

Abstract
One of the challenges of incorporating molecularly targeted drugs into multi-agent chemotherapy (backbone) regimens is defining dose limiting toxicities (DLTs) of the targeted agent against the background of toxicities of the backbone regimen. An international panel of 22 pediatric acute lymphocytic leukemia (ALL) experts addressed this issue (www.ALLNA.org). Two major questions surrounding DLT assessment were explored: 1) how toxicities can be best defined, assessed, and attributed; and 2) how effective dosing of new agents incorporated into multi-agent ALL clinical trials can be safely established in the face of disease- and therapy-related systemic toxicities. The consensus DLT definition incorporates tolerance of resolving Grade 3 and some resolving Grade 4 toxicities with stringent safety monitoring. This functional DLT definition is being tested in two Children’s Oncology Group (COG) ALL clinical trials.
Keywords: ALL, ALL relapse, developmental therapeutics, dose-limiting toxicity, maximum tolerated dose
INTRODUCTION
Remarkable advances have been made in the past 40 years in the cure rates for children with newly diagnosed acute lymphoblastic leukemia (ALL), with average 5-year event-free survival (EFS) of >85% (1). However, high-risk patients and those who experience disease relapse have a very guarded prognosis despite aggressive chemotherapy and stem cell transplant (2). Incorporation of molecularly targeted agents into reinduction and consolidation therapy for these children is necessary to improve outcome.
Molecular targets relevant to the biology of ALL, and for which drugs are in early clinical trials, include (but are not limited to) novel nucleoside analogues, inhibitors of FLT-3, BCR-ABL, mTOR, Bcl-2, ribonucleotide reductase, and the proteasome complex (3–7). Currently available inhibitors of these targets have the potential to increase treatment efficacy, and usually have non-overlapping toxicities with standard cytotoxic chemotherapy agents, making them attractive agents for addition to chemotherapy induction and consolidation regimens for patients with relapsed ALL (8).
Although many new agents undergo single-agent drug testing to determine their maximal tolerated dose (MTD) in pediatric solid tumors, accrual of pediatric ALL patients to single-agent phase 1 trials is challenging. Because complete response (CR) rates in single agent phase 1 trials have historically been no better than 8–10% (9), and a variety of multi-agent regimens have reported CR rates of 40% for children with multiply relapsed ALL (2), it is common practice to treat children with relapsed ALL using combinations of well-known cytotoxic agents to achieve rapid disease control and, hopefully, a complete remission. One possible approach to Phase 1 testing of new agents for ALL that may overcome difficulties in accruing patients to single-agent phase 1 studies is to define the MTD of a novel agent in the context of a multi-agent “backbone” chemotherapy regimen.
There are advantages to testing targeted agents in the context of a well-known backbone regimen. This approach offers the possibility of disease control even if the novel agent proves to be inactive. Moreover, clinical application of active novel agents is likely to be in the context of multi-agent regimens, so defining the MTD in that setting is more clinically relevant than in a trial as a single agent. This approach allows definition of a tolerable dose for new agents in the context of multi-agent chemotherapy, potentially enhancing the therapeutic efficacy of the backbone regimen.
However, this approach presents several challenges. Toxicities of the novel agent are confounded not only by the toxicities of the underlying disease, but also by the substantial toxicity of the cytotoxic backbone regimen. Although standardized supportive care measures can minimize many frequent toxicities (electrolyte abnormalities, nausea, vomiting, febrile neutropenia), toxicity attributions are often difficult to discern.
The frequency of severe systemic toxicities associated with multi-agent ALL reinduction chemotherapy is high; clinicians and patients routinely accept substantial morbidity from the therapy in order to obtain potential benefit. Toxicity rates often exceed conventional rates of dose-limiting toxicity for single agents tested in solid tumors and thus could compromise dose-finding trials. For example, many single-agent phase 1 studies use the 3+3 design (reviewed in 10) in order to determine the MTD of an agent, typically defined as that which is associated with a dose-limiting toxicity (DLT) rate of 33% or less (10). Since standard definitions of DLT include most non-hematologic Grade 3 or 4 toxicities, the incidence of DLTs in most reinduction regimens for ALL would routinely exceed this rate of severe dose-limiting toxicities if the standard DLT definition were applied (Table I). Although variable reporting of toxicities in phase 2 and 3 clinical trials often underestimates the true incidence of toxicities, conventional phase 1 dose escalation rules would likely inappropriately limit dose escalation of a new drug. In many cases, it is likely that no dose of a potentially useful agent would meet conventional standards in the context of an intensive cytotoxic regimen.
