

Abstract
Diabetic retinopathy shares many characteristics features of a low grade chronic inflammatory disease. Its progression resists arrest when good metabolic control is re-established after a period of poor metabolic control, suggesting a ‘metabolic memory’ phenomenon. The aim of this study is to investigate the effect of reversal of high glucose to normal glucose on the inflammatory mediators in pericytes, the site of histopathology in diabetic retinopathy. Bovine retinal pericytes were incubated in high glucose (20mM) for 2 days followed by normal glucose (5mM) for 4 days (2→4), or in high glucose for 4 days followed by normal glucose for 4 days (4→4) or 8 days (4→8). Pericytes incubated in continuous normal or high glucose for 2-12 days served as controls. Continuous high glucose exposure for 2-12 days significantly elevated gene expressions and protein concentrations of IL-1β, NF-kB, VEGF, TNF-α, TGF-β and ICAM-1 in retinal pericytes. Four days of normal glucose that followed 2 days of high glucose (2→4) had marginal, but significant, beneficial effect on the increases in these inflammatory mediators. Four days of normal glucose in 4→4 group failed to reverse increases in inflammatory mediators and cell apoptosis remained elevated, but addition of dexamethasone during normal glucose exposure ameliorated such increases. However, when normal glucose exposure, after 4 days of high glucose was extended to 8 days (4→8), increases in these mediators were significantly decreased. Hyperglycemia-induced elevations in inflammatory mediators in retinal microvascular cells resist reversal after re-institution of normal glucose conditions. Both, the duration of the initial exposure to high glucose, and normal glucose that follows high glucose, are critical in determining the outcome of the alterations in the inflammatory mediators.
Keywords: Diabetic retinopathy, Inflammation, Metabolic memory, Pericytes, Retina
Introduction
Diabetes is a significantly growing public health problem and retinopathy is one of its very serious and blinding complications. A number of hyperglycemia-induced metabolic abnormalities have been shown to contribute to the pathogenesis of diabetic retinopathy but the exact mechanism remains unclear. Studies suggest that diabetic retinopathy has many characteristics of a low grade inflammatory disease; the levels of pro-inflammatory cytokines are increased in the retina and vitreous in diabetes, and retinal capillaries become nonperfused and ischemic with increased number of platelet-fibrin thrombi (Boeri et al., 2001; Joussen et al., 2001; Joussen et al., 2004; Kowluru and Odenbach 2004a&b; Kern 2007; Chan et al., 2008).
Capillaries of the retina are lined with equal numbers of pericytes and endothelial cells; pericytes provide vascular stability and control endothelial proliferation (Haefliger et al., 1994). Retinal pericytes present various metabolic abnormalities that are implicated in the development of diabetic retinopathy (Kowluru and Koppolu 2002; Kowluru et al., 2003; Miller et al., 2006; Zhang et al., 2008). The loss of pericyte is considered as one of the earliest morphological changes (Hammes et al., 2002), and the possible mechanisms include apoptosis (Mizutani et al., 1996) and migration signalled via Ang-2/Tie-2 pathway (Pfister et al., 2008). However, what triggers pericytes loss in diabetic environment is not completely elucidated.
Good glycemic control remains one of the major plausible therapeutic options in reducing the incidence and progression of diabetic retinopathy. Clinical trials have demonstrated that the advantages of intensive glycemic control persist beyond the duration of maintenance of tight glycemic control, and that reestablishment of good glycemic control after a profound period of poor control does not immediately benefit the progression of retinopathy (Diabetes Control and Complications Trial Research Group 1993; Diabetes Control and Complications Trial Research Group 1998; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group 2008). This imprinted effect of normal or high glucose levels on the health of the retina that could either result in the long lasting benefits of good glycemic control, or the resistance of pathology to halt, is commonly termed as the “metabolic memory” phenomenon in diabetic retinopathy (Roy et al., 1990; LeRoith et al., 2005). Animal models of diabetic retinopathy, including chemically induced diabetic rodents and dogs, have successfully mimicked metabolic memory phenomenon (Engerman and Kern 1987; Kowluru 2003). We have shown that if the rats are allowed to maintain good glycemic control soon after induction of diabetes, the retina escape from diabetes-induced metabolic and histopathological alterations (Kowluru 2003; Kowluru et al., 2004; Kowluru et al., 2007; Kanwar and Kowluru 2009). How the duration of poor control before initiation of good control, and also the duration of good control after a period of poor control affects retinal metabolism remains elusive.
Using rat model of diabetes, we have recently shown that the pro-inflammatory mediators, which are postulated to play a role in the development of diabetic retinopathy, resist arrest after reestablishment of good glycemic control (Chan et al., 2008), and microvascular cells continue to undergo apoptosis (Kowluru and Chan 2009). The purpose of this study is to elucidate the role of inflammation in the metabolic memory in the retinal pericyte, the target of histopathology, and to investigate the effect of the duration of high glucose exposure before initiating normal glucose. We have also investigated the effect of duration of normal glucose exposure after a period of high glucose on the metabolic memory phenomenon, and dexamethasone supplementation, which is shown to inhibit protein and gene expressions of proinflammatory mediators (Holz et al. 2005), during the reversal of high glucose on the inflammatory mediators.
