

Abstract
Adoptively transferred T cells possess anticancer activities partially mediated by T-cell FasL engagement of Fas tumor targets. However, antigen-induced T-cell activation and clonal expansion, which stimulates FasL activity, is often inefficient in tumors. As a gene therapy approach to overcome this obstacle, we have created oncoretroviral vectors to overexpress FasL or non-cleavable FasL (ncFasL) on murine T cells of a diverse T-cell receptor repertoire. Expression of c-FLIP was also engineered to prevent apoptosis of transduced cells. Retroviral transduction of murine T lymphocytes has historically been problematic, and we describe optimized T-cell transduction protocols involving CD3/CD28 co-stimulation of T cells, transduction on ice using concentrated oncoretrovirus, and culture with IL-15. Genetically modified T cells home to established prostate cancer tumors in vivo. Co-stimulated T cells expressing FasL, ncFasL and ncFasL/c-FLIP each mediated cytotoxicity in vitro against RM-1 and LNCaP prostate cancer cells. To evaluate the compatibility of this approach with current prostate cancer therapies, we exposed RM-1, LNCaP, and TRAMP-C1 cells to radiation, mitoxantrone, or docetaxel. Fas and H-2b expression were upregulated by these methods. We have developed a novel FasL-based immuno-gene therapy for prostate cancer that warrants further investigation given the apparent constitutive and inducible Fas pathway expression in this malignancy.
Keywords: apoptosis, c-FLIP, docetaxel, Fas, gene transfer, mitoxantrone
Introduction
Adoptive cell transfer of T lymphocytes has demonstrated successful tumor regression in clinical trials.1,2 In mouse models of prostate cancer, adoptive cell transfer of genetically engineered T cells showed a potential for tumor eradication and increased survival in mice.3 Although such responses are promising, one obstacle to T-cell therapy is the requirement for T-cell activation through the TCR with subsequent clonal expansion not only to generate sufficient numbers for effective tumor rejection but to activate effector molecules such as FasL to become involved in actual tumor killing. This may pose an obstacle to effective T-cell therapy, as in some cases a suitable tumor antigen may not be available for TCR activation or T-cell tolerance to the tumor may be induced4 and in other cases, T cells may undergo antigen-induced cell death rather than clonal expansion prior to effector molecule expression.5
T cells exert their cytotoxicity through the release of perforin and granzymes, or by inducing apoptosis mediated by Fas ligand (FasL). Signaling through the FasL lytic pathway is triggered when low or high affinity T-cell receptor engagement of antigen occurs, whereas perforin/granzyme-mediated killing is dependent on high affinity TCR–antigen interactions.6 As tumor antigens are often weakly immunogenic, the role of FasL in T-cell-mediated tumor killing may be vitally important. In fact, FasL expression on T lymphocytes is necessary for their full cytotoxic activity against lung tumors in vivo7 and renal and prostate tumors in vitro.8 Furthermore, when cell surface density of FasL is increased on T cells by inhibiting its proteolytic cleavage to a soluble form, T-cell cytotoxicity through FasL is enhanced.9 As well, CD4−CD8− lymph node-derived cells from lpr mice, which lack functional Fas and naturally express higher than normal levels of FasL, have been shown to induce killing in Fas+ target cells whereas lymph node-derived cells from wild-type mice did not exert a similar killing effect.10
FasL has been demonstrated to have therapeutic efficacy in several prostate cancer models. Cisplatin-treated DU145 cells undergo Fas-mediated killing by patient-derived tumor infiltrating lymphocytes.8 Delivery of FasL cDNA by a prostate-restricted replicative adenovirus inhibited prostate tumor growth in mice.11 Furthermore, we have recently demonstrated that primary human prostate cancer cell lines are sensitive to killing by FasL-expressing K562 cells.12 Although systemic distribution of soluble FasL (sFasL) proteins or anti-Fas antibodies are lethal in vivo,13 cell surface-expressed FasL does not seem to share this potency. Injection of CD4−CD8− lymph node-derived cells from lpr mice with high FasL expression into tumor-bearing mice did not induce measurable toxicity.10
Based on these observations, we hypothesize that the antiprostate cancer potency of T cells may be improved by genetically modifying these cells to overexpress FasL in a stable context. We designed oncoretroviral vectors to engineer the expression of FasL or a modified, non-cleavable form of FasL (ncFasL). ncFasL has been reported to possess high local biological activity and to limit toxicity from systemic distribution of sFasL.14 This immuno-gene therapy method uses a polyclonal population of T cells generated ex vivo through anti-CD3 and anti-CD28 co-stimulation. This approach offers the following potential advantages: (1) such co-stimulation results in an activated T-cell phenotype that persists in vivo and maintains a capacity for tissue homing; (2) the polyclonal nature of the T-cell population obviates the need for clonal expansion and requisite long-term culture propagation; (3) FasL expression can be optimized to achieve supra-physiological levels of effector molecule function; and (4) novel gene engineering methods can be used to enhance the survival of the gene-modified T cells. Specifically, we reasoned that survival of T cells overexpressing FasL might be reduced due to suicidal or fratricidal Fas/FasL interaction. To overcome this potential obstacle, an additional construct engineers co-expression of both ncFasL and c-FLIPL. c-FLIPL has been shown to protect cells from Fas-mediated apoptosis15 without inducing accumulation of activated or autoreactive T cells when overexpressed in the lymphocyte compartment.16
To combine adoptive cell transfer approaches with overexpression of FasL in appropriate animal models, transduction of primary murine T lymphocytes is required. Genetically modified lymphocytes are a valuable tool under development for broad applications in cancer therapy. Although human T lymphocytes are amenable to retroviral transduction, their murine counterparts have proven more difficult to work with, as demonstrated by the limited number of studies that make use of this pre-clinical model. Several studies report optimizations to murine T-cell transduction, including the use of ecotropic viral particles,17–19 an optimized T-cell stimulation period prior to infection,17,19 and a centrifugation step during transduction (‘spinoculation’).19,20 Several published protocols make use of ‘ping-pong’ methods18–20 or co-culture17 to achieve high viral tires. These approaches present unacceptable safety risks due to the potential of cross-contamination of T-cell cultures with virus-producing cells and creation of replication-competent virus. We have further attempted to develop a reproducible and convenient method for primary murine T-cell transduction by comparing infection by ‘spinoculation’, on fibronectin-coated plates, at low temperature and using concentrated viral preparations. As well, the use of multiple transductions and various stimulation conditions are compared. As T cells activated by co-stimulation with anti-CD3/CD28 antibodies are highly functional, we developed a transduction protocol using these cells that combines efficiency with ease of handling for large numbers of T cells.
Prostate cancer is regularly treated with radiation therapy. Although chemotherapy is not curative, agents such as docetaxel and mitoxantrone are often used for palliation of symptoms. The DNA damage and stress responses induced by chemotherapeutic agents often stimulate increased surface expression of Fas receptor in tumor models.21–23 This result is not universal, as mitoxantrone induces Fas expression on LNCaP cells but not on PC-3 or DU145 cells.24 Although combined treatment of tumor cells with chemotherapeutic agents and FasL has instigated synergistic killing in some tumor models,22,25 no benefit is perceived in other situations.21,26 We investigated the potential for such synergistic prostate cancer cell killing in our FasL-immunotherapy model.
This report describes our optimized protocol for oncoretroviral transduction of murine T lymphocytes, and the impact of overexpressing FasL on T-cell capacity to lyse RM-1 and LNCaP prostate cancer cell lines. We also examine the effect of common prostate cancer therapeutics on Fas expression, major histocompatibility complex (MHC) expression and Fas-mediated killing.
Materials and methods
Cells and culture conditions
RM-1 cells were kindly provided by Dr T Thompson (Baylor College of Medicine, Houston, Texas) and cultured in DMEM (Sigma, Oakville, Canada) with 10% FCS, 10 mm HEPES (Invitrogen, Burlington, Canada), 100 U ml−1 penicillin and 100 mg ml−1 streptomycin (Sigma). GP + E86 cells27 were cultured in complete DMEM (10% FCS, 100 μg ml−1 penicillin and 100 mg ml−1 streptomycin, 2 mm l-glutamine). All other cell lines were obtained from ATCC (Rockville, MD). LNCaP cells were cultured in complete RPMI, NIH 3T3 cells in complete DMEM and TRAMP-C1 cells in high glucose DMEM (4.5 g l−1 glucose without sodium pyruvate, Sigma), with 5% FCS, 5% NuSerum IV culture supplement (BD Biosciences, Mississauga, Canada), penicillin, streptomycin, 10−8 M DHT and 5 μg ml−1 bovine insulin.