Table I.
Non-hematologic Grade 3 and 4 toxicities that occurred in more than 5% of the 109 patients receving VPLD* in COG AALL01P2.
| Grade 3 or 4 toxicity | % of patients with toxicity |
|---|---|
| Hypertension | 7% |
| Low fibrinogen | 32% |
| Pancreatits | 7% |
| Stomatitis | 6% |
| Hemorhage | 6% |
| Hyperbilirubinemia | 6% |
| Hypoalbuminemia | 9% |
| Elevated ALT | 13% |
| Febrile Neutropenia | 58% |
| Hyperglycemia | 11% |
| Hypocalcemia | 8% |
| Hypokalemia | 7% |
| Elevated lipase | 8% |
vincristine, prednisone, L-asparaginase, and doxorubicin
In designing regimens that assess the addition of a novel agent to existing chemotherapies, the novel agent may add to the toxicity of the backbone cytotoxic regimen. If the toxicities of the backbone therapy and the experimental agent are completely non-overlapping, one can use a standard escalation design with a definition of DLT limited to toxicities associated only with the experimental agent. However, partial or complete overlap of toxicities is more likely and interactions cannot be excluded. Thus, investigators must be willing to accept greater, but not excessive, overall toxicity for an experimental agent when added to a multi-agent regimen, and dose escalation rules and the definition of DLT need to be modified accordingly.
An international panel of 22 pediatric ALL experts from 6 countries (Canada, Australia, Italy, the Netherlands, United Kingdom, and the United States) and 9 cooperative cancer groups (COG, POETIC, TACL, DFCI, AIEOP, CCLG, DCOG, ITCC, and I-BRM SG New Agents Group) was convened at an ALL New Agents (ALLNA; www.ALLNA.org) meeting on February 2–4, 2008 to discuss DLT definitions for novel agents incorporated into multi-agent clinical trials. Results of this meeting reflect the opinions of the coauthors and do not represent an official position of the cooperative groups.
DEFINING THE ISSUE
The standard re-induction chemotherapy regimen VPLD (Vincristine, Prednisone, L-Asparaginase, Doxorubicin) is an example of a backbone into which a new drug might be integrated for patients with relapsed ALL. The safety and efficacy of a novel agent X, may be evaluated by adding X to VPLD (VPLD+X). For example, the COG has added the anti-CD22 monoclonal antibody epratuzumab to this backbone regimen in a pilot phase 2 clinical trial. To establish the safety and maximum tolerated dose (MTD) of the new agent in combination with VPLD, it is necessary to determine the attribution of all toxicities.
However, when VPLD is administered as a reinduction regimen for children with relapsed ALL, it is associated with significant toxicities that can confound efforts to define the MTD of X in this setting. Table I summarizes the more frequent severe toxicities associated with a variant of VPLD (weekly PEG-asparaginase was substituted for thrice-weekly L-asparaginase) in a recent Children’s Oncology Group (COG) study for relapsed ALL (AALL01P2) (11). The frequency of individual non-hematologic Grade 3 and 4 toxicities ranged up to 58% (41% if febrile neutropenia is excluded as a DLT), and 67% of patients would have experienced a DLT if a conventional phase 1 definition (Table II) were applied without modification. Therefore, most patients treated with VPLD experienced one or more severe toxicities that would commonly be considered dose-limiting even in the absence of an experimental agent X.
Table II.
Conventional non-hematologic DLT definition
| Although the DLT definitions used by different clinical trial consortia vary, many are similar to the one used in the U.S. by the Children’s Oncology Group (COG): |
| Non-hematologic dose-limiting toxicity: Any Grade 3 or greater non-hematological toxicity attributable to the investigational drug. |
Common exclusions:
|
Figure 1A shows the theoretical probability of rejecting all tested doses of a new agent as unacceptably toxic, as a function of the background DLT rate attributable to the combination therapy exclusive of new agent X, and where the contribution of X to the toxicity rate is negligible. As Figure 1A shows, a background DLT rate of 24% or higher would result in >50% of trials rejecting any dose of an innocuous new agent X. Figure 1B shows the probability of selecting different doses in a trial when DLTs due to agent X are additive on top of a 20% background DLT rate. Here, 52% of trials would result in no acceptable dose of X being selected. Thus, the challenge is to establish a DLT definition that will enable defining an appropriate dose of a new agent without either under-dosing due to background toxicity, or overdosing resulting in an unacceptable increase in morbidity.