Methods
Preparation of pericytes and incubation conditions
Pericyte were prepared from calf eyes using the methods routinely employed in our laboratory (Kowluru and Koppolu 2002; Kowluru et al., 2003). Briefly, the retina was removed aseptically, homogenized and passed through 210 and 86 sieves. The cells collected on the sieve were incubated with collagenase (type I) for one hour at 37°C, washed with Dulbecco's Modified Eagle's Medium (DMEM), and plated into gelatin-coated 60mm petridishes. The cells were cultured in the pericyte growth media (DMEM containing 20% fetal bovine serum and 1% antibiotic/antimycotic mixture).
To investigate the effect of the duration of the initial and post glucose exposure on the inflammatory mediators, pericytes from 4th-6th passages (60-80% confluent) were randomly divided into three treatment conditions. The first group consisted of a short duration (2 days) of initial insult with high glucose (20mM) followed by 4 days of normal (5mM) glucose (2d-4d). In the second group, the cells were exposed to 20mM glucose for 4 days, and that was followed with 5mM glucose incubation for 4 additional days (4d-4d). The third group had 4 days of 20mM glucose exposure followed by long duration (8 days) of 5mM glucose exposure (4d-8d). Each group had parallel controls in which the cells were incubated in 5mM glucose or in 20mM glucose for the entire duration of experiment. The cells received fresh media every 48 hours. At the end of the desired initial insult of high glucose (2 or 4 days), the cells were rinsed with DMEM before changing the medium. The incubation was terminated by rinsing the cells with PBS followed by scraping. The cells incubated in 20mM mannitol served as osmotic control.
The effect of direct inhibition of inflammation on the metabolic imprinting phenomenon was determined by incubating pericytes in 20mM glucose for 4 days, followed by incubation in 5mM glucose medium containing 1 μmM dexamethasone for 4 additional days (4d-4d+Dex). Additional controls included the cells incubated in 5mM or 20mM glucose medium with dexmethasone (1μM) for 4 to 8 days.
Protein extraction and concentration determination
Pericytes were suspended in lysis buffer (30mM Tris-HCl, 10mM EGTA, 5mM EDTA, 1% Triton X-100, 250mM sucrose, 1mM NaF, 1mM phenylmethylsulfonyl fluoride, 1mM Na3VO4 and protease inhibitors; 15μg/ml aprotenin, 5μg/ml leupeptin and 5μg/ml pepstatin), and sonicated 3×5 seconds. For ELISA, pericytes form one 60mm petridish were sonicated in 75-100μl phosphate buffered saline containing protease inhibitors, and centrifuged at 1000×g for 5 minutes to remove cell debris. Protein concentration was quantified using the bicinchoninic acid assay (Sigma-Aldrich, St Louis, MO).
Total RNA isolation and cDNA synthesis and PCR
Total RNA was isolated using TRIZol reagent (Invitrogen, Carlsbad, CA), as routinely used in our laboratory (Chan et.al., 2008; Kowluru and Chan, 2009). The concentration and integrity of total RNA was determined with UV spectrophotometry. RNA (1μg) was converted to single stranded cDNA using the High Capacity cDNA Reverse Transcription Kit with RNase inhibitor (Applied Biosystems, ABI, Foster City, CA, USA). cDNA was synthesized in a total volume of 20μl using the GeneAmp PCR system 9700 thermal cycler (ABI). The single stranded cDNA was quantified spectrophotometerically.
Quantitative real-time PCR (RT-PCR) for interleukin 1β (IL-1β), nuclear transcriptional factor-kB (NF-kB, p65 subunit) tumor necrosis factor-α (TNF-α), vascular endothelial growth factor (VEGF), intercellular adhesion molecule-1 (ICAM-1) and transforming growth factor beta (TGF-β) was performed with 50ng cDNA template in 96-well plates using the ABI-7500 sequence detection system, and 18sRNA was used as a housekeeping control. The gene expression of p65 subunit of NF-kB was quantified because our previous studies have shown that in diabetes its protein expression is increased in the retina and its capillary cells (Kowluru et al., 2003). Each sample was measured in triplicate. Genbank accession numbers of TaqMan primers (bovine, made to order or custom) for IL-1β, NF-kB, VEGF, ICAM-1, TGF-β, TNF-α and 18s are NM1_74093, DQ35511, NM_174216, NM_001166068, NM_174348, AF3848421 and X03205 respectively. The cDNA templates were combined with the TaqMan assay probes and TaqMan Universal PCR Master Mix in a total volume of 20μl and amplified. The standard PCR conditions included 2 minutes at 50°C and 10 min at 95°C followed by 40 cycles of extension at 95°C for 15 seconds and one minute at 60°C. Threshold lines were automatically adjusted to intersect amplification lines in the linear portion of the amplification curves and cycle to threshold (Ct) were recorded automatically. Replicated data were normalized with 18sRNA and the fold change in gene expression relative to normal was calculated using the ddCt method.