Splenocytes were collected from 3 to 7-month-old C57Bl/6 or B6.MRL-Faslpr/J mice (Jackson Laboratories, Bar Harbor, ME), separated through a 30 μm mesh, and RBCs lysed using red blood cell lysis buffer (Sigma). The T-cell population was enriched by depleting B cells with IgG-coated magnetic beads (Qiagen Biomag, Mississauga, Canada). Unless otherwise specified, T cells were cultured in RPMI-1640 medium containing 10% FCS, 2 mm l-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μm 2-mercaptoethanol, 3.3 μm N-acetyl cysteine (Sigma), 20 U ml−1 recombinant human IL-2 (Roche, Laval, Canada) and 20 ng ml−1 recombinant human IL-7 (Peprotech, Rocky Hill, NJ). Stimulation was performed using anti-CD3/anti-CD28-conjugated magnetic beads (CD3/CD28 beads) at a 3:1 bead-to-cell ratio. Beads were prepared by incubating anti-CD3 and anti-CD28 monoclonal antibodies (BD Biosciences) with M450 Dynabeads (Dynal ASA, Oslo, Norway) as described.28 Where indicated, IL-2 was substituted with 20 or 100 IU ml−1 recombinant human IL-15 (ebiosciences, San Diego, CA).
Construction of oncoretroviral vectors and stable virus-producing cell lines
Human FasL cDNA was ligated into the pCI-vector (Promega, Madison, WI). An NcoI restriction site was introduced at the 5′ end of FasL by site-directed mutagenesis (Stratagene, La Jolla, CA) to create pCI-FasL/NcoI. To produce ncFasL, the metalloproteinase cleavage site at aa 103–136 of FasL was removed14 from the pCI-Fas/NcoI vector by introducing PmeI and AscI sites around the cleavage site followed by digestion and blunt-end ligation, creating pCI-ncFasL/NcoI. c-FLIPL cDNA was PCR amplified from CLL cell mRNA. pSV-ncFasL/IRES/c-FLIP was constructed by ligating the encephalomyocarditis virus internal ribosomal entry site (IRES) sequence and c-FLIPL cDNA downstream of ncFasL. An NcoI/NotI digest was used to introduce the transgene fragments from these three vectors into the Moloney murine leukemia virus-based backbone pUMFG29 resulting in pUMFG-FasL, pUMFG-ncFasL and pUMFG-ncFasL/c-FLIP vectors. Integrity of the constructs was confirmed by sequencing (UHN Research DNA Sequencing Facility).
pUMFG vectors were co-transfected along with the neomycin resistance-containing vector pGT-N28 into the ecotropic virus packaging cell line GP + E8630 using CaCl2 in the presence of 25 μm chloroquine.27 Transfected cells were selected with G418. Double-transfected, FasL-expressing populations were enriched by fluorescence-activated flow sorting on a Modulator Flow Cytometer (MoFlo, Dako, Glostrup, Denmark). Transgene expression was then re-confirmed by flow cytometry. The resulting virus particles carry the Moloney murine leukemia virus envelope protein. Titer was determined by transduction of naive NIH 3T3 cells with serial dilutions of viral supernatant in the presence of 8 μg ml−1 protamine sulfate and found to range from 2.5 × 105 to 2 × 106 infectious particles per ml (ip ml−1).
T-cell transduction
Supernatants were collected from the various E86 cell lines and passed through a 0.45 μm filter to remove cellular debris. Supernatants were used immediately or concentrated 100-fold by 12 h centrifugation at 9000 g at 4 °C in a Sorvall RCSCPlus centrifuge (Mandel Scientific, Guelph, Canada). Viral pellets were resuspended in 10% TNE (10 mm Tris-HCl, pH 7.2, 0.1 M NaCl, 1 mm EDTA) in PBS. Concentrated viral titers typically ranged from 1.3 × 107 to 1.5 × 108 ip ml−1. Viral preparations were used immediately to transduce murine splenic T lymphocytes.
T cells were stimulated for 24–48 h with 3:1 anti-CD3/CD28 beads, 2.5 μg ml−1 PHA (Sigma), or 5 μg ml−1 ConA (Sigma), as indicated. For T-cell transduction, typically 3 × 106 T cells were resuspended in 100 μl T-cell medium and 300 μl-concentrated virus (MOI ≈ 15) in 1.5 ml Eppendorf tubes and left on ice for 3 h, as described.31 Following this incubation, 400 μl T-cell medium and 8 μg ml−1 protamine sulfate were added to each tube and cells transferred to 6-well plates for a further 6 h at 37 °C, with 5% CO2 before an additional 1 ml of T-cell medium and 8 μg ml−1 protamine sulfate were added. For transduction on fibronectin, 6-well plates were first coated with 50 μg ml−1 fibronectin (Roche, Laval, Canada) for 45 min before addition of 3 × 106 T cells, 2 ml viral supernatant and 8 μg ml−1 protamine sulfate. For spinoculation protocols, cells were centrifuged for 1 h at 1000 g and 32 °C in 6-well plates at the beginning of the transduction period. The following day cells were washed and placed in fresh T-cell medium. Transgene expression was confirmed on the transduced cells by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA) for eGFP or FasL; the latter detected with PE-conjugated anti-human FasL antibody, (clone NOK-1; ebiosciences). T-cell phenotype was determined by flow cytometry using antibodies against murine CD3ε (ebiosciences), CD4 (ebiosciences), CD8a (BD Biosciences) and FoxP3 (ebiosciences). Where indicated, 20 μm Z-VAD-FMK (BD PharMingen, Mississauga, Canada) was added to T-cell cultures at the time of transduction and supplemented with subsequent media changes. Cells were carefully washed in PBS prior to use in cytotoxicity assays.
3H-incorporation assays
To determine relative rates of cell growth, 105 T cells were plated in triplicate in 96-well plates in 100 μl total volume. 3H-thymidine (GE Healthcare, Baie d'Urfe, Canada) was added at 1 μCi per well and cells were incubated 6 h prior to collection with a cell harvester (PHD, Cambridge Technologies, Cambridge, MA) onto glass-filter paper (Brandel Inc., Gaithersburg, MD). Samples were analyzed on a Beckman scintillation counter. For assays involving multiple time points, cells were all plated on the same day, but 3H-thymidine was added on the day of analysis.
Monitoring in vivo T-cell trafficking
Nine to 11-week-old C57Bl/6 mice were injected with 3 × 105 cloned RM-1 cells subcutaneously (s.c.) in the flank. Prior to this, T cells were transduced with pUMFG-eGFP viral vectors, resulting in 7% eGFP marking. T cells were also labeled by 10 min incubation with 5 μm CFSE (Invitrogen). FCS was then added to a final concentration of 25% for 3 min before washing cells in PBS. All anti-CD3/CD28 beads were magnetically removed and 107 T cells were injected either intravenously (i.v.) (n = 6) or s.c. into the tumor site (n = 3) in mice bearing 6 days RM-1 tumors. T cells were also given i.v. to two tumor-free control mice. At 16 and 40 h after T-cell injection, mice were killed and peripheral blood, spleens, axillary and mesenteric lymph nodes, lungs, and tumors were collected and single-cell suspensions were made by finely mincing organs and passing suspensions through a 30 μm cell strainer (BD Falcon, Mississauga, Canada). RBCs were lysed using red blood cell lysing buffer (Sigma) in the lung, spleen, and peripheral blood samples to minimize cell number. Organs were also collected from untouched control mice. Samples from each organ were left unstained or labeled with anti-CD3ε or isotype-matched control antibody (BD Biosciences) and 105 events were analyzed by flow cytometry using CellQuest Pro software. Data is presented as the percentage of CD3+ cells that were also labeled with CFSE.
51Cr-release assays
Cytotoxicity induced by gene-modified T cells was assessed by 51Cr-release assays. Target RM-1 cells were labeled with 110 μCi Na2[51Cr]O4/106 cells (Perkin Elmer, Boston, MA) for 2 h. Cells were plated at 1–2 × 104 cells per well in round-bottom 96-well plates (BD Falcon). T cells were added at 1:1, 10:1, 25:1 or 50:1 ratios. Control wells contained labeled RM-1 cells in T-cell media alone (background release) or in media with 1% Triton X-100 (maximum release). Plates were centrifuged briefly and incubated at 37 °C with 5% CO2 for 6 h. 100 μl of cell supernatants were collected and analyzed on a 1277 GammaMaster gamma counter (Perkin Elmer). Percent cytotoxicity was calculated as (experimental c.p.m.—background c.p.m.) / (maximum release c.p.m.—background c.p.m.). In some groups, 4 mm EGTA and 3 mm MgCl2 (Fisher Scientific, Nepean, Canada) were added to T cells 1 h prior to 51Cr-release assays to prevent exocytosis-mediated killing.32 Fas-mediated killing was blocked by the addition of 1–40 μg ml−1 NOK-1 anti-FasL antibody.