Figure 1.
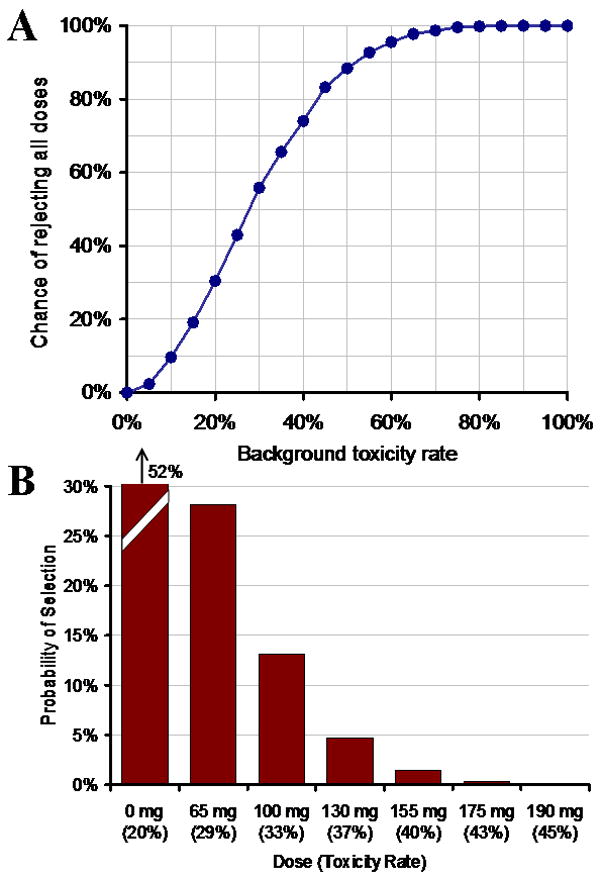
(A) Probability of rejecting all doses of a new agent X when agent X is added to a backbone chemotherapy regimen but adds negligible toxicity to the combination, as a function of background toxicity rate. (B) Probability of selecting a dose of agent X which adds no toxicity over a background rate of 20%. Computations are based on standard 3+3 cohort design with 6 dose levels. With the high background rates of DLT in the backbone regimen, there is a high probability that no dose of X will be found acceptable in combination with standard chemotherapy.
POTENTIAL APPROACHES FOR DEFINING A NOVEL AGENT DLT
Several different approaches can be used to assess novel agent DLTs when novel agent X is incorporated into a backbone regimen:
Itemized exclusion approach: Table I quantified the percentage of many of the toxicities in a previous reinduction regimen. Grade 3 and 4 toxicities occurring at a high frequency in the backbone chemotherapy regimen could be excluded from the DLT definition. As an example, a study incorporating novel agent X with VPLD could exclude 1) all toxicities listed in Table I, and 2) all Grade 3 and 4 nausea or vomiting, resolving metabolic abnormalities due to tumor lysis syndrome, and resolving transaminase elevations. However, if these exclusions were employed, the baseline toxicity rate would be reduced to only 33%. As shown in Figure 1A, this would mean that about 60% of trials would result in no dose of agent X being acceptable.
Limiting toxicities included as DLTs: Another strategy (also referred to as the “don’t ask, don’t tell” approach) is to limit toxicity reporting to only Grade 4 (life threatening) non-hematologic, non-metabolic toxicities. Using the VPLD example (Table I), if toxicity reporting were limited to Grade 4 toxicities, and if Grade 4 nausea, vomiting, anorexia, fatigue, fever, infection, resolving metabolic abnormalities, and hypofibrinogenemia are excluded, the baseline toxicity rate decreases to 10%, which appears to be an acceptable number in terms of trial design (Figure 1A).
