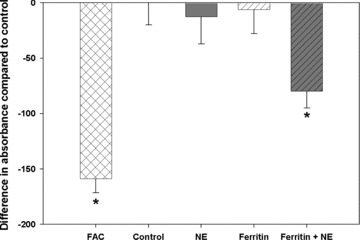
Calcein‐AM fluorescence assay for intracellular nonheme iron. Normal human bronchial epithelial cells were preloaded with calcein‐AM for 1 hour and then exposed to control vehicle, NE (100 nM), ferritin (500 ng/mL), or ferritin and NE (4 hours, 37°C). Ferric ammonium citrate (FAC, 200 μM, 1 hour) treatment was used as a positive control. Data are expressed as quenching of fluorescence compared to control treatment conditions. Data represent mean ± SEM, n= 6–9 samples from 3 separate experiments. *Ferritin and NE, or FAC are significantly different from control, NE, or ferritin alone (p < 0.05).
Figure 5.

57Fe‐ferritin assay for NE‐induced iron uptake. Apoferritin was loaded with 57Fe which was confirmed by ICPMS. NHBE cells were exposed to control vehicle or to 57Fe‐ferritin (100 ng/mL) in the presence or absence of NE (100 nM, 4 hour). Cellular uptake of 57Fe was determined by ICPMS of cell lysates. In the presence of 57Fe‐ferritin alone, there was some uptake of 57Fe reflecting a degradation of ferritin (*p < 0.05). However, there was significantly more intracellular 57Fe following incubation of 57Fe‐ferritin and NE (**p < 0.01). Data represent mean ± SEM, n= 6, 2 separate experiments.
NE degrades ferritin
To test whether ferritin could function as a target for the proteolytic action of NE, in vitro incubations of NE and ferritin were conducted. A human ferritin ELISA, based on two mouse monoclonal antibodies, was used to measure intact ferritin concentrations. Significant decrements in intact human spleen ferritin concentrations recognized by ELISA were noted after 1 hour with 150 or 250 nM neutrophil elastase, supporting ferritin degradation ( Figure 6A ). To confirm that NE degrades ferritin, potentially releasing the metal, degradation of horse spleen ferritin was detected by western analysis using a rabbit affinity purified antihorse spleen ferritin antibody. Western analysis results confirm ELISA results and demonstrate that ferritin is digested by NE from a large multimer (approximately 250 kD) to at least two degradation products (approximately 30 and 25 kD) following NE treatment for 1 hour ( Figure 6B and C ). This was associated with a decrease in the density of the multimer band. Together, these results support a role for NE to act in the extracellular milieu to degrade iron‐containing proteins, releasing iron and potentially increasing concentrations of nonheme iron available for cellular uptake.
Figure 6.
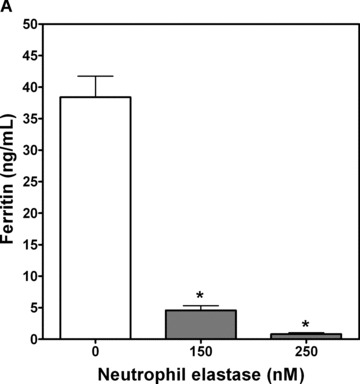
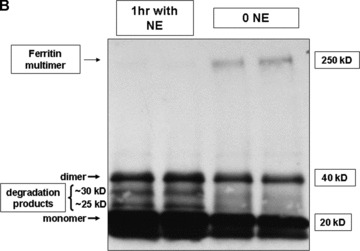
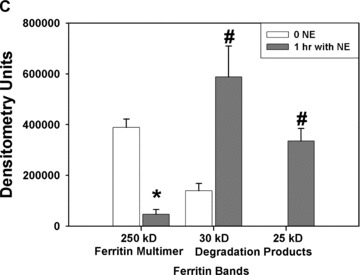
In vitro assays for ferritin degradation by neutrophil elastase. (A) Human spleen ferritin (50 ng/mL) was incubated for 1 hour with control vehicle or NE (150 and 250 nM). Intact ferritin concentrations were then measured by enzyme immunoassay. Significant decreases in intact human spleen ferritin concentrations were observed at both concentrations of NE tested (*significantly different from control ferritin levels, mean ± SEM, p < 0.05, n= 9/treatment group from two experiments). (B and C) Horse spleen ferritin (1 μg/mL) was incubated for 1 hour with NE (1 μM) or control vehicle. The reaction mixture was mixed with SDS‐PAGE loading buffer, but not boiled, prior to applying the sample to a 4% to 15% SDS‐polyacrylamide gel. Following transfer to nitrocellulose, the membrane was incubated with rabbit affinity‐purified antihorse spleen ferritin antibody and secondary goat antirabbit HRP‐conjugated secondary antibody, followed by ECL plus development. Western analysis is representative of four experiments (B). Protein bands were quantitated by densitometry, and the relative band densities following control treatment or NE treatment for the ferritin multimer (approximately MW 250 kD) and the degradation products (approximately 30 kD and 25 kD) are summarized graphically (C), mean ± SEM, n= 4 to 8 from four separate experiments. *NE 250 kD ferritin multimer significantly less than control, p < 0.05. #NE 30 kD and 25 kD degradation products significantly greater than control, p < 0.05.
Discussion
A1AT‐deficient patients have increased pulmonary iron and ferritin