To investigate the effect of inhibition of dexamethasone on inflammatory mediators, semi-quantitative PCR was performed for VEGF using Gene Amp PCR System 9700 (Applied Biosystems) for PCR amplification. VEGF gene was amplified using the forward primer (ACA AGA TCC GCA GAC GTG TAA A) and the reverse primer (GGC GGC TAT GGG TAG TTC TGT) corresponding to the nucleotide sequences 384-405 and 563-583 of the bovine VEGF-A mRNA (Genbank accession: M31836.1). Equal volumes of reaction mixture from each sample were loaded onto 1.2% agarose gels. The images were digitally captured for analysis of intensity with Un-Scan-It software. Levels of the target gene mRNA was normalized relative to β-actin in the same sample.
Enzyme-linked immunoassay
NF-kB concentration was quantified by TransAM NF-kB kit (Active Motif; Carlsbad, CA) as previously reported by us (Kowluru and Odenbach 2004c) using 5-15μg protein. The samples were loaded on a 96-well plate to which oligonucleotide containing the NF-kB binding site has been immobilized. The activated NF-kB present in the samples that bound to this nucleotide was detected by a secondary antibody conjugated to HRP. The standard concentrations used in the ELISA ranged from 8pg/μl to 125pg/μl, and our samples were routinely in the range of 16 to 40pg/μg protein.
Similarly, IL-1β was quantified by IL-1β ELISA kit (#RLB00; R& D Systems, Minneapolis, MN), as previously described by us (Kowluru and Odenbach 2004 a&b). The samples (30-50μg protein) were loaded into a microplate pre-coated with rat polyclonal antibody for IL-1β. Good linearity (R=0.99) was obtained using human antibody (as provided in the ELISA kit) and bovine IL-1β (US Biological, Swampscott, MA) as standard (62.5-500pg/ml). After washing the plates to remove unbound substances, enzyme-linked rat IL-1β polyclonal antibody was added to the plate and washed again and the substrate was added and the color was measured at 450nm. ELISA experiments were performed in duplicate to ensure reproducibility of the data. The values varied from 5-7% within each assay and in between two assays.
The amount of VEGF was quantified using 30-75μg protein by solid-phase ELISA using serial dilutions of the recombinant human VEGF as standard (25pg/ml to 250pg/ml) (Chan et al., 2008), and our samples ranged from 35-60pg/ml.
Western blot
Protein (20-30μg) was separated by SDS-PAGE on a 10% gel and blotted to nitrocellulose membrane. The membranes were blocked in 5% nonfat milk suspended in Tris-buffered saline containing 0.02% Tween 20, and incubated with the primary antibody (NF-kB p65 and NF-kB phosphor p65; Abcam, Cambridge, MA) for one hour at room temperature. The blots were washed and incubated with appropriate horseradish peroxidase-coupled secondary antibody for one hour. The target proteins were enhanced by ECL reagent (Santa Cruz Biotechnology), and determined by autoradiography.
Cell death analysis
Cell death was determined by performing ELISA using Cell Death Detection ELISAPLUS (Roche Diagnostics) as routinely used in our laboratory (Kowluru and Odenbach 2004b). Relative amounts of mono- and oligonucleosomes generated from apoptotic cells were quantified using monoclonal antibodies directed against DNA and histones respectively. After separation of the cytoplasmic fraction from pericytes, the nuclear pellet was suspended in 50mM sodium phosphate buffer (pH 7.5) containing 2mM NaCl, DNA was measured in this fraction, and used to normalize apoptosis.
Statistical analysis
Each assay was run in duplicate/triplicate using 3 or more samples, and each experiment was repeated in at least 3 different cell preparations. Data are reported as mean ± SD, and experimental groups were compared using the nonparametric Kruskal-Wallis test followed by Mann-Whitney test. Similar conclusions were also reached by using ANOVA with Fisher or Tukey tests. P values <0.05 were considered statistically significant.
Results
Effect of high glucose on the inflammatory mediators in the retinal pericytes
Incubation of retinal pericytes in 20mM glucose increased the gene expressions of IL-1β by 35% (Figure 1a), NF-kB by 45% (Figure 2a), VEGF by about 100% (Figure 3a), and TNF-α, TGF-β and ICAM-1 by 60-400% (Figures 4a-c). In the same pericyte preparations the concentrations of IL-1β, NF-kB and VEGF were increased by 30-70% compared to the pericytes incubated in 5mM glucose (Figures 1b, 2b and 3b), and NF-kB was activated as evidenced by increased expression of phospho-p65 (Figure 2c). To confirm their secreted levels in the media, we measured the absolute amount of IL-1β in the media. The results obtained from the cells and media complemented each other, but the cellular levels of these pro-inflammatory mediators were 2-5 lower than their secreted levels. However, it is important to point out that our results show that these mediators were significantly elevated in the pericytes incubated in 20mM glucose compared to the pericytes incubated in 5mM glucose. Glucose-induced increase in the inflammatory mediators was not influenced by the duration of incubation in continuous high glucose, the values obtained after 2, 4, 8 and 12 days of 20mM glucose exposure were comparable. Incubation of pericytes in 20mM mannitol instead of glucose had no effect on any of the proinflammatory mediators investigated in our study.
