

Abstract
There is a biomedical need to develop molecular recognition systems that selectively target the interfaces of protein and lipid aggregates in biomembranes. This is an extremely challenging problem in supramolecular chemistry because the biological membrane is a complex dynamic assembly of multifarious molecular components with local inhomogeneity. Two simplifying concepts are presented as a framework for basing molecular design strategies. The first generalization is that association of two binding partners in a biomembrane will be dominated by one type of non-covalent interaction which is referred to as the keystone interaction. Structural mutations in membrane proteins that alter the strength of this keystone interaction will likely have a major effect on biological activity and often will be associated with disease. The second generalization is to view the structure of a cell membrane as three spatial regions, that is, the polar membrane surface, the midpolar interfacial region and the non-polar membrane interior. Each region has a distinct dielectric, and the dominating keystone interaction between binding partners will be different. At the highly polar membrane surface, the keystone interactions between charged binding partners are ion-ion and ion-dipole interactions; whereas, ion-dipole and ionic hydrogen bonding are very influential at the mid-polar interfacial region. In the non-polar membrane interior, van der Waals forces and neutral hydrogen bonding are the keystone interactions that often drive molecular association. Selected examples of lipid and transmembrane protein association systems are described to illustrate how the association thermodynamics and kinetics are dominated by these keystone noncovalent interactions.
Keywords: membranes, molecular recognition, phospholipid, electrostatics, hydrogen bonding, ion-dipole interactions, van der Waals Forces
Introduction
Our current understanding of the structure and function of cell membranes has grown considerably since the fluid mosaic model was articulated by Singer and Nicholson in 1972.1 There is a consensus that biomembranes are highly dynamic molecular assemblies and that both the lateral and transmembrane (TM) distributions of membrane components are not homogeneous. While the number of different molecular components can be quite high (perhaps several hundred) there are basically two major classes of constituent compounds, polar lipids and proteins, and thus, there are three primary intermolecular association partners: protein-protein, protein-lipid, and lipid-lipid (Figure 1). These association systems are crucial components in the cell life cycle, and mutations that alter the binding dynamics and binding selectivities are often associated with disease.2, 3 Furthermore, the advent of proteomics,4 and lipidomics,5 has greatly increased our awareness of the large number of molecular targets in a cell membrane that have potential therapeutic value.6 To be clear, this article is not concerned with small “drug-like” molecules that target membrane proteins, of which there are an increasing number of well-characterized examples.7 Rather, the focus is on molecular recognition strategies that target the polar lipids or the interfaces between protein and lipid aggregates in biomembranes. These molecules may act as agonists or antagonists of biological activity and thus they can be used for various applications, such as chemical tools to study signal transduction pathways, sensing and imaging agents for diagnostic measurements, or as chemotherapeutic agents.
Figure 1.

Noncovalent interactions play critical roles in maintaining cell membrane structure and facilitating membrane function. The membrane components; polar lipids, proteins, cholesterol, and inorganic ions, associate via a composite of hydrogen bonding, van der Waals contacts, electrostatic attractions, and ion-dipole interactions.
The structural and dynamic complexity of the cell membrane makes the task of selective targeting a very challenging problem. A practically useful strategy, which is often employed in science, is to make some general assumptions that simplify a complex problem so it becomes tractable with the technology that is currently available. This article presents two simplifying concepts that are connected. The first generalization is that association of two binding partners in a biomembrane will be dominated by one type of non-covalent interaction which we refer to as the keystone interaction.* Structural mutations that alter the strength of this keystone interaction will likely have a major effect on biological activity and often will be associated with disease. The second generalization is to view the structure of a cell membrane as three spatial regions, each with a distinct dielectric, and to appreciate that the dominating keystone interaction(s) between binding partners will be different in each of the three membrane regions. Identifying these keystone interactions will likely facilitate the process of designing synthetic molecules to control association events in biomembranes.
The Three Regions of a Cell Plasma Membrane
It is helpful to view a biological membrane as a collection of proteins embedded in a bilayer of phospholipids. Furthermore, the phospholipid bilayer can be simplified as a fluid phase with three distinct regions, the nonpolar hydrocarbon interior, the midpolar interfacial region containing the uncharged phospholipid acyl ester groups, and the highly polar membrane surface that is exposed to water and contains the charged phospholipid head groups (Figure 2). An ion or small molecule that migrates from the surface of a phospholipid bilayer to the interior experiences a dramatic decrease in polar solvation and dielectric constant. Indeed, the low polarity of the hydrocarbon interior prevents charged or polar species from penetrating beyond the acyl region, and most proteins that span a bilayer membrane have a sequence of nonpolar residues that match the thickness of the hydrocarbon region. Additional complicating features, not discussed in this article, are the asymmetric TM distribution of phospholipids and the ubiquitous presence of TM electrochemical gradients. Because of these features, a biological membrane is not a symmetrical structure, and targeting molecules are likely to interact very differently with the opposite surfaces of a biomembrane.