Our results confirm that A1AT‐deficient patients have elevated iron and ferritin levels in the BAL. These results are similar to those found in CF patients. 10 , 20 Interestingly, both patient groups are characterized by excessive NE activity and protease capacity in the airway surface lining fluid. These observations raise the possibility that there is a link between high levels of airway protease and high concentrations of nonheme iron in the airway. To test this relationship directly, we exposed rats to intratracheal NE and found persistent elevations in iron concentrations of the lavage and lung.
NE and increased nonheme iron level
We then evaluated in vitro for a mechanism by which NE protease activity could increase available iron levels. Our experiments demonstrate that NE degrades iron‐containing proteins in the extracellular fluid resulting in release of nonheme iron. The action of NE to release protein‐bound iron is not unique to NE. Other proteases, serine and nonserine proteases, including pseudomonas elastase, porcine pancreatic elastase, and trypsin can degrade transferrin 3 and lactoferrin 4 releasing iron.
This nonheme iron is likely taken up by cells resident in the lower respiratory tract including epithelial cells and macrophages. Cell import is predicted to be both transferrin and nontransferrin dependent. 21 This transport likely results in an increase in the intracellular labile iron pool, a cytosolic domain of nonheme, catalytically active iron. 22 Increased iron in the labile iron pool is sensed by iron regulatory proteins that upregulate ferritin translation. Ferritin is a major intracellular storage protein that sequesters iron, diminishing catalytic activity. 21 However, ferritin can be released from macrophages into the extracellular space. 23 This mechanism may explain the increased level of ferritin in the lining fluid and sputum of patients with A1AT‐deficiency and CF. Such sequestration of iron by ferritin in the lower respiratory tract does not absolutely block the potential to mount an oxidative stress as ferritin can present an additional target for proteases or reductants to release iron. 23
NE and oxidative stress
NE has been reported to increase reactive oxygen species by several mechanisms including increased mitochondrial oxidant release, 24 or activation of oxidoreductases such as dual oxidase (DUOX) 25 and NADP(H) quinone oxidoreductase 1 (NQO1). 26 In this report, we present evidence that NE also increases iron concentrations in cells and this will contribute to oxidative stress. Nonheme iron is a catalyst for the generation of reactive oxygen species via the Haber–Weiss reaction including superoxide, hydrogen peroxide, and hydroxyl radical. 21
Conclusion
Impact of iron on pulmonary health
Iron is essential for life but must be strictly sequestered in heme or iron‐binding proteins to prevent host injury. Iron participates in numerous lung injuries including those after exposure to pollutants including silica dust, coal dust, oil fly ash, welding dust, tobacco smoke, and diesel particles. 27 In addition, available iron can participate in lung disease 27 such as acute respiratory distress syndrome, postcardiopulmonary bypass, pulmonary hemorrhage, and pulmonary alveolar proteinosis. 14 Importantly, increased airway iron promotes infection with microorganisms including bacteria, protozoans, and fungus. Iron is scavenged from the host using siderophores/receptors and ferrireductases present on microbial membranes. Host sources of iron, such as transferrin and lactoferrin, can then be utilized as sources of metal required for microbial proliferation. 2 Therefore, host responses to increased airway nonheme iron are critical to protect against oxidative stress and to prevent infection. Our report suggests that NE is another participant in the host battle for essential iron. Elastase in the lower respiratory tract may degrade iron‐containing proteins including ferritin, lactoferrin, and transferrin, releasing catalytically active iron. This results in an alteration of iron homeostasis, accumulation of metal required for microbial proliferation, and increased risk of infection. Furthermore, the release of the iron by degradation of the storage proteins promotes extracellular‐to‐intracellular oxidative stress in the airway epithelium. Importantly, our findings suggest a new therapeutic target in the treatment of A1AT deficiency.