Figure 1.

Effect of variant glucose on IL-1β: Retinal pericytes were incubated with 20mM glucose for 2 or 4 days, followed by 5mM glucose for 4 or 8 additional days. The cells incubated in continuous 5mM or 20mM glucose served as controls. (a) IL-1β mRNA was determined by RT-PCR method using the TaqMan Assays on Demand (ABI), and (b) its concentration was quantified by ELISA using a kit from R& D Systems. Each measurement was made in duplicate using 3 or more different cell preparations. The values are represented as mean ±SD, and the numbers obtained in continuous 5mM are considered as 100%. 5mM or 20mM= exposure of cells to continuous 5mM glucose or 20mM glucose; 2d-4d=2days of 20mM followed by 4days of 5mM glucose; 4d-4d= 4days of 20mM followed by 4days of 5mM glucose, and 4d-8d= 4days of 20mM followed by 8days of 5mM glucose. *P<0.05 compared to continuous 5mM glucose, and #P<0.05 compared to continuous 20mM glucose.
Figure 2.
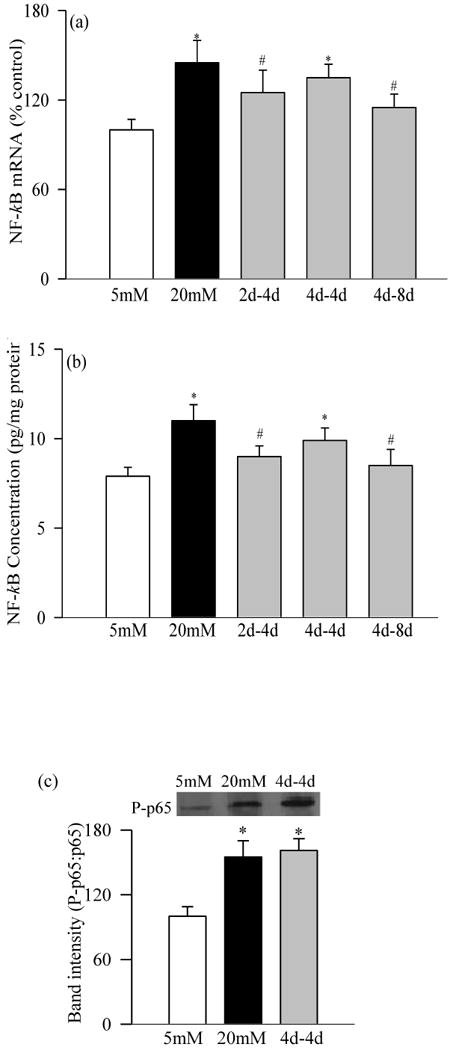
Effect of variant glucose exposure on NF-kB in retinal pericytes: As stated in the experimental section, at the end of the desired incubation, RNA and proteins were extracted from pericytes. (a) Gene expression of NF-kB was determined by RT-PCR, (b) its concentration by ELISA (TransAM NF-kB) using bovine NF-kB as standard, and (c) activation by western blot technique using antibodies against p65-NF-kB and phspho p65-NF-kB; the histogram represents the band intensity of phospho p65 (P-p65) adjusted to that of p65. *P<0.05 compared to continuous 5mM glucose, and #P<0.05 compared to continuous 20mM glucose.
Figure 3.

Glucose exposure and VEGF: (a) Gene expression of VEGF was quantified by RT-PCR method using 18s as a loading standard. 1=5mM glucose, 2=4d-8d, 3=2d-4d, 4=4d-4d and 5=20mM glucose (b) ELISA kit (R &D System) was used to quantify its concentration. The values obtained in continuous 5mM are considered as 100%, and are mean of results obtained from 3-4 different cell preparations. 5mM glucose, and #P<0.05 compared to continuous 20mM glucose.
Figure 4.

Effect of variant glucose exposure on gene expressions of (a) TNF-α and (b) TGF-β in retinal pericytes: At the end of the desired incubation, RNA was extracted from pericytes, and the gene expressions of TNF-α, TGF-β and ICAM-1 were quantified by RT-PCR. Each measurement was made in duplicate in the 3 cell preparations. 1=5mM glucose, 2=20mM glucose, 3=2d-4d, 4=4d-8d and 5=4d-4d. In the histogram continuous 5mM glucose is considered as 100%. *P<0.05 compared to continuous 5mM glucose, and #P<0.05 compared to continuous 20mM glucose.