Figure 2.
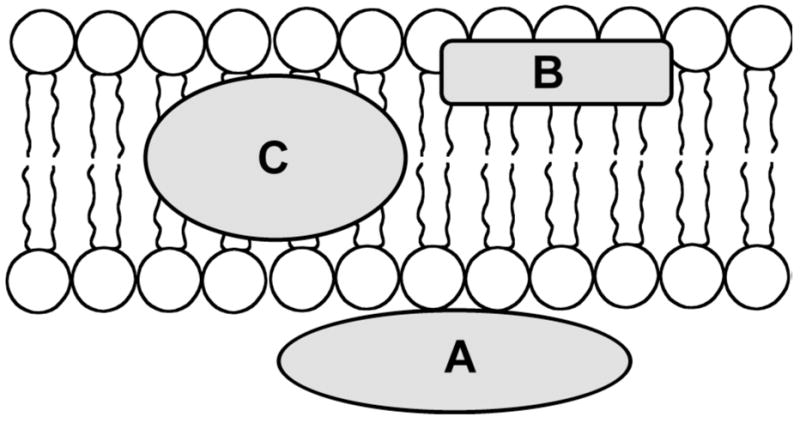
The bilayer membrane can be simplied as a fluid phase with three chemically distinct regions, (A) the polar membrane surface, (B) the midpolar interfacial region, and (C) the non-polar membrane interior.
The concept of simplifying the bilayer membrane as three spatial regions of distinct polarity has been presented before, primarily as a way of rationalizing the depth of membrane penetration by peptide and protein sequences.8 The hydrocarbon core of the membrane is around 15 Å, and it is flanked on either side by interfacial and surface regions that have a comparable width (i.e., total membrane width is about 45Å). The propensity of the amino acid side-chains to partition into a biomembrane has been quantified (hydrophobicity scale) and compared to partition constants with simple organic solvents.9 The correlation is not linear for all amino acids and the exceptions have been explained by peptide folding and aggregation processes, as well as specific interactions between functional groups on the amino acid side-chains and the phospholipids.
In the following sections, we discuss selected examples of association partners in each of the three membrane regions and show how the thermodynamics and kinetics of association are dominated by a keystone noncovalent interaction. Furthermore, we propose that each membrane region is dominated by a different type of keystone interaction and that it drives association (entropy effects are ignored). At the highly polar head group surface, the keystone interactions between charged binding partners are ion-ion and ion-dipole interactions; whereas, ion-dipole and ionic hydrogen bonding are very influential at the mid-polar interfacial region. In the non-polar membrane interior, van der Waals forces and neutral hydrogen bonding are the keystone interactions that control molecular association. Each section concludes with a short discussion of a synthetic molecular recognition system that utilizes a keystone interaction.
Keystone Interactions at the Polar Membrane Surface (Ion-Ion and Ion-Dipole Interactions)
The extracellular domains of many TM proteins display charged amino acids on the membrane surface, which frequently serve as electrostatic contacts to position the proteins correctly for ligand binding and other biochemical activity. Attractive interactions between oppositely charged proteins and a cell membrane or between TM proteins and their binding partners are often responsible for initiating protein-membrane or protein-protein binding. This important initial interaction often positions the protein to subsequently form additional stabilizing contacts with complementary functional groups at the membrane surface.
The human growth hormone receptor (hGHR) is a single-pass TM protein with an extracellular ligand binding domain projecting from the membrane surface. Human growth hormone is a peptide that binds to hGHR at a rate approximately 10,000 fold slower than the diffusion limit of the hormone, but approximately 1,000 times faster than expected if hormone-hGHR binding required the hormone to collide with the receptor in the correct orientation for binding.10 This enhanced value for the association rate, kon, arises from electrostatic attractions between four key Arg residues on the hormone and negatively charged groups on the hGHR that causes the hormone to approach the hGHR in the proper orientation for binding.11 This charge-charge interaction specifically orients the hormone for subsequent formation of stabilizing contacts that form in the ligand binding site. This electrostatic interaction is estimated to enhance kon by approximately a factor of 20.