Flow cytometric analysis for Fas and H-2b expression
Levels of cell surface Fas expression were tracked over time following exposure of RM-1, TRAMP-C1, or LNCaP cells to 1–1000 nm mitoxantrone (Sigma), 1–100 nm docetaxel, or 2–8 Gy irradiation (Gamma Cell Irradiator 37Cs source; MDS Nordion, Ottawa, Canada) as indicated below. Expression of H-2b was also followed on RM-1 cells. Following 4 to 72 h incubation, cells were harvested with EDTA-containing buffer and blocked with 10% mouse serum. Cells were labeled with PE-anti-mouse Fas (ebiosciences), APC-anti-human Fas (clone DX2; BD Biosciences), biotin-anti-mouse H-2 Kb/H-2 Db (BD Biosciences) followed by streptavidin-PE labeling (BD Biosciences), or isotype-matched control antibody and analyzed by flow cytometry. Live cells were distinguished by 7-AAD exclusion. Results were interpreted as the mean fluorescence intensity (MFI) of (antibody-labeled cells) – (isotype-labeled cells) and normalized to that of untreated control cells.
Statistical analysis
Data is presented as mean ± s.d. Sample means were compared using two-tailed unpaired t-tests, with a significance level of P < 0.05 unless otherwise indicated.
Results
Construction and generation of pUMFG-FasL, pUMFG-ncFasL and pUMFG-ncFasL/c-FLIP recombinant oncoretroviral transfer plasmids
Two monocistronic oncoretroviral vectors were constructed to engineer expression of human FasL or a modified non-cleavable FasL (ncFasL). A third, bicistronic construct was synthesized to contain ncFasL followed by c-FLIPL under translational control of an IRES element to allow for simultaneous expression of both ncFasL and c-FLIPL. Expression of all transgenes was controlled by the viral LTR. Vector plasmids were transfected into the GP + E86 retroviral producer cell lines30 yielding the E86 FasL, E86 ncFasL and E86 ncFasL/c-FLIP cell lines. As a control, an oncoretroviral plasmid encoding for eGFP was also transfected in GP + E86 cells. Recombinant oncoretroviruses generated from these cell lines were used to transduce primary murine T lymphocytes. Human FasL has been shown to efficiently cross-react with the murine Fas receptor.33
Optimization of transduction of primary murine T lymphocytes
Oncoretroviral transduction of murine T lymphocytes has historically been challenging, and much effort has been invested into optimizing this procedure.17–20 To obtain satisfactory levels of transgene expression in splenocyte-derived T cells in our hands, a further multi-parameter optimization was performed. We compared the levels of transduction under several conditions, including transduction by: ‘spinoculation’, in the presence of fibronectin, and by co-culture with virus-producing cells. Following 48 h stimulation with anti-CD3/CD28 beads, T cells underwent two rounds of transduction with the E86 ncFasL viral supernatant. Infection in the presence of protamine sulfate or fibronectin resulted in transduction of 34 ± 8% and 41 ± 14% of cells, respectively. When a centrifugation step was included at the initiation of transduction (‘spinoculation’),34 the most reproducible results were achieved with 46 ± 1.4% of cells functionally transduced (Figure 1a). T-cell transduction by co-culturing murine lymphocytes with virus-producing cells has been reported to lead to satisfactory rates of transduction,17,35 but similar results were not obtained using our activation conditions and transduction frequencies of less than 10% were routinely seen after a 3-day co-culture (Figure 1a). This may result from the poor viability of virus-producing cells by the end of the co-culture period, seemingly induced by the T cells themselves.
Figure 1.
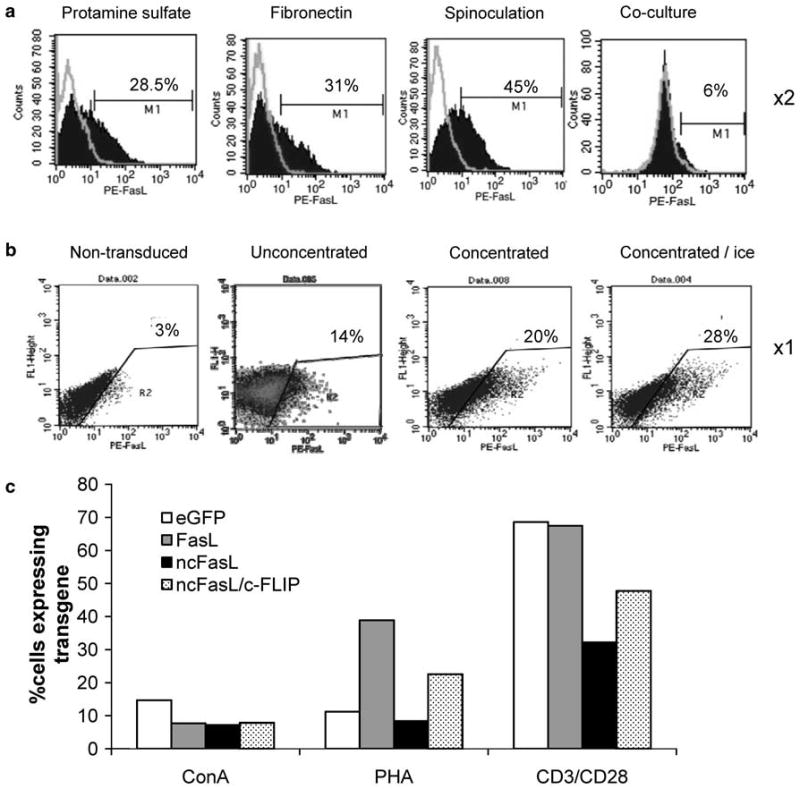
Optimization of T-cell transduction. T cells were transduced with E86-ncFasL virus under various conditions and transduction efficiency determined. (a) Representative plots showing ncFasL-expression on T cells following transduction with oncoretroviral supernatant in the presence of protamine sulfate with: no additional manipulation; transduction performed on fibronectin-coated plates; transduction by spinoculation at 1000 g for 1 h, 32 °C; or by co-culture with E86 ncFasL virus-producing cells for 3 days. With the exception of co-culture transduced cells, T cells were transduced two times. Filled histograms represent anti-FasL-labeled cells, outline histograms represent matched isotype control staining. (b) Representative flow cytometry plots showing FasL expression on non-transduced (NT) T cells or T cells following one round of transduction with: unmanipulated retroviral supernatant; concentrated retroviral supernatant; or concentrated retroviral supernatant with a 3 h incubation on ice. In parts (a) and (b), T cells were stimulated with anti-CD3/CD28 beads. (c) Effect of the T-cell stimulation method on transduction efficiency. T cells were stimulated for 48 h with 5 μg ml−1 ConA, 2.5 μg ml−1 PHA, or anti-CD3/CD28 beads at an initial bead-to-cell ratio of 3:1, and subjected to retroviral transduction with concentrated supernatant and incubation on ice. Transgene expression levels are shown on day 3 post-transduction for E86-eGFP, E86-FasL, E86-ncFasL and E86-ncFasL/c-FLIP transduced T cells, as determined by flow cytometry.
Although ‘spinoculation’ yielded adequate transduction rates in our hands, when scaled-up for large numbers of T cells transduced with multiple different transgenes the protocol becomes time-consuming and cumbersome because of the need for many centrifugation steps. As well, though multiple rounds of transduction may improve transgene expression, T-cell viability is negatively impacted. As such, we sought an alternative transduction protocol. High virus titers are important for efficient retroviral transduction and we next tested the impact of concentrating our viral preparations by low-speed centrifugation. Following a single transduction, the use of 100-fold concentrated viral supernatants led to higher levels of transgene expression (20%) than unconcentrated viral preparations (14%). When cells were incubated for 3 h on ice at the start of the transduction period,31 transgene expression was further improved by 40% (Figure 1b). As cells must be dividing for effective oncoretroviral transduction, we also investigated the impact of T-cell stimulation methods on transgene expression levels. Cells were stimulated for 48 h with ConA, PHA, or anti-CD3/CD28 beads and transduced with concentrated oncoretrovirus on ice. We reproducibly observed substantial increases in the percentage of cells expressing the transgene product when CD3/CD28 stimulation was used compared with the other mitogens. Improvements in transduction were several-fold in most cases (Figure 1c). Finally, using our optimized transduction protocol with concentrated, high-titer viral supernatants, stimulation of T cells with anti-CD3/CD28 beads and incubation on ice, we routinely achieved transgene expression of 25–45% in primary murine T lymphocyte pools (data not shown).