Effect of short high glucose insult (2 days) followed by 4 days of normal glucose (2d-4d group)
Four days of normal glucose that followed 2 days of high glucose had marginal, but statistically significant (P<0.05 compared with 20mM glucose), beneficial effect on the increases in IL-1β, NF-kB, VEGF and TGF-β; their gene transcripts and concentrations were reduced by 15-30% in the pericytes incubated in 20mM glucose for 2 days followed by 5mM glucose for 4 additional days compared to the pericytes incubated in 20mM glucose for the entire six days (Figures 1, 2, 3a and 4). However, the gene transcripts of ICAM-1 remained significantly elevated, and the values were significantly higher compared to those obtained from the cells incubated in continuous 5mM glucose (Figure 4c).
Effect of 4 days of high glucose followed by 4 days of normal glucose (4d-4d group)
When the period of exposure of high glucose was extended to 4 days, which was then followed by normal glucose for 4 days, normal glucose exposure had no beneficial effect on any of the pro-inflammatory mediators investigated, the levels of IL-1β, NF-kB, VEGF, TNF-α, TGF-β and ICAM-1 remained elevated as compared to those obtained from the pericytes incubated in continuous 5mM glucose medium for 8 days, and the values obtained in 4→4day group were not different from those obtained from the cells incubated in 20mM glucose for the entire duration of the experiment (P>0.05; Figures 1-4).
Effect of long recovery after 4 days of high glucose (4-8d group)
Extension of the duration of normal glucose exposure after 4 days of high glucose exposure to 8 days partially ameliorated glucose-induced alterations in the gene transcripts of these inflammatory markers. Levels of IL-1β, NF-kB, VEGF, TNF-α and ICAM-1 were significantly lower compared to those obtained from the cells exposed to continuous 20mM glucose for 8-12 days (P<0.05), but these values still remained 15-25% higher compared to the cells exposed to continuous normal glucose for the entire duration (Figures 1-4). Eight days of normal glucose after 4 days of high glucose did not provide much beneficial effect on the TGF-β levels, and the values remained significantly elevated (Figure 4b).
Effect of reversal of high glucose exposure on the apoptosis of retinal pericytes
Apoptosis levels determined by measuring the cytoplasmic nucleosomal DNA, as expected, were elevated by over 70% in the pericytes incubated in 20mM glucose for 8 days (Figure 5). However, 4 days of 5mM glucose incubation that had followed 4 days of 20mM glucose (4d-4d group) failed to provide any protection to the retinal pericytes from undergoing apoptosis.
Figure 5.

Apoptosis was measured by performing ELISA for cytoplasmic histone-associated-DNA-fragments using an assay kit from Roche Diagnostics. The values are adjusted to the total DNA in each sample, and numbers obtained from the cells incubated in continuous 5mM glucose are considered 100%. 4d-4d+Dex= cells incubated in 20mM glucose for 4 days followed by 5mM glucose containing 1μM dexmethasone for 4 additional days.
Effect of inhibition of dexamethasone on inflammatory mediators in the retinal pericytes
To determine the effect of direct inhibition of inflammation on the metabolic memory phenomenon, dexamethasone was included during normal glucose incubation period that had followed 4 days of high glucose exposure (4d-4d+Dex), and NF-kB and VEGF gene transcripts were quantified by RTPCR and semiquantitative PCR methods. As shown in figure 6, addition of dexamethasone for the entire duration of the experiment (8 days) attenuated glucose-induced increases in these inflammatory mediators. In addition, when dexamethasone was included during the normal glucose exposure (4 days) that had followed 4 days of high glucose (4d-4d+Dex group), NF-kB and VEGF levels were significantly decreased compared to the values obtained from the cells that were incubated without dexamethasone during these 4 days of the reversal period (4d-4d group). Our goal here was to show ‘in principle’ that direct inhibition of inflammation during reversal phase will have better effect than just the reversal of high glucose alone; we did not quantify the effect of dexamethasone on IL-1β, ICAM-1 and TGF-β. Similarly, dexamethasone inhibited glucose-induced apoptosis of retinal pericytes (Figure 5), and also prevented cells from undergoing accelerated apoptosis when their high glucose exposure was reversed.
Figure 6.

Effect of direct inhibition of inflammation on the metabolic memory phenomenon: Pericytes were incubated in 20mM glucose for 4 days, followed by 5mM glucose containing 1 μmol/L dexamethasone for 4 days. Gene expression of (a) VEGF was determined by semiquantitative PCR, and (b) that of NF-kB by real time- PCR. The values obtained in continuous 5mM glucose are considered 100%.