Acknowledgment
This study was financially supported by National Institute of Health grants HL082504 (JAV) and HL081763 (BMF).
References
- 1. Weinberg ED. Iron and infection. Microbiol Rev. 1978; 42(1): 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marx JJ. Iron and infection: competition between host and microbes for a precious element. Best Pract Res Clin Haematol. 2002; 15(2): 411–426. [PubMed] [Google Scholar]
- 3. Miller RA, Britigan BE. Protease‐cleaved iron‐transferrin augments oxidant‐mediated endothelial cell injury via hydroxyl radical formation. J Clin Invest. 1995; 95(6): 2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Britigan BE, Hayek MB, Doebbeling BN, Fick RB Jr. Transferrin and lactoferrin undergo proteolytic cleavage in the Pseudomonas aeruginosa‐infected lungs of patients with cystic fibrosis. Infect Immun. 1993; 61(12): 5049–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okazaki I, Brinckerhoff CE, Sinclair JF, Sinclair PR, Bonkowsky HL, Harris ED Jr. Iron increases collagenase production by rabbit synovial fibroblasts. J Lab Clin Med. 1981; 97(3): 396–402. [PubMed] [Google Scholar]
- 6. Brumlik MJ, Storey DG. Zinc and iron regulate translation of the gene encoding Pseudomonas aeruginosa elastase. Mol Microbiol. 1992; 6(3): 337–344. [DOI] [PubMed] [Google Scholar]
- 7. Shigematsu T, Fukushima J, Oyama M, Tsuda M, Kawamoto S, Okuda K. Iron‐Mediated regulation of alkaline proteinase production in Pseudomonas aeruginosa. Microbiol Immunol. 2001; 45(8): 579–590. [DOI] [PubMed] [Google Scholar]
- 8. Gardi C, Arezzini B, Fortino V, Comporti M. Effect of free iron on collagen synthesis, cell proliferation and MMP‐2 expression in rat hepatic stellate cells. Biochem Pharmacol. 2002; 64(7): 1139–1145. [DOI] [PubMed] [Google Scholar]
- 9. Lucey EC, Stone PJ, Christensen TG, Breuer R, Snider GL. An 18‐month study of the effects on hamster lungs of intratracheally administered human neutrophil elastase. Exp Lung Res. 1988; 14(5): 671–686. [DOI] [PubMed] [Google Scholar]
- 10. Reid DW, Lam QT, Schneider H, Walters EH. Airway iron and iron‐regulatory cytokines in cystic fibrosis. Eur Respir J. 2004; 24(2): 286–291. [DOI] [PubMed] [Google Scholar]
- 11. Stites SW, Plautz MW, Bailey K, O’Brien‐Ladner AR, Wesselius LJ. Increased concentrations of iron and isoferritins in the lower respiratory tract of patients with stable cystic fibrosis. Am J Respir Crit Care Med. 1999; 160(3): 796–801. [DOI] [PubMed] [Google Scholar]
- 12. Rabinovitz M, Gavaler JS, Kelly RH, Van Thiel DH. Association between heterozygous alpha 1‐antitrypsin deficiency and genetic hemochromatosis. Hepatology. 1992; 16(1): 145–148. [DOI] [PubMed] [Google Scholar]
- 13. Bonkovsky HL, Lambrecht RW. Iron‐induced liver injury. Clin Liver Dis. 2000; 4(2): 409–429, vi–vii. [DOI] [PubMed] [Google Scholar]