Transduction does not alter T-cell phenotype
To determine the phenotype of the transduced T-cell populations, the percentage of CD3+, CD4+ and CD8+ T cells in cultures was assessed 3 days after transduction. In all cases, cultures were composed of >98% CD3+ cells. The percentage of CD4+ cells ranged from 9–24% and of CD8+ cells from 69–91% over multiple experiments. Representative plots are shown in Figure 2a. The overall distribution of CD4+ and CD8+ cells was similar in non-transduced T cells and ncFasL-T cells (Figure 2a). Culture of T cells with IL-2 not only supports the proliferation of activated T cells but is also important in the homeostasis and suppressive function of regulatory T cells.36 To determine whether our T-cell cultures contained significant levels of regulatory T cells, we also measured Foxp3 expression on CD4+ cells. We were unable to detect any CD4+ Foxp3+ cells over background isotype staining in our T-cell cultures (Figure 2b).
Figure 2.
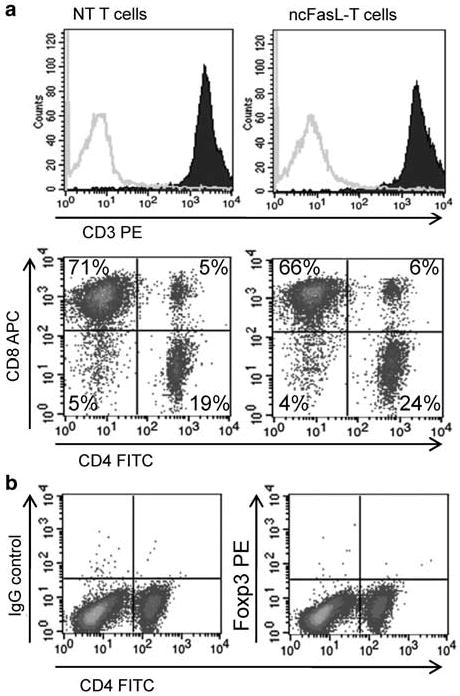
Phenotype of non-transduced (NT) and ncFasL T cells. (a) Expression of CD3, CD4 and CD8 was assessed on T cells following 4 days in culture (3 days after transduction). Filled histograms represent anti-CD3-labeled cells, outline histograms represent matched isotype control-labeled cells. (b) Non-transduced T cells were co-labeled with anti-CD4 and anti-Foxp3 antibodies to detect regulatory T cells. Similar results were obtained when ncFasL-T cells were analyzed.
Transgene expression is lost from transduced populations of T cells over time
Although initial rates of transduction are satisfactory using this optimized procedure, we observed that subsequent culture of these cells led to a decrease in the percentage of productively transduced cells over time (Figure 3a). In some cases, even when the percentage of transgene-positive cells remained constant, the MFI of these cells was reduced over time, demonstrating a decline in transgene expression levels (Figure 3b). Reduced rates of T-cell activity have been shown to correlate with decreased transcriptional activity from oncoretroviruses,37,38 but the overall loss of transgene-expressing murine T cells may be a consequence of impeded growth compared with non-transduced (NT) T cells. Here, we observed that pools of transduced cells proliferate at a significantly slower rate than NT control cells by day 5 post-transduction when assessed by 3H-thymidine incorporation assays (4.8 × 104 ± 0.5 × 104 c.p.m. vs 25.4 × 104 ± 2.1 × 104 c.p.m., respectively, P < 0.05; Figure 3c). Whether this phenomenon involving murine T cells resulted from the transduction process or is a response to oncoretroviral transduction and integration remains to be seen. Alternatively, loss of transgene-expressing cells could result from the toxicity of the FasL transgene product itself.
Figure 3.

Reduction of T-cell transgene expression over time in culture is slowed by IL-15. (a) Transgene expression on T cells stimulated with anti-CD3/CD28 beads and transduced with oncoretroviruses is shown relative to expression levels at day 3, as measured by flow cytometry. Data are representative of two independent experiments. (b) Where the percentage of eGFP-expressing T cells remains stable by day 3 post-transduction, the MFI of transduced cells is decreased with time. MFI is measured by flow cytometry and is calculated on events falling in the gated area R4. (c) Transduced cells have a reduced proliferative capacity compared with non-transduced control cells over time, as measured by 3H-thymidine incorporation assays. Cells (105) were plated per well on day 3 and growth rates were assessed on day 3, day 4, and day 5 post-transduction. Transduced cells were 46% eGFP positive on day 3. Data are representative of two experiments, each with n = 3. (d) When ncFasL T cells are cultured with IL-15 rather than IL-2, loss of ncFasL transgene expression is slowed.
Culturing T cells in IL-15 has been reported to protect CD8+ T cells from apoptosis.39 When T cells cultured with IL-15 were transduced with the E86-ncFasL viral supernatant, transgene maintenance was indeed improved (Figure 3d), however, cell expansion was retarded as the proliferative effect of this cytokine is mainly limited to memory cells in short-term culture.40 The maintenance of FasL-expressing cells in a less actively dividing T-cell population again suggested transduced cells may be at a growth disadvantage. To rule out Fas-mediated killing of the transduced populations, we repeated transduction experiments using Fas-deficient T cells from lpr mice and observed similar rates of transgene loss, although overall viability of cultures was improved (data not shown). T cells cultured in IL-15 show more potent antitumor activity in in vivo adoptive cell transfer studies because of its support of memory phenotype CD8+ T cells.41 However, cells of an effector phenotype are more reactive in vitro,41 hence we continued to use IL-2 in our cultures. As such, all further assays used T cells at day 3 post-transduction when transgene expression was high.
Ex vivo-manipulated T cells circulate and reach tumor sites in vivo
To assess whether ex vivo culture and transduction may alter the homing patterns of T cells, an in vivo trafficking experiment was carried out in RM-1 tumor-bearing mice. Splenocytes were transduced with E86-eGFP viral supernatant. In this experiment, cells were additionally labeled with CFSE dye to improve detection of manipulated cells. The percentage of CD3+ cells which were co-labeled with CFSE were assessed in cell suspensions derived from the spleen, tumor, lymph node and lung at 16 and 40 h after injection of T cells into mice harboring day 6 RM-1 tumors. Injected T cells initially accumulated in the lung (2.1 ± 0.6% at 16 h vs 0.6 ± 0.3 at 40 h, P < 0.05) then began to collect in the lymph node over time (0.6 ± 0.4% at 16 h vs 2.1 ± 1.4% at 40 h; Figure 4). Levels of i.v. injected, CD3+CFSE+ cells remained constant in the spleen over the period assessed. The pattern of T-cell distribution was similar in tumor-bearing and non-tumor bearing control mice. In tumor-bearing mice, CFSE+ T cells were detected in the tumor at 16 and 40 h after injection, but the number of these cells in the tumor had decreased by 40 h. Higher numbers of CFSE+ T cells were detected in the tumor when delivered by i.v. rather than s.c. injection into the tumor site (Figure 4). Negative control values from tumor-bearing mice that did not receive injections of T cells remained below 0.3%. Manipulated T cells were undetectable in the peripheral blood by flow cytometry.
Figure 4.
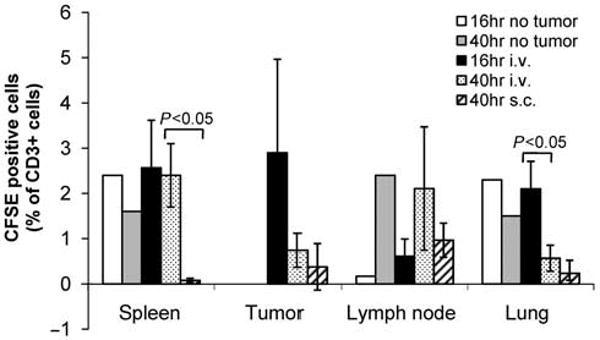
Ex vivo manipulated T cells reach tumor sites in vivo. Splenic T cells were transduced with E86-eGFP virus and labeled with CFSE to aid in visualization. After 4 days in culture, 107 cells were injected i.v.(n = 6) or s.c. into the tumor area (n = 3) of C57Bl/6 mice bearing 6 days established RM-1 tumors. T cells were also injected i.v. into non-tumor-bearing mice as a control (n = 2). The percentage of cells that co-labeled with anti-CD3 and CFSE in each organ at 16 or 40 h after T-cell injection are shown. All s.c.-injected mice were analyzed at 40 h; i.v.-injected and control mice were split into two equal groups for analysis at 16 and 40 h. The number of manipulated T cells is presented as the percentage of CD3+ cells in the lung, lymph node, spleen and tumor that were also CFSE+, as measured by flow cytometry and based on 105 events.