We recognize that the ELISA kits for IL-1β and VEGF employed are for rat and NF-kB is for human, and there is a possibility that the concentrations of these pro-inflammatory mediators could either be underestimated or overestimated. ELISA results for IL-1β, NF-kB and VEGF were complemented by their gene expression using bovine primers for real time PCR (TaqMan custom primers). As presented above, our experiments using both, ELISA and mRNA measurements under 5-6 different incubation conditions (5mM, 20mM, 2 or 4 days of 20mM followed by 4 or 8 days of 5mM with or without dexamethasone) resulted in complementary conclusions.
Discussion
Our results demonstrate that retinal pericytes, the site of histopathology associated with diabetic retinopathy, experience metabolic memory phenomenon, and increased levels of inflammatory markers persist for some time after cessation of high glucose exposure. Further, we demonstrate that both the duration of the initial exposure to high glucose (pre-good glycemic control), and the duration of normal glucose that follows high glucose exposure (post- poor glycemic control), are critical in determining the outcome of the alterations in inflammatory mediators. These results could have immense clinical implications because they suggest that the consequences of the cessation of poor glycemic control on the progression of diabetic retinopathy are associated with how long poor glycemic control is maintained before initiating good glycemic control, and also long duration of post- good glycemic control will have better results in preventing the proinflammatory mediators and the progression of diabetic retinopathy.
Pericytes wrap around the capillary endothelium to provide vascular stability and regulate capillary blood flow. The retina has the highest number of pericytes in the body (Motiejunaite and Kazlauskas 2007), and their loss, an early, specific and selective event, is considered a hallmark of retinopathy in animal models of diabetic retinopathy (Kuwabara and Cogan 1965; Hammes et al., 2002; Hammes 2005). Pericytes contribute to the barrier function of microvessels and help in regulating inflammatory events, such as leakage of plasma proteins (Sims 2000). In the pathogenesis of diabetic retinopathy, pericytes lost is considered to be mainly an apoptotic phenomenon that occurs before vascular histopathology can be seen (Mizutani et al., 1996; Kern et al., 2000). Diabetes has been shown to upregulate various proinflammatory mediators in the retina, including, ICAM-1, VEGF, NF-kB, iNOS, and TGF-β, and localized inflammatory processes is considered to play a role in the development of diabetic retinopathy (Joussen et al., 2001; Kowluru and Odenbach 2004a; Kern 2007; Chan et al., 2008). Data presented here clearly show high glucose significantly elevates various pro-inflammatory mediators in retinal pericytes suggesting that increased inflammation is an important contributor to their accelerated apoptosis and progressive loss during the development of diabetic retinopathy. In support, our previous studies using an inhibitor of NF-kB or IL-1β receptor antagonist have shown that these inflammatory mediators have a major role in glucose-induced apoptosis of retinal capillary cells (Kowluru and Odenbach 2004a&b).
Advantages of intensive glycemic control persist beyond the duration of maintenance of tight glycemic control, and the reestablishment of good glycemic control after a profound period of poor control does not immediately benefit the progression of retinopathy (Diabetes Control and Complications Trial Research Group 1993; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group 2008). Metabolic abnormalities including oxidative stress, nitrative stress and advanced glycation end products resist reversal when good glycemic control is reinstituted in rats after a period of poor glycemic control (Kowluru 2003; Kowluru et al., 2004; Ihnat et al., 2007; Kowluru et al., 2007; Chan et al., 2008; Kanwar and Kowluru 2009; Kowluru and Chan 2009), and dysregulation of epigenetic histone modifications is postulated as one of the underlying mechanism for sustained proinflammatory phenotype of diabetic cells (Villeneuve et al., 2008). We have demonstrated that pro-inflammatory cytokines and adhesion molecules that are implicated in the development of diabetic retinopathy, also resist suppression after institution of good glycemic control in rats (Chan et al., 2008), but the limitation of our study was the use of the whole retina homogenate, and that restricted us from identifying the specific retinal cell types that continued to have increased insult. Here, using pericytes isolated from the retina, we show that the levels of IL-1β, NF-kB, VEGF, TNF-α, TGF-β and ICAM-1, once activated by high glucose, do not immediately become normal after glucose insult is terminated, implicating that prior glucose leaves an imprint on the pericytes. In addition, the continued deleterious effects of glucose on these mediators suggest that inflammation is important in the continued pericyte loss, ultimately, in the progression of diabetic retinopathy. In support, our in vivo results have shown that retinal microvascular cells continue to undergo apoptosis even after hyperglycemia is terminated (Kowluru and Chan 2009).
The duration of initial glucose insult is an important factor in the outcome of the effects of the normal glucose exposure that follows. Exposure of pericytes to normal glucose for 4 days after 2 days of high glucose partially attenuates increases in IL-1β, NF-kB, TGF-β and VEGF suggesting that good glucose control has potential to produce beneficial effects if the cells are exposed to high glucose for a shorter duration. However, when the high glucose exposure is extended to 4 days, 4 days of normal glucose that follows provides no beneficial effect on any of the pro-inflammatory mediators, and the pericytes continue to undergo apoptosis, suggesting that the pre-good glycemic control duration has a major role in dictating the benefits of good control that follows. The results implicate that the inflammatory process, which is initiated in high glucose conditions, becomes less amicable with time to revert by normal glucose, and the early glucose exposure is crucial in metabolic imprinting. This is in consistent with the findings reported in DCCT studies (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group 2008), and with the studies using islet transplants in diabetic rats (Hammes et al., 1993). Thus, our results clearly demonstrate that early metabolic control has potential to offset the deleterious effects of high glucose on inflammatory mediator, and to slow down the progression of diabetic retinopathy.