- 14. Ghio AJ, Stonehuerner JG, Richards JH, Crissman KM, Roggli VL, Plantadosi CA, Carraway MS. Iron homeostasis and oxidative stress in idiopathic pulmonary alveolar proteinosis: a case‐control study. Respir Res. 2008; 9: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghio AJ, Carter JD, Samet JM, Quay J, Wortman IA, Richards JH, Kennedy TP, Devlin RB. Ferritin expression after in vitro exposures of human alveolar macrophages to silica is iron‐dependent. Am J Respir Cell Mol Biol. 1997; 17(5): 533–540. [DOI] [PubMed] [Google Scholar]
- 16. Cabantchik ZI, Glickstein H, Milgram P, Breuer W. A fluorescence assay for assessing chelation of intracellular iron in a membrane model system and in mammalian cells. Anal Biochem. 1996; 233(2): 221–227. [DOI] [PubMed] [Google Scholar]
- 17. Polanams J, Ray AD, Watt RK. Nanophase iron phosphate, iron arsenate, iron vanadate, and iron molybdate minerals synthesized within the protein cage of ferritin. Inorg Chem. 2005; 44(9): 3203–3209. [DOI] [PubMed] [Google Scholar]
- 18. Taraseviciene‐Stewart L, Voelkel NF. Molecular pathogenesis of emphysema. J Clin Invest. 2008; 118(2): 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol. 2002; 26(4): 447–452. [DOI] [PubMed] [Google Scholar]
- 20. Stites SW, Walters B, O’Brien‐Ladner AR, Bailey K, Wesselius LJ. Increased iron and ferritin content of sputum from patients with cystic fibrosis or chronic bronchitis. Chest. 1998; 114(3): 814–819. [DOI] [PubMed] [Google Scholar]
- 21. Turi JL, Yang F, Garrick MD, Piantadosi CA, Ghio AJ. The iron cycle and oxidative stress in the lung. Free Radic Biol Med. 2004; 36(7): 850–857. [DOI] [PubMed] [Google Scholar]
- 22. Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic Biol Med. 2002; 33(8): 1037–1046. [DOI] [PubMed] [Google Scholar]
- 23. Wesselius LJ, Nelson ME, Skikne BS. Increased release of ferritin and iron by iron‐loaded alveolar macrophages in cigarette smokers. Am J Respir Crit Care Med. 1994; 150(3): 690–695. [DOI] [PubMed] [Google Scholar]
- 24. Aoshiba K, Yasuda K, Yahui S, Tamaoki J, Nagai A. Serine proteases increase oxidative stress in lung cells. Am J Physiol Lung Cell Mol Physiol. 2001; 281: L556–L564. [DOI] [PubMed] [Google Scholar]
- 25. Shao MX, Nadel JA. Dual oxidase 1‐dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proc Natl Acad Sci USA. 2005; 102(3): 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng S, Byrd AS, Fischer BM, Grover AR, Ghio AJ, Voynow JA. Regulation of MUC5AC expression by NAD(P)H:quinone oxidoreductase 1. Free Rad Biol Med. 2007; 42(9): 1398–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quinlan GJ, Evans TW, Gutteridge JM. Iron and the redox status of the lungs. Free Radic Biol Med. 2002; 33(10): 1306–1313. [DOI] [PubMed] [Google Scholar]