RM-1 tumor cells are killed by FasL-expressing T cells
To assess the efficiency of our FasL- or ncFasL-expressing T lymphocytes in tumor cell killing, we used 51Cr-release assays to measure induced cytotoxicity. FasL-, ncFasL-, ncFasL/c-FLIP- or NT control T cells were co-incubated with murine RM-1 prostate cancer cells at various ratios for 6 h. A dose-dependent cytotoxic response was observed (Figure 5a). Although levels of cell killing were low after 6 h, cytolysis induced by the FasL-expressing T cells exceeded that induced by NT control cells. When the co-incubation period was increased to 22 h, killing by FasL-T cells reached 33 ± 5.7% (Figure 5b). Typically, responses ranged from 6 to 28% cell killing by 6 h. Human prostate cancer LNCaP cells were also more efficiently killed by ncFasL-expressing cells than by control NT- or eGFP-T cells in a dose-dependent response that reached 13.3 ± 0.5% in 6 h (Figure 5c). We observed that the induction of high levels of RM-1 cell killing depended more on the health of the T cells than on the level of transgene expression (data not shown). As c-FLIP did not appear to mediate the predicted protective effect on populations of transduced cells in our experience, Z-VAD-FMK, a pan caspase inhibitor was added to T cell cultures to improve the viability of FasL-expressing cells and allow for a more robust cytotoxic response (Figures 5c and 6b second panel).
Figure 5.

RM-1 cells can be killed by FasL-overexpressing T cells. RM-1 cells undergo significant cytotoxicity following (a) 6 h or (b) 22 h co-incubation with FasL, ncFasL, or ncFasL/c-FLIP-expressing T cells, compared with non-transduced (NT) control T cells. (c) LNCaP cells are killed in a dose-dependent manner when co-incubated with FasL-expressing T cells in a similar 6 h cytotoxicity assay. In this assay, T cells were cultured in the presence of 20 μm Z-VAD-FMK. *P < 0.05 compared with NT control cell killing at same ratio.
Figure 6.
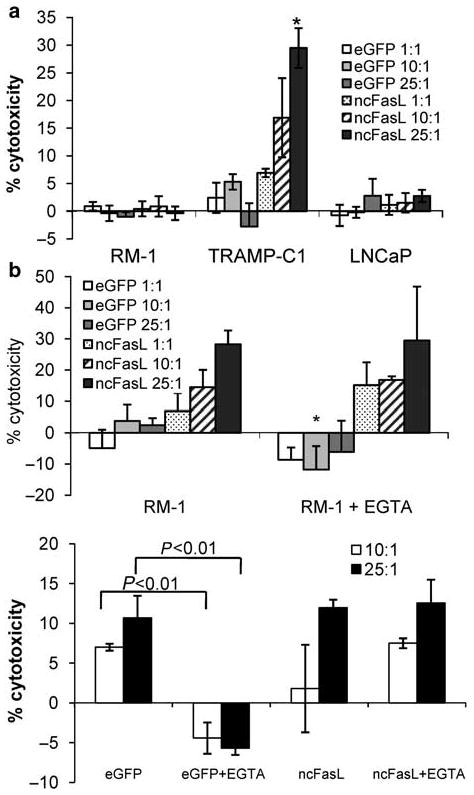
Prostate cancer cell killing is T-cell-mediated but independent of exocytosis pathways. (a) 51Cr-release assays were repeated using clonal K562 cells expressing either ncFasL or control eGFP as effector cells and RM-1, TRAMP-c1 and LNCaP tumor cell targets at the effector/target cell ratios indicated. Data is representative of two independent experiments; n = 3 for all points. (b) Cytotoxicity assays were repeated in the presence of 3 mm MgCl2 and 4 mm EGTA to inhibit cytolysis through exocytosis pathways. The assay in the second panel was performed using T cells cultured in the presence of Z-VAD-FMK. Z-VAD-FMK was removed prior to assay. *P < 0.05 compared with culture without MgCl2 and EGTA.
Efficient prostate cancer cell killing is mediated by T cells but is independent of perforin/granzyme mechanisms
To assess the relative efficiency of cell killing by our pool of FasL-T cells (∼30% FasL+), we compared cytotoxicity against that induced by a clonal population of K562-ncFasL cells. We have previously shown that K562-ncFasL cells induce cytolysis in primary human prostate cancer epithelial cells.12 However, when FasL-expressing T cells were substituted with ncFasL-expressing K562 cells in similar killing assays, RM-1 and LNCaP cells failed to undergo any measurable cytotoxicity (Figure 6a). Although this suggests a T-cell-specific function, cytotoxicity induced by ncFasL-expressing T cells was independent of the perforin/granzyme pathway, as the cytotoxic response was unaltered by the addition of MgCl2 and EGTA and remained at 28% cytolysis (Figure 6b). In contrast, levels of eGFP-T-cell-induced killing were significantly reduced indicating that background cell killing was caused by an exocytosis pathway (Figure 6b lower panel).
Fas and MHC expression is upregulated on prostate cancer cell lines by mitoxantrone, docetaxel and irradiation
DNA-damaging agents have been reported to upregulate Fas receptor expression on tumor cells25 and in some cases to sensitize these cells to apoptosis through the Fas pathway.22 We have previously shown that treatment of primary prostate cancer cells with mitoxantrone or docetaxel not only increases Fas expression levels on these cells but improves Fas-mediated killing of drug-treated cells by up to 3-fold.12 As basal levels of Fas were low on RM-1 cells compared with murine TRAMP-C1, human LNCaP, and PC-3 cells (Figure 7a), we sought to increase the expression of this receptor. We exposed RM-1 cells to the antiprostate cancer agents mitoxantrone (1–1000 nm), docetaxel (1–100 nm), or γ-irradiation (2–8 Gy) and observed dose- and time-dependent increases in Fas receptor expression by flow cytometry. Following 1000 nm mitoxantrone treatment for 24 h, the MFI for Fas staining increased 3.9 ± 0.5-fold above basal levels (Figure 7b). Similarly, Fas receptor expression was enhanced by docetaxel and irradiation in a dose-dependent manner (Figure 7b). Upregulation following docetaxel treatment peaked at 3.3 ± 0.1-fold after 48-h treatment with 100 nm, and following irradiation with 8 Gy at 4.3 ± 0.5-fold in Fas MFI 48 h post-irradiation (data not shown). Treatment of TRAMP-C1 or LNCaP cells with these anticancer agents also led to the enhancement of Fas receptor expression although induction of Fas in TRAMP-C1 cells was somewhat muted compared with RM-1 or LNCaP cells (Figures 7c and d).
Figure 7.
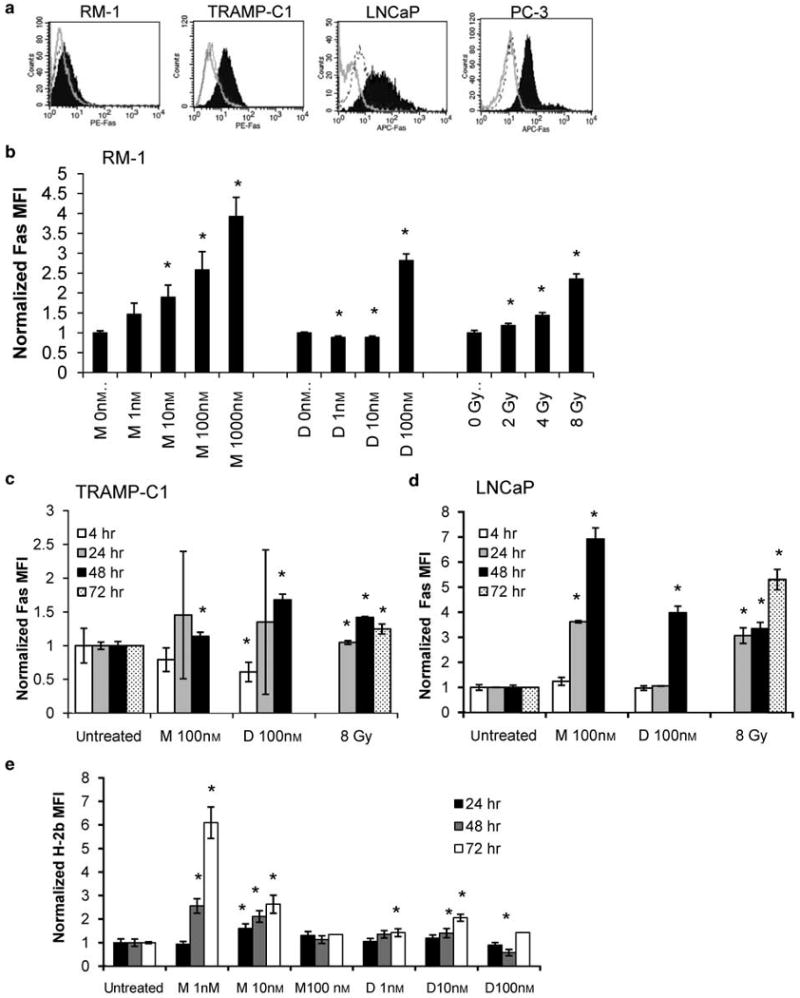
Fas and major histocompatibility complex (MHC) expression are upregulated on prostate cancer cell lines by treatment with mitoxantrone, docetaxel or irradiation. (a) Basal levels of Fas expression on RM-1, TRAMP-C1, LNCaP and PC-3 cells are shown, as measured by flow cytometry. Filled histograms = anti-Fas antibody-labeled cells, outline histograms = unstained cells, dashed histograms = isotype control-labeled cells. (b) The MFI of anti-Fas antibody staining is increased several-fold above basal levels when RM-1 cells are treated with 1–1000 nm mitoxantrone (n = 2 to 4 for each data point); 1–100 nm docetaxel (n = 3); or 2–8 Gy irradiation (n = 2) for 24 h. Increases in Fas expression are also seen when (c) TRAMP-C1 and (d) LNCaP cells are exposed to 100 nm mitoxantrone (n = 3 or 4), 100 nm docetaxel (n = 3), or 8 Gy irradiation (n = 2). (e) MFI of anti-H-2 Kb and H-2 Db antibody-labeled RM-1 cells 24, 48 and 72 h after exposure to 1, 10, or 100 nm mitoxantrone or docetaxel was measured by flow cytometry. n = 3 for each data point except cells treated for 72 h with 100 nm docetaxel or mitoxantrone, where cells from three replicate wells were pooled and analyzed together. *P < 0.05. M = mitoxantrone, D = docetaxel. Results are presented as fold-change in MFI compared with untreated control cells.