Dexamethasone, a corticosteroid, inhibits the expressions (both, protein and gene) of proinflammatory mediators (Holz et al. 2005), and in retinal pericytes it is shown to inhibit high glucose-induced or cytokine-induced release of inflammatory and angiogenic proteins (Nehmé and Edelman 2008). Here we demonstrate that the supplementation with dexamethasone during the normal glucose exposure (reversal period) that has followed high glucose significantly ameliorates elevations in proinflammatory mediators, and the levels of these inflammatory mediators are significantly lower compared to those obtained without dexamethasone supplementation during the 4 days of the reversal period. These results imply that supplementation with agents that can target increased inflammation during good glycemic control should provide greater benefits of glycemic control in preventing pericytes loss than just the good glycemic control itself. This could have great clinical implications because, along with maintenance of good glycemic control in diabetic patients, inhibition of inflammation with therapies could prevent/retard retinopathy to further progress.
Retinal pathology remains unchanged in patients after re-establishment of good control, and the longer patients can maintain a good control the greater their protection is from complications. Here we show that when the duration of normal glucose is increased to 8 days after 4 days of high glucose insult, increases in pro-inflammatory mediators are partially ameliorated. This suggests that the benefits of normal glucose begin to appear when the duration of normal glucose exposure is extended; increases in inflammatory mediators brought out in high glucose conditions begin to halt, and is consistent with the results from EDIC studies (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group 2005; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group 2008).
Thus, our study highlights the importance of inflammation in retinal pericytes apoptosis and in the metabolic memory phenomenon, and pinpoints the importance of the duration of the reversal in its outcome. We have provided exciting results showing that the early good control has great benefit on the accumulation of proinflammatory mediators, once accumulated; these mediators resist arrest for sometime before beginning to subside. The results are expected to have tremendous clinical significance, and suggest that the heath care professionals should advise patients that the benefits of the maintaining good glycemic control could take a little longer, and prescribed additional therapies to directly retard inflammation.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health (R01-EY17313), Juvenile Diabetes Research Foundation (1-2006-0616), The Thomas Foundation, and Research to Prevent Blindness.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boeri D, Maiello M, Lorenzi M. Increased prevalence of microthromboses in retinal capillaries of diabetic individuals. Diabetes. 2001;50:1432–1439. doi: 10.2337/diabetes.50.6.1432. [DOI] [PubMed] [Google Scholar]
- Chan PS, Kanwar M, Kennedy A, Kowluru RA. Resistance of retinal inflammatory mediators to suppress after re-institution of good glycemic control: Novel mechanism for metabolic memory. J Diabetes Complications. 2008 doi: 10.1016/j.jdiacomp.2008.10.002. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Grou. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Group. Early worsening of diabetic retinopathy in the diabetes control and complication trial. Arch Ophthalm. 1998;116:874–886. doi: 10.1001/archopht.116.7.874. [DOI] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Prolonged effect of intensive therapy on the risk of retinopathy complications in patients with type 1 diabetes mellitus: 10 years after the Diabetes Control and Complications Trial. Arch Ophthalmol. 2008;126:1707–1715. doi: 10.1001/archopht.126.12.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–812. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- Haefliger IO, Zschauer A, Anderson DR. Relaxation of retinal pericyte contractile tone through the nitric oxide-cyclic guanosine monophosphate pathway. Invest Ophthalmol Vis Sci. 1994;35:991–997. [PubMed] [Google Scholar]
- Hammes HP. Pericytes and the pathogenesis of diabetic retinopathy. Horm Metab Res. 2005;37:39–43. doi: 10.1055/s-2005-861361. [DOI] [PubMed] [Google Scholar]
- Hammes HP, Lin J, Renner O, Shani M, Lundqvist A, Betsholtz C, Brownlee M, Deutsch UJ. Pericytes and the pathogenesis of diabetic retinopathy. Diabetes. 2002;51:3107–3112. doi: 10.2337/diabetes.51.10.3107. [DOI] [PubMed] [Google Scholar]
- Hammes HP, Hammes HP, Klinzing I, Wiegand S, Bretzel RG, Cohen AM, Federlin K. Islet transplantation inhibits diabetic retinopathy in the sucrose-fed diabetic Cohen diabetic rat. Invest Ophthalmol Vis Sci. 1993;34:2092–2096. [PubMed] [Google Scholar]