Functional DLT approach: A third approach, which the ALLNA consensus panel favored, is to report both Grade 3 and 4 toxicities, but to consider Grade 3 toxicities as dose-limiting only if they substantially delay delivery of backbone chemotherapy. With certain exceptions (nausea, vomiting, anorexia, fatigue, infection, fever with or without neutropenia, resolving metabolic abnormalities, and hypofibrinogenemia), Grade 4 toxicities would be considered DLTs. Using this approach, the baseline toxicity rate in the AALL01P2 historical control study was between 9% (grade 3 toxicities known to have delayed delivery of the backbone chemotherapy regimen) and 16% (includes Grade 3 toxicities that may have delayed therapy). A functional DLT approach allows for the differences in significant reporting bias between phase 3 studies and the intensive reporting required for trials that include investigational agents. Since the incidence of Grade 3 and 4 toxicities is often substantially underreported in historical control studies, and centers are more likely to report all Grade 3 and 4 toxicities when careful monitoring is required, there is likely to be a substantial increase in the perceived occurrence of severe toxicities.
Pragmatic DLT approach: This approach would be limited to regimens in which there are incomplete data on the toxicity of the backbone regimen. Similar to the functional DLT approach, Grade 4 toxicities, as well as Grade 3 toxicities that delay administration of the backbone regimen, would be considered DLTs. For all DLTs possibly attributed to the investigational agent, an assigned committee would determine if each patient had experienced a DLT. Although this approach was considered more subjective than other approaches, it would provide a framework for DLT assessment in circumstances when novel agents are being tested in combination with a backbone regimen for which the expected toxicity rate is not well defined.
ADVANTAGES AND DISADVANTAGES OF DIFFERENT APPROACHES FOR DEFINING NOVEL AGENT DLT
Each of the above approaches for DLT assessment has advantages and disadvantages:
-
Itemized exclusion approach: Although itemization of specific exclusions is a precise approach, it does not account for severe toxicities that occur at low frequency. Table I lists the 13 toxicities occurring at a frequency of >5% in the COG AALL01P2 study Block 1; however, an additional 12 Grade 3 and 4 toxicities occurred at a frequency of 1–5%. Although these individual toxicities were infrequent, in aggregate, they accounted for a 33% background toxicity rate. This implies that if all Grade 3 or 4 toxicities occurring at a rate of >5% were excluded, the background toxicity rate would still be 33%. As shown in Figure 1A, a 33% toxicity incidence in the backbone regimen makes it unlikely that any dose of a novel agent would be acceptable in 60% of cases.
This approach also ignores the potential of a novel agent to increase the frequency of a known toxicity of the cytotoxic reinduction regimen, since these toxicities are excluded from the DLT definition. Thus, this approach is unlikely to be able to exclude enough toxicities to reduce the backbone toxicity rate to an acceptable level, and is also likely to miss additive toxicity from combining the novel agent and the cytotoxic backbone regimen. It also does not take into account that a significant increase in the frequency of some of the common grade 3 or 4 toxicities associated with the backbone regimen (i.e. pancreatitis) would be considered intolerable.
Limiting toxicities included as DLTs: This DLT definition has the significant disadvantage of not counting Grade 3 toxicities as DLTs, regardless of tolerability, duration or severity. Some Grade 3 toxicities, such as non-resolving grade 3 hypersensitivity reactions or non-resolving nodal arrhythmias, would be of sufficient severity to be considered intolerable. This definition also does not take into account novel agent exacerbations of Grade 3 toxicities related to the backbone regimen.
-
Functional DLT approach: This approach has several advantages. Life-threatening Grade 4 toxicities, with a few exceptions (nausea/vomiting, anorexia, fatigue, fever with or without neutropenia, and hypofibrinogenemia) would count as DLTs. This definition also includes non-resolving Grade 3 toxicities of sufficient severity to result in delay or omission of administration of the backbone chemotherapy regimen for >7 days. Although the COG has collected limited information about the resolution of Grade 3 toxicities on previous high-risk ALL induction regimens such as AALL01P2, most grade 3 toxicities (with the exception of neutropenic fever) with reported resolution dates resolved or decreased in severity to <grade 3 within 7–10 days.