An important requirement for tumor antigen recognition by T cells is the presentation of these antigens in the context of the MHC. Tumors frequently downmodulate their expression of MHC proteins as a means of immune escape, rendering them less visible to circulating immune cells.42 Agents such as IFNγ and cisplatin have been reported to increase the levels of MHC class I in prostate and colorectal tumor models, respectively.43,44 We assessed the impact of mitoxantrone and docetaxel on RM-1 cell MHC expression. Following exposure to increasing doses of these drugs, up to a 6.1 ± 0.7-fold increase in the MFI of H-2b was seen (Figure 7e). The increase seen in H-2b expression was most strongly induced by the lowest dose of mitoxantrone tested, however, time-dependent increases in H-2b expression were noted for 10 nm mitoxantrone and 1 to 10 nm docetaxel as well. Upregulation of MHC expression was also seen when cell cultures derived from primary human prostate cancer cells were treated with mitoxantrone or docetaxel (data not shown).
Discussion
We have shown that genetic modification of T lymphocytes to express higher than physiological levels of FasL contributes to the active killing of RM-1 and LNCaP prostate cancer tumor targets. Of note, this killing was achieved in the absence of cycloheximide, a translation inhibitor regularly used in FasL-based assays to improve the killing of target cells.45,46 To facilitate high level expression of functional FasL on T cells, polyclonal stimulation using anti-CD3/CD28 beads was essential, as was the use of highly concentrated viral preparations, allowing efficient transgene expression following a single transduction cycle. Transduced T cells maintained their ability to home to tumor cells in vivo. In addition, we have shown induction of Fas receptor expression using current clinical interventions. These results set a solid groundwork for next generation studies in animal disease models of prostate cancer.
To generate pools of murine lymphocytes expressing our transgenes, it was necessary to optimize the T-cell transduction procedure. Other groups have reported similar findings that ‘spinoculation’ is an effective method for murine T-cell transduction, and that high-titer virus is important for efficient transduction.19 In contrast to other reports,19 we found that multiple rounds of oncoretroviral vector transductions improved transgene expression in murine T cells. However, T-cell viability was hindered by repeated transductions. We prefer to subject our cells to one round of infection with highly concentrated viral preparations by incubation on ice due to the ease of handling large sample sizes and many groups. Despite these optimizations of murine T-cell transduction, these cells are not robust for long-term in vitro studies, however, because of the losses in transgene expression over time. Nevertheless, recent studies show that genetically modified murine T cells are maintained at high levels in the spleens of mice in vivo47 and demonstrate efficacy in tumor regression in mouse models.35 These findings suggest that complications in maintaining genetically modified T cells may be caused by in vitro culture conditions. In contrast, human T lymphocytes are easier to manipulate in culture and several clinical studies are underway which make use of genetically modified human T cells.
Treatment of RM-1 cells with mitoxantrone, docetaxel, or irradiation induces an upregulation of both H-2b and Fas receptor expression. Despite this increase in the number of Fas receptors on the cell surface, we have not observed any improvement in FasL-T-cell-mediated killing of RM-1 cells following pre-treatment with 10 or 100 nm mitoxantrone or 10 nm docetaxel for 48 h (data not shown). Several studies report the sensitization of tumor cells to FasL following similar regimens,22,25 and treatment of RM-1 cells with IFNγ has been reported to improve Fas-mediated killing of these cells both in vitro and in vivo.48 In an in vitro primary prostate cancer cell system, we have shown that both mitoxantrone and docetaxel are able to improve Fas-mediated killing of these primary cells.12 However, in accordance with our current data, a lack of sensitization has been reported in other models.24,26 Generalizations about the co-operative effects of chemotherapeutic agents and Fas-mediated killing should thus be avoided, especially for commonly studied prostate cancer cell lines. A lack of correlation between basal Fas expression and susceptibility to Fas-mediated killing has been seen in a number of solid tumor models,49 and as such the degree of Fas upregulation by DNA-damaging agents may not be the best marker to predict changes in susceptibility to apoptosis following combination treatments.
The low levels of RM-1 cell killing induced by NT and eGFP-expressing control T cells was shown to involve perforin and granzymes whereas cell killing by transgenic FasL-expressing T cells was found to be independent of exocytosis pathways. However, FasL-T-cell-mediated killing was not inhibited by the antagonistic anti-human FasL antibody NOK-1,23 suggesting that the mechanism of this killing was in fact not Fas-mediated (data not shown). Even so, perforin-independent induction of RM-1 cell cytotoxicity was limited to FasL or ncFasL-expressing T cells. We have shown that K562 cells expressing ncFasL are effective inducers of apoptosis in primary prostate cancer cells.12 yet ncFasL-K562 cells were unable to induce killing of RM-1 or LNCaP cells under the same conditions. This lack of killing is in spite of uniform expression of ncFasL on the K562 cells compared with moderate levels of FasL expression on the T cells (∼30% of cells expressed FasL or ncFasL). This suggests that FasL overexpression on T cells is more efficacious than its expression on the relatively inert K562 cells and that the observed cytotoxic response is a T-cell-specific effect. The complete resistance of RM-1 cells to killing by ncFasL-K562 cells also highlights the intrinsic insensitivity of RM-1 cells to Fas-mediated killing. RM-1 cells are derived from an in vivo murine prostate carcinoma model where overexpression of ras and myc oncogenes were engineered in urogenital sinus epithelial cells.50 Ras has been shown to limit not only the basal expression of Fas receptor on ras-transformed cells, but to limit the induction of this surface protein following DNA-damaging treatments51 and is a potent antiapoptotic protein.
The mechanism of killing induced by our FasL-T cells is undefined at this time, but may involve reverse signaling through the cytoplasmic tail of FasL. The transduction of co-stimulatory signals through the intracellular domain of this molecule in T cells has been described in conjunction with T-cell receptor stimulation. These signals are proliferative52 and initiate signaling through the Akt, ERK1/2 and JNK pathways, activate transcription through NFAT and AP-1 and stimulate IFNγ production.53 Alternatively, as an anti-human antibody was used to block killing through this pathway, endogenous mouse FasL may be contributing to the observed toxicity in conjunction with the exogenous human FasL.