- Holz LE, Jakobsen KP, Van Snick J, Cormont F, Sewell WA. Dexamethasone inhibits IL-9 production by human T cells. J Inflamm (Lond) 2005;2:3. doi: 10.1186/1476-9255-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihnat MA, Thorpe JE, Kamat CD, Szabó C, Green DE, Warnke LA, Lacza Z, Cselenyák A, Ross K, Shakir S, Piconi L, Kaltreider RC, Ceriello A. Reactive oxygen species mediate a cellular ‘memory’ of high glucose stress signalling. Diabetologia. 2007;50:1523–1531. doi: 10.1007/s00125-007-0684-2. [DOI] [PubMed] [Google Scholar]
- Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152. doi: 10.1016/S0002-9440(10)63952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- Kanwar M, Kowluru R. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes. 2009;58:227–234. doi: 10.2337/db08-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern TS, Tang J, Mizutani M, Kowluru R, Nagraj R, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: Comparison of diabetes and galactosemia. Invest Ophthalmol, Vis Sci. 2000;41:3972–3978. [PubMed] [Google Scholar]
- Kowluru RA. Effect of re-institution of good glycemic control on retinal oxidative stress and nitrative stress in diabetic rats. Diabetes. 2003;52:818–823. doi: 10.2337/diabetes.52.3.818. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Chakrabarti S, Chen S. Re-Institution of good metabolic control in diabetic rats on the activation of caspase-3 and nuclear transcriptional factor (NF-kB) in the retina. Acta Diabetologica. 2004;44:194–199. doi: 10.1007/s00592-004-0165-8. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Chan PS. Metabolic memory in diabetes - from in vitro oddity to in vivo problem: Role of Apoptosis. Brain Res Bull. 2009 May 20; doi: 10.1016/j.brainresbull.2009.05.006. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Kanwar M, Kennedy A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp Diabetes Res. 2007;2007:2196. doi: 10.1155/2007/21976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Koppolu P. Diabetes-induced activation of caspase-3 in retina: Effect of antioxidant therapy. Free Radical Res. 2002;36:993–999. doi: 10.1080/1071576021000006572. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res. 2003;37:1169–1180. doi: 10.1080/10715760310001604189. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Odenbach S. Role of interleukin-1beta in the development of retinopathy in rats: effect of antioxidants. Invest Ophthalmol Vis Sci. 2004a;45:4161–4166. doi: 10.1167/iovs.04-0633. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. British J Ophthalmol. 2004b;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Odenbach S. Effect of long-term administration of alpha lipoic acid on retinal capillary cell death and the development of retinopathy in diabetic rats. Diabetes. 2004c;53:3233–3238. doi: 10.2337/diabetes.53.12.3233. [DOI] [PubMed] [Google Scholar]
- Kuwabara T, Cogan DG. Retinal vascular patterns. VII. Acellular change. Invest Ophthalmol Vis Sci. 1965;4:1049–1064. [PubMed] [Google Scholar]
- LeRoith D, Fonseca V, Vinik A. Metabolic memory in diabetes-focus on insulin. Diabetes Metab Res Rev. 2005;21:85–90. doi: 10.1002/dmrr.530. [DOI] [PubMed] [Google Scholar]
- Miller AG, Smith DG, Bhat M, Nagaraj RH. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J Biol Chem. 2006;281:11864–11871. doi: 10.1074/jbc.M513813200. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiejunaite R, Kazlauskas A. Pericytes and ocular diseases. Exp Eye Res. 2007;86:171–177. doi: 10.1016/j.exer.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Nehmé A, Edelman J. Dexamethasone inhibits high glucose-, TNF-alpha-, and IL-1beta- induced secretion of inflammatory and angiogenic mediators from retinal microvascular pericytes. Invest Ophthalmol Vis Sci. 2008;49:2030–2038. doi: 10.1167/iovs.07-0273. [DOI] [PubMed] [Google Scholar]
- Pfister F, Feng Y, Vom Hagen F, Hoffmann S, Molema G, Hillebrands JL, Shani M, Deutsch U, Hammes HP. Pericyte migration: A novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes. 2008;57:2495–2502. doi: 10.2337/db08-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Sala R, Cagliero E, Lorenzi M. Overexpression of fibronectin induced by diabetes or high glucose: phenomenon with a memory. Proc Natl Acad Sci USA. 1990;87:404–408. doi: 10.1073/pnas.87.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims DE. Diversity within pericytes. Clin Exp Pharmacol Physiol. 2000;27:842–846. doi: 10.1046/j.1440-1681.2000.03343.x. [DOI] [PubMed] [Google Scholar]
- Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A. 2008;105:9047–9052. doi: 10.1073/pnas.0803623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang J, Dashti A, Wilson K, Szweda L, Zou MH, Lyons T, Ma JX. Pigment epithelium-derived factor (PEDF) mitigates inflammation and oxidative stress in retinal pericytes exposed to oxidized-LDL. J Mol Endocrinol. 2008;41:135–143. doi: 10.1677/JME-08-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]