The functional DLT definition approach also accounts for the potential of novel agent X to exacerbate backbone-related toxicities. This is an important advantage since it is not possible to know a priori which toxicities might be exacerbated by a novel agent. Of the three approaches discussed to define a novel agent DLT in the presence of known toxicity data from the backbone chemotherapy, only this approach accounted for the potential of novel agent X to potentiate toxicity as well as enhance disease control.
Pragmatic DLT approach: This approach should work similarly to the functional approach. However, since baseline toxicities are not known, it would require review of all toxicities by an appointed team. This team would have to be impartial and not involved with the design or conduct or the trial, analogous to an independent data safety and monitoring committee. This approach has the disadvantage of potentially adding significant bias in the evaluation of toxicity attributions, but could be an acceptable alternative in the absence of toxicity data from a historical or concurrent control group.
DEFINITON OF HEMATOLOGIC DLT FOR A NOVEL AGENT IN ALL
It is commonly accepted that the cytotoxic agents used to treat relapsed leukemia can frequently cause severe myelosuppression. Based on data collected from induction regimens in high-risk ALL, it is possible to calculate mean time to platelet and neutrophil recovery in patients without progressive disease. From this information, a hematologic DLT can be defined as any novel agent that increases the expected recovery period beyond two standard deviations above the mean platelet/ANC recovery time in the absence of infection or persistent leukemia.
In patients responding to therapy, this definition of hematologic DLT can be implemented as a standard DLT in a phase 1 dose-escalation study. However, hematologic DLTs cannot be assessed until the required time period has passed for each dose level of novel agent X, which may result in a several week delay between dose escalations of the novel agent and undue prolongation of the dose escalation portion of the study. Another method of implementation, which may be particularly useful for agents that are not expected to be markedly myelosuppressive, is to employ a stopping rule that dose de-escalates novel agent X if more than an acceptable percentage of patients experience a clinically significant delay in delivery of backbone chemotherapy due to myelosuppression. Severe infections and bleeding could also be monitored as indications of excessive myelosuppression.
FUNCTIONAL DLT ALGORITHM FOR INCORPORATING NOVEL AGENTS INTO ALL THERAPY
Employing the functional DLT definition, the following algorithm was constructed for evaluating novel agent DLTs when combined with cytotoxic chemotherapy regimens.
First, tolerable toxicities are delineated as exclusion sets:
Exclusion set A: Acceptable Grade 3 or greater non-hematologic toxicities expected to occur with backbone chemotherapy alone at a frequency of 7–10% or greater.
Exclusion set B: Acceptable Grade 3 or greater non-hematologic toxicities expected to occur with novel agent alone at a frequency of 10% or greater.
Exclusion set C: Grade 3 toxicities that are acceptable and result in delay/omission of subsequent backbone chemotherapy for ≥ 7 days and occur with a frequency of ≥10% (for example: Grade 3 or 4 infections).
Next, a DLT is defined as any non-excluded Grade 4 toxicity, and any non-excluded Grade 3 toxicity that delays delivery of the backbone chemotherapy by a period that is felt to be unacceptable clinically. In some cases, such as in reinduction therapy, a short window would be allowed before needing to restart therapy; in these cases no more than a 1 week delay would be considered tolerable. In other cases, such as consolidation in a newly-diagnosed patient, a longer delay, such as 1–3 weeks, would be considered acceptable:
DLT definition:
Any Grade 4, non-heme toxicity possibly, probably or definitely related to study drug, with the exception of exclusion set A and B
Any Grade 3 non-heme toxicity that results in omission/delay of the subsequent course of chemotherapy for > 7 days (number of days could vary from 7–21), with the exception of exclusion set C.
In addition to a DLT definition, an additional safety mechanism is the inclusion of a stopping rule to account for an increased frequency of severe toxicity or increased toxic death rate above that expected from the backbone chemotherapy regimen. This provides another measure of safety and allows for cessation of a study if unanticipated severe toxicities are encountered.
APPLICATION OF THE FUNCTIONAL DLT ALGORITHM IN TWO ALL CLINICAL TRIALS
Two current COG studies employ the functional DLT algorithm for patients with ALL at high risk of treatment failure. These studies provide insight into how this approach can be applied to the testing of novel molecularly targeted agents in the context of multi-agent ALL chemotherapy regimens used for reinduction in relapsed ALL (AALL07P1) or as post-induction consolidation therapy in newly diagnosed infant ALL (AALL0631).