The relative effectiveness of each of the pUMFG-FasL, pUMFG-ncFasL and pUMFG-ncFasL/c-FLIP constructs in inducing prostate cell killing varied between assays and no distinct pattern emerged which would allow us to discern which construct is more efficacious in this model. We included the c-FLIP cDNA in our third oncoretroviral construct with the hypothesis that expression of this factor would preserve the viability of transduced T cell cultures. However, we failed to detect any survival benefit in our pools of transduced cells. We have not ruled out the possibility that the cytotoxicity observed was that of non-transduced T cells. Owing to technical difficulties in sorting active, transduced murine T cells (our observations and see Abad et al.54), we were unable to assess the impact of c-FLIP expression in pure populations of cells to definitively answer this question. Nonetheless, evidence exists suggesting that rather than preventing apoptosis, c-FLIPL can activate caspase 8 at the DISC, leading to increased basal levels of apoptosis in T cells engineered to express high levels of c-FLIPL.55 Alternative antiapoptotic approaches may prove more fruitful, such as using the c-FLIP short-form which does not activate caspase 8,56 introducing the viral caspase inhibitor CrmA57 or by using siRNA against the Fas receptor.58
This report presents a T-cell transduction protocol that is broadly applicable to a variety of T-cell-based gene therapy strategies in murine models. We have described a FasL-based gene therapy approach with the potential for prostate cancer therapy. Future studies will combine this strategy with antigen-specific targeting of T cells. Our laboratory has previously generated prostate-specific antigen -reactive T cells using a dendritic cell immunotherapy technique.59 Combination of FasL-T cells with antigen-specific targeting and activity may be important in vivo to improve delivery of therapy to the tumor area. Further investigations into the efficacy of this approach in an in vivo setting are warranted, where the impact of pre-treatment of targets with pro-inflammatory anticancer therapies may prove synergistic.
Acknowledgments
We thank Anton Neschadim and Dr Richard Miller for helpful discussions about transductions and T-cell biology. This work is supported by a grant from the Prostate Cancer Research Foundation of Canada.
References
- 1.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA. 2004;101:14639–14645. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gade TPF, Hassen W, Santos E, Gunset G, Saudemont A, Gong MC, et al. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080–9088. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 4.Kershaw MH, Teng MW, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5:928–940. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 5.Lu B, Finn OJ. T-cell death and cancer immune tolerance. Cell Death Differ. 2008;15:70–79. doi: 10.1038/sj.cdd.4402274. [DOI] [PubMed] [Google Scholar]
- 6.Kessler B, Hudrisier D, Schroeter M, Tschopp J, Cerottini JC, Luescher IF. Peptide modification or blocking of CD8, resulting in weak TCR signaling, can activate CTL for Fas-but not perforin-dependent cytotoxicity or cytokine production. J Immunol. 1998;161:6939–6946. [PubMed] [Google Scholar]
- 7.Dobrzanski MJ, Reome JB, Hollenbaugh JA, Hylind JC, Dutton RW. Effector cell-derived lymphotoxin α and Fas ligand, but not perforin, promote Tc1 and Tc2 effector cell-mediated tumor therapy in established pulmonary metastases. Cancer Res. 2004;64:406–414. doi: 10.1158/0008-5472.can-03-2580. [DOI] [PubMed] [Google Scholar]
- 8.Frost P, Caliliw R, Belldegrun A, Bonavida B. Immuno-sensitization of resistant human tumor cells to cytotoxicity by tumor infiltrating lymphocytes. Int J Oncol. 2003;22:431–437. [PubMed] [Google Scholar]
- 9.Schulte M, Reiss K, Lettau M, Maretzky T, Ludwig A, Hartmann D, et al. ADAM10 regulates FasL cell surface expression and modulates FasL-induced cytotoxicity and activation-induced cell death. Cell Death Differ. 2007;14:1040–1049. doi: 10.1038/sj.cdd.4402101. [DOI] [PubMed] [Google Scholar]
- 10.Shimizu M, Takeda Y, Yagita H, Yoshimoto T, Matsuzawa A. Antitumor activity exhibited by Fas ligand (CD95L) overexpressed on lymphoid cells against Fas+ tumor cells. Cancer Immunol Immunother. 1998;47:143–148. doi: 10.1007/s002620050514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Liu YH, Zhang YP, Zhang S, Pu X, Gardner TA, et al. Fas ligand delivery by a prostate-restricted replicative adenovirus enhances safety and antitumor efficacy. Clin Cancer Res. 2007;13:5463–5473. doi: 10.1158/1078-0432.CCR-07-0342. [DOI] [PubMed] [Google Scholar]
- 12.Symes JC, Kurin M, Fleshner NE, Medin JA. Fas-mediated killing of primary prostate cancer cells is increased by mitoxantrone and docetaxel. Mol Cancer Ther. 2008;7:3018–3028. doi: 10.1158/1535-7163.MCT-08-0335. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka M, Suda T, Yatomi T, Nakamura N, Nagata S. Lethal effect of recombinant human Fas ligand in mice pretreated with Propionibacterium acnes. J Immunol. 1997;158:2303–2309. [PubMed] [Google Scholar]
- 14.Aoki K, Akyurek LM, San H, Leung K, Parmacek MS, Nabel EG, et al. Restricted expression of an adenoviral vector encoding Fas ligand (CD95L) enhances safety for cancer gene therapy. Mol Ther. 2000;1:555–565. doi: 10.1006/mthe.2000.0076. [DOI] [PubMed] [Google Scholar]
- 15.Mezzanzanica D, Balladore E, Turatti F, Luison E, Alberti P, Bagnoli M, et al. CD95-mediated apoptosis is impaired at receptor level by cellular FLICE-inhibitory protein (long form) in wild-type p53 human ovarian carcinoma. Clin Cancer Res. 2004;10:5202–5214. doi: 10.1158/1078-0432.CCR-03-0537. [DOI] [PubMed] [Google Scholar]
- 16.Van Parijs L, Refaeli Y, Abbas AK, Baltimore D. Autoimmunity as a consequence of retrovirus-mediated expression of C-FLIP in lymphocytes. Immunity. 1999;11:763–770. doi: 10.1016/s1074-7613(00)80150-8. [DOI] [PubMed] [Google Scholar]
- 17.Hagani AB, Riviere I, Tan C, Krause A, Sadelain M. Activation conditions determine susceptibility of murine primary T-lymphocytes to retroviral infection. J Gene Med. 1999;1:341–351. doi: 10.1002/(SICI)1521-2254(199909/10)1:5<341::AID-JGM58>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Pouw NM, Westerlaken EJ, Willemsen RA, Debets R. Gene transfer of human TCR in primary murine T cells is improved by pseudo-typing with amphotropic and ecotropic envelopes. J Gene Med. 2007;9:561–570. doi: 10.1002/jgm.1047. [DOI] [PubMed] [Google Scholar]
- 19.Zhang T, Tsang TC, Harris DT. Efficient transduction of murine primary T cells requires a combination of high viral titer, preferred tropism, and proper timing of transduction. J Hematother Stem Cell Res. 2003;12:123–130. doi: 10.1089/152581603321210208. [DOI] [PubMed] [Google Scholar]
- 20.Annenkov AE, Daly GM, Chernajovsky Y. Highly efficient gene transfer into antigen-specific primary mouse lymphocytes with replication-deficient retrovirus expressing the 10A1 envelope protein. J Gene Med. 2002;4:133–140. doi: 10.1002/jgm.258. [DOI] [PubMed] [Google Scholar]
- 21.Sheard MA, Uldrijan S, Vojtesek B. Role of p53 in regulating constitutive and X-radiation-inducible CD95 expression and function in carcinoma cells. Cancer Res. 2003;63:7176–7184. [PubMed] [Google Scholar]
- 22.Bagnoli M, Balladore E, Luison E, Alberti P, Raspagliesi F, Marcomini B, et al. Sensitization of p53-mutated epithelial ovarian cancer to CD95-mediated apoptosis is synergistically induced by cisplatin pretreatment. Mol Cancer Ther. 2007;6:762–772. doi: 10.1158/1535-7163.MCT-06-0357. [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee D, Schmitz I, Krueger A, Yeung K, Kirchhoff S, Krammer PH, et al. Induction of apoptosis in 9-nitrocamptothecin-treated DU145 human prostate carcinoma cells correlates with de novo synthesis of CD95 and CD95 ligand and down-regulation of c-FLIP(short) Cancer Res. 2001;61:7148–7154. [PubMed] [Google Scholar]
- 24.Liu QY, Rubin MA, Omene C, Lederman S, Stein CA. Fas ligand is constitutively secreted by prostate cancer cells in vitro. Clin Cancer Res. 1998;4:1803–1811. [PubMed] [Google Scholar]