Example 1
Childhood relapsed ALL
The COG pilot phase 2 clinical trial (AALL07P1) incorporates the proteasome inhibitor bortezomib into 2 sequential blocks of cytotoxic chemotherapy previously used in the AALL01P2 pilot study (9). Both non-hematologic and hematologic toxicities for AALL01P2 have been well characterized (Table I), including the mean time to platelet and ANC recovery. Although severe toxicities were very common, few toxicities (with the exception of febrile neutropenia or infection) resulted in therapy delay beyond 1 week. In the AALL07P1 study DLT is defined as shown in Table III.
Table III.
Non-hematologic DLT definition for a COG ALL study (AALL07P1)incorporating bortezomib into combination chemotherapy for relapsed ALL
|
Example 2
Infant ALL
The COG phase 3 infant ALL trial (AALL0631), randomizes very high-risk infants to treatment with multi-agent chemotherapy +/− the FLT3 inhibitor lestaurtinib (12). A predecessor infant ALL study (POG 9407) reported severe (> Grade 3) and unexpected toxicities that occurred with a frequency of ≥ 10%. Toxicities included: stomatitis (24%), diarrhea (17%), transaminase elevations (17%), and neurologic toxicities (10%) (unpublished data). In addition, these infants also experienced a >10% incidence of expected toxicities, including constitutional symptoms, transient laboratory abnormalities, transient hypertension or hypotension, skin toxicities, and tumor lysis syndrome. These toxicities were assigned to Exclusion Set A. Most of these toxicities, particularly Grade 3 toxicities, resolved within 7 days. However, Grade 3 infectious complications were common in this study and frequently (>10% of events) delayed chemotherapy for >7 days; febrile neutropenia and infections were considered exclusion set C.
The toxicities of the FLT3 inhibitor lestaurtinib had been previously assessed in adults (13;14) and in two pediatric phase 1 clinical trials (COG-ADVL0631 and New Approaches to Neuroblastoma Therapy (NANT) N2007-001). Lestaurtinib-related toxicities included nausea, vomiting, anorexia, and transient transaminase elevations (unpublished data). These were considered as exclusion set B.
Combining these exclusion sets, the study incorporates the DLT definition as shown in Table IV. As demonstrated, lestaurtinib DLTs are limited to intolerable Grade 4 toxicities caused by lestaurtinib and intolerable Grade 3 toxicities that delay administration of the backbone chemotherapy by more than three weeks. The study also incorporates a stopping rule for any statistically significant increase in toxic death rate.
Table IV.
DLT definition for the COG AALL0631 study incorporating lestaurtinib into multi-agent chemotherapy for infant ALL
|
In both studies, the definition of hematologic DLT was constructed as a stopping rule. Since the mean time from start of therapy to ANC recovery was several weeks in both in the pediatric relapsed ALL study AALL01P2 (49 days) and in infant study POG-9407 (21 days) waiting for hematologic DLT assessment prior to dose escalation could significantly delay study progression. To avoid these delays, hematologic toxicity is assessed at the completion of therapy in the first 12 patients. The experimental agent would be dose reduced for hematologic DLT if 4 or more of the first 12 patients experience prolonged ANC or platelet recovery.
CONCLUSIONS
Defining appropriate DLTs in studies incorporating novel agents into known cytotoxic backbone regimens used to treat ALL is challenging and requires a method to account for toxicities of the disease state and the cytotoxic chemotherapy backbone into the DLT assessment. Several potential approaches to defining DLTs have been presented; each of these approaches has advantages and disadvantages. Modifying the standard DLT definition will enable evaluation of new drugs within multi-agent clinical trials for children with ALL at doses that are safe and effective. This second generation DLT algorithm recommended by the consensus conference, will be tested in two ongoing COG trials incorporating new agents for the treatment of ALL.
Acknowledgments
The authors wish to thank the USC-CHLA Institute for Pediatric Clinical Research for its financial support of the 2008 ALLNA conference, and Kate vonWahlde for her assistance in the organization and conduct of the conference.
Support for the 2008 Acute Lymphoblastic Leukemia New Agents Conference was provided by the USC-CHLA Institute for Pediatric Clinical Research, the Monroe Carell Jr. Children’s Hospital at Vanderbilt, and the Vanderbilt-Ingram Cancer Center. SPH is the Ergen Family Chair in Pediatric Cancer. Support provided by K23 CA113775 (TMH) and the Ladies Leukemia League.