- 25.Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med. 1998;188:2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romano C, De Fanis U, Sellitto A, Chiuraxxi F, Guastafierro S, Giunta R, et al. Induction of CD95 upregulation does not render chronic lymphocytic leukemia B-cells susceptible to CD95-mediated apoptosis. Immunol Lett. 2005;97:131–139. doi: 10.1016/j.imlet.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 27.Liang SB, Yoshimitsu M, Poeppl A, Rasaiah VI, Cai J, Fowler DH, et al. Multiple reduced-intensity conditioning regimens facilitate correction of Fabry mice after transplantation of transduced cells. Mol Ther. 2007;15:618–627. doi: 10.1038/sj.mt.6300075. [DOI] [PubMed] [Google Scholar]
- 28.Jung U, Foley JE, Erdmann AA, Eckhaus MA, Fowler DH. CD3/CD28-costimulated T1 and T2 subsets: differential in vivo allosensitization generates distinct GVT and GVHD effects. Blood. 2003;102:3439–3446. doi: 10.1182/blood-2002-12-3936. [DOI] [PubMed] [Google Scholar]
- 29.Takenaka T, Qin G, Brady RO, Medin JA. Circulating alpha-galactosidase A derived from transduced bone marrow cells: relevance for corrective gene transfer for Fabry disease. Hum Gene Ther. 1999;10:1931–1939. doi: 10.1089/10430349950017293. [DOI] [PubMed] [Google Scholar]
- 30.Markowitz D, Goff S, Bank A. A safe packaging line for gene transfer: separating viral genes on two different plasmids. J Virol. 1988;62:1120–1124. doi: 10.1128/jvi.62.4.1120-1124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niederman TM, Ghogawala Z, Carter BS, Tompkins HS, Russell MM, Mulligan RC. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci USA. 2002;99:7009–7014. doi: 10.1073/pnas.092562399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouvier E, Luciani MF, Golstein P. Fas involvement in Ca(2+)-independent T cell-mediated cytotoxicity. J Exp Med. 1993;177:195–200. doi: 10.1084/jem.177.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi T, Tanaka M, Inazawa J, Abe T, Suda T, Nagata S. Human Fas ligand: gene structure, chromosomal location and species specificity. Int Immunol. 1994;6:1567–1574. doi: 10.1093/intimm/6.10.1567. [DOI] [PubMed] [Google Scholar]
- 34.Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Sci USA. 1995;92:7739–7743. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darcy PK, Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, et al. Redirected perforin-dependent lysis of colon carcinoma by ex vivo genetically engineered CTL. J Immunol. 2000;164:3705–3712. doi: 10.4049/jimmunol.164.7.3705. [DOI] [PubMed] [Google Scholar]
- 36.Turka LA, Walsh PT. IL-2 signaling and CD4+ CD25+ Foxp3+ regulatory T cells. Front Biosci. 2008;13:1440–1446. doi: 10.2741/2773. [DOI] [PubMed] [Google Scholar]
- 37.Pollok KE, van der Loo JC, Cooper RJ, Kennedy L, Williams DA. Costimulation of transduced T lymphocytes via T cell receptor-CD3 complex and CD28 leads to increased transcription of integrated retrovirus. Hum Gene Ther. 1999;10:2221–2236. doi: 10.1089/10430349950017202. [DOI] [PubMed] [Google Scholar]
- 38.Quinn ER, Lum LG, Trevor KT. T cell activation modulates retrovirus-mediated gene expression. Hum Gene Ther. 1998;9:1457–1467. doi: 10.1089/hum.1998.9.10-1457. [DOI] [PubMed] [Google Scholar]
- 39.Wallace DL, Berard M, Soares MV, Oldham J, Cook JE, Akbar AN, et al. Prolonged exposure of naive CD8+ T cells to interleukin-7 or interleukin-15 stimulates proliferation without differentiation or loss of telomere length. Immunology. 2006;119:243–253. doi: 10.1111/j.1365-2567.2006.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. 2003;170:5018–5026. doi: 10.4049/jimmunol.170.10.5018. [DOI] [PubMed] [Google Scholar]
- 41.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Algarra I, Garcia-Lora A, Cabrera T, Ruiz-Cabello F, Garrido F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. Cancer Immunol Immunother. 2004;53:904–910. doi: 10.1007/s00262-004-0517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bander NH, Yao D, Liu H, Chen YT, Steiner M, Zuccaro W, et al. MHC class I and II expression in prostate carcinoma and modulation by interferon-alpha and -gamma. Prostate. 1997;33:233–239. doi: 10.1002/(sici)1097-0045(19971201)33:4<233::aid-pros2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 44.Ohtsukasa S, Okabe S, Yamashita H, Iwai T, Sugihara K. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J Cancer Res Clin Oncol. 2003;129:719–726. doi: 10.1007/s00432-003-0492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watermann I, Gerspach J, Lehne M, Seufert J, Schneider B, Pfizenmaier K, et al. Activation of CD95L fusion protein prodrugs by tumor-associated proteases. Cell Death Differ. 2007;14:765–774. doi: 10.1038/sj.cdd.4402051. [DOI] [PubMed] [Google Scholar]
- 46.Herrmann T, Grosse-Hovest L, Otz T, Krammer PH, Rammensee HG, Jung G. Construction of optimized bispecific antibodies for selective activation of the death receptor CD95. Cancer Res. 2008;68:1221–1227. doi: 10.1158/0008-5472.CAN-07-6175. [DOI] [PubMed] [Google Scholar]
- 47.Westwood JA, Murray WK, Trivett M, Shin A, Neeson P, MacGregor DP, et al. Absence of retroviral vector-mediated transformation of gene-modified T cells after long-term engraftment in mice. Gene Therapy. 2008;15:1056–1066. doi: 10.1038/gt.2008.47. [DOI] [PubMed] [Google Scholar]
- 48.Selleck WA, Canfield SE, Hassen WA, Meseck M, Kuzmin AI, Eisensmith RC, et al. IFN-gamma sensitization of prostate cancer cells to Fas-mediated death: a gene therapy approach. Mol Ther. 2003;7:185–192. doi: 10.1016/s1525-0016(02)00040-0. [DOI] [PubMed] [Google Scholar]
- 49.Owen-Shaub L, Radinsky R, Kruzel E, Berry K, Yonehara S. Anti-Fas on nonhematopoietic tumors: levels of Fas/APO-1 and bcl-2 are not predictive of biological responsiveness. Cancer Res. 1994;54:1580–1586. [PubMed] [Google Scholar]
- 50.Baley PA, Yoshida K, Qian W, Sehgal I, Thompson TC. Progression to androgen insensitivity in a novel in vitro mouse model for prostate cancer. J Steroid Biochem Mol Biol. 1995;52:403–413. doi: 10.1016/0960-0760(95)00001-g. [DOI] [PubMed] [Google Scholar]
- 51.Fenton RG, Hixon JA, Wright PW, Brooks AD, Sayers TJ. Inhibition of Fas (CD95) expression and Fas-mediated apoptosis by oncogenic Ras. Cancer Res. 1998;58:3391–3400. [PubMed] [Google Scholar]
- 52.Suzuki I, Fink PJ. Maximal proliferation of cytotoxic T lymphocytes requires reverse signaling through Fas ligand. J Exp Med. 1998;187:123–128. doi: 10.1084/jem.187.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun M, Ames KT, Suzuki I, Fink PJ. The cytoplasmic domain of Fas ligand costimulates TCR signals. J Immunol. 2006;177:1481–1491. doi: 10.4049/jimmunol.177.3.1481. [DOI] [PubMed] [Google Scholar]
- 54.Abad JD, Wrzensinski C, Overwijk W, De Witte MA, Jorritsma A, Hsu C, et al. T-cell receptor gene therapy of established tumors in a murine melanoma model. J Immunother. 2008;31:1–6. doi: 10.1097/CJI.0b013e31815c193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dohrman A, Russell JQ, Cuenin S, Fortner K, Tschopp J, Budd RC. Cellular FLIP long form augments caspase activity and death of T cells through heterodimerization with and activation of caspase-8. J Immunol. 2005;175:311–318. doi: 10.4049/jimmunol.175.1.311. [DOI] [PubMed] [Google Scholar]
- 56.Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276:20633–20640. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 57.Smith KG, Strasser A, Vaux DL. CrmA expression in T lymphocytes of transgenic mice inhibits CD95 (Fas/APO-1)-transduced apoptosis, but does not cause lymphadenopathy or autoimmune disease. EMBO J. 1996;15:5167–5176. [PMC free article] [PubMed] [Google Scholar]
- 58.Dotti G, Savoldo B, Pule M, Straathof KC, Biagi E, Yvon E, et al. Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood. 2005;105:4677–4684. doi: 10.1182/blood-2004-08-3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Medin J, Liang SB, Hou J, Kelley LS, Peace DJ, Fowler DH. Efficient transfer of PSA and PSMA cDNAs into DCs generates antibody and T cell antitumor responses in vivo. Cancer Gene Ther. 2005;12:540–551. doi: 10.1038/sj.cgt.7700810. [DOI] [PubMed] [Google Scholar]