*Consortium abbreviations
- COG
Children’s Oncology Group
- POETIC
Pediatric Oncology Experimental Therapeutics Investigators
- TACL
Therapeutic Advances in Childhood Leukemia
- DFCI
Dana-Farber Cancer Institute ALL Consortium
- AIEOP
Italian Association for Paediatric Hematology Oncology
- CCLG
Childhood Cancer and Leukemia Group, UK
- DCOG
Dutch Childhood Oncology Group
- ITCC
Innovative Therapies for Children with Cancer
- I-BFM SG
International Berlin-Frankfurt-Münster Study Group
Footnotes
Conflict of Interest Statements:
1. Pamela Kearns: I have received speakers honoraria from Genzyme and have been on advisory boards for Genzyme, BMS, Johnson & Johnson and Wyeth.
2. Paul Gaynon: I have research funding (>10,000/12 months) and have received honoraria from Genzyme (< 10,000/12 months).
References
- 1.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006 Jan 12;354(2):166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 2.Gaynon PS. Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol. 2005 Dec;131(5):579–87. doi: 10.1111/j.1365-2141.2005.05773.x. [DOI] [PubMed] [Google Scholar]
- 3.Larson RA. Three new drugs for acute lymphoblastic leukemia: nelarabine, clofarabine, and forodesine. Semin Oncol. 2007 Dec;34(6 Suppl 5):S13–S20. doi: 10.1053/j.seminoncol.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Brown P, Smith FO. Molecularly targeted therapies for pediatric acute myeloid leukemia: progress to date. Paediatr Drugs. 2008;10(2):85–92. doi: 10.2165/00148581-200810020-00003. [DOI] [PubMed] [Google Scholar]
- 5.Stubbs MC, Armstrong SA. FLT3 as a therapeutic target in childhood acute leukemia. Curr Drug Targets. 2007 Jun;8(6):703–14. doi: 10.2174/138945007780830782. [DOI] [PubMed] [Google Scholar]
- 6.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009 doi: 10.1158/1078-0432.CCR-08-0144. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCloskey SM, McMullin MF, Walker B, Irvine AE. The therapeutic potential of the proteasome in leukaemia. Hematol Oncol. 2008 Jun;26(2):73–81. doi: 10.1002/hon.848. [DOI] [PubMed] [Google Scholar]
- 8.Grant S, Dent P. Simultaneous interruption of signal transduction and cell cycle regulatory pathways: implications for new approaches to the treatment of childhood leukemias. Curr Drug Targets. 2007 Jun;8(6):751–9. doi: 10.2174/138945007780830764. [DOI] [PubMed] [Google Scholar]
- 9.Shah S, Weitman S, Langevin AM, Bernstein M, Furman W, Pratt C. Phase I therapy trials in children with cancer. J Pediatr Hematol Oncol. 1998 Sep;20(5):431–8. doi: 10.1097/00043426-199809000-00005. [DOI] [PubMed] [Google Scholar]
- 10.Skolnik JM, Barrett JS, Jayaraman B, Patel D, Adamson PC. Shortening the timeline of pediatric phase I trials: the rolling six design. J Clin Oncol. 2008 Jan 10;26(2):190–5. doi: 10.1200/JCO.2007.12.7712. [DOI] [PubMed] [Google Scholar]
- 11.Raetz EA, Borowitz MJ, Devidas M, Linda SB, Hunger SP, Winick NJ, et al. Reinduction platform for children with first marrow relapse in acute lymphoblastic lymphoma. J Clin Oncol. 2008 Aug 20;26(24):3971–8. doi: 10.1200/JCO.2008.16.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown P, Smith FO. Molecularly targeted therapies for pediatric acute myeloid leukemia: progress to date. Paediatr Drugs. 2008;10(2):85–92. doi: 10.2165/00148581-200810020-00003. [DOI] [PubMed] [Google Scholar]
- 13.Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, Chopra R, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006 Nov 15;108(10):3262–70. doi: 10.1182/blood-2006-04-015560. [DOI] [PubMed] [Google Scholar]
- 14.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004 May 15;103(10):3669–76. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]