Similar electrostatic driving forces are observed in the binding of phospholipid membranes by certain phospholipases.12 For example, the human group IIa secreted phospholipase A2 (PLA2), which preferentially binds anionic membranes, has an association rate constant, kon, for membrane binding that is 1,000–10,000 fold greater than random diffusion would predict for a specific protein-ligand binding event.10, 13, 14 The importance of electrostatics to this enhancement were revealed in charge-reversal mutants of PLA2,15 where the cationic residues surrounding the PLA2 active site were mutated to anionic residues. Such mutants exhibited a decline in kon of greater than ten fold. An even more dramatic illustration of the crucial role of electrostatic interactions is binding of the protein Myristoylated Alanine-Rich C-Kinase Substrate (MARCKS) to anionic phospholipid membranes, particularly those enriched in the anionic lipid phosphatidylinositol-4,5-bisphosphate (PIP2).16 The effector domain of MARCKS, residues 151–175, contains 13 basic residues, making MARCKS an ideal binding partner for anionic membranes. The interaction between MARCKS and anionic membranes is so favorable that the rate of association, kon, has been approximated as diffusion limited based on experiments using truncated versions of the protein with an intact effector domain.17
The formation of weakly bound protein-membrane or protein-protein complexes by electrostatic forces is frequently followed by formation of specific contacts between the interacting proteins or between protein residues and chemical functionality at the membrane surface (Figure 3). These interactions normally stabilize the complex by decreasing the rate of dissociation, koff, resulting in a tightly-bound protein-protein or protein-membrane complex. MARCKS-membrane binding illustrates how important the post-binding formation of specific contacts can be to protein-membrane complex stability and protein activity. Upon membrane binding, five Phe residues in the effector domain of MARCKS partition deeply into the bilayer, pulling the MARCKS protein down onto the membrane-water interface and forcing the protein backbone into the membrane interfacial region.18 In the Phe to Ala mutant, where all five Phe residues have been mutated to Ala, the protein associates with the membrane, but remains separated from the surface by a distance of approximately 10 Å, held in place purely by long-range Coulombic interactions.19
Figure 3.

Electrostatic interactions initiate the binding of many proteins to the membrane surface. (A) The oppositely charged membrane and protein experience an attractive coulombic interaction resulting in, (B) the formation of a weakly-bound protein-membrane complex. (C) Upon association with the membrane surface, key amino acid side-chains form specific contacts, via hydrophobic partitioning, hydrogen bonding, or cation-π interactions, that stabilize the protein-membrane complex, leading to tight protein-membrane association.
The protein and the plasma membrane do not necessarily need to be of opposite charge for electrostatics to play a key role in membrane binding. Often a protein and membrane can form a tightly-bound complex via a bridging metal ion, especially Ca2+ (Figure 4). The C2 domain is a phospholipid binding domain originally observed in protein kinase C and commonly found in a number of phospholipases.20 This domain binds to anionic membranes through a bridging Ca2+ in a cooperative manner,21 where neither the protein nor the membrane have a strong affinity for Ca2+, but when all three components come together into a three-component-assembly, a stable protein-Ca2+-membrane complex is formed. The cooperativity of membrane binding by the C2 domain of synaptotagmin I has been described, and two Ca2+ ions are known to be bound by the protein in the protein-ion-lipid assembly. The first Ca2+ is bound tightly, while the second occupies a binding site of weaker Ca2+ affinity. A phosphoryl oxygen of a phospholipid headgdroup is required for tight Ca2+ binding and complex formation. Studies of the C2 domain in protein kinase C α (PKCα) have uncovered similar binding mechanisms, where bridging Ca2+ ions anchor the phospholipid,22 especially phosphatidylserine (PS), into the binding site of the C2 domain. Importantly, the presence of a bridging Ca2+, and not simply a center of cationic charge, is absolutely required for binding. PKC mutants with cationic residues introduced into the C2 domain where Ca2+ would normally be found were unable to bind anionic membranes.23 Identical observations have also been made using the PS-binding protein Annexin V,24 which requires at least two Ca2+ ions to simultaneously coordinate the carboxylate and the phosphate of PS (Figure 5). These Ca2+ ions form a bridge that links Annexin V and PS in a tightly-bound three-component assembly, a membrane binding mechanism that is highly conserved among the Annexin family of proteins.
Figure 4.
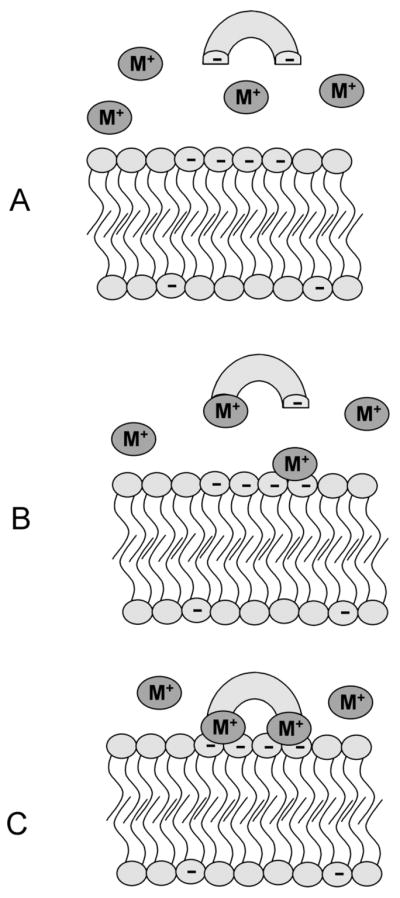
Metal cation mediates formation of a protein-membrane interaction. (A) The metal cation binding affinity of both the negatively-charged protein and the anionic membrane surface is weak in the absence of the third complexation partner; (B) and (C) formation of a highly stable three-component assembly process, where metal cation(s) bridge the protein and membrane surface.
Figure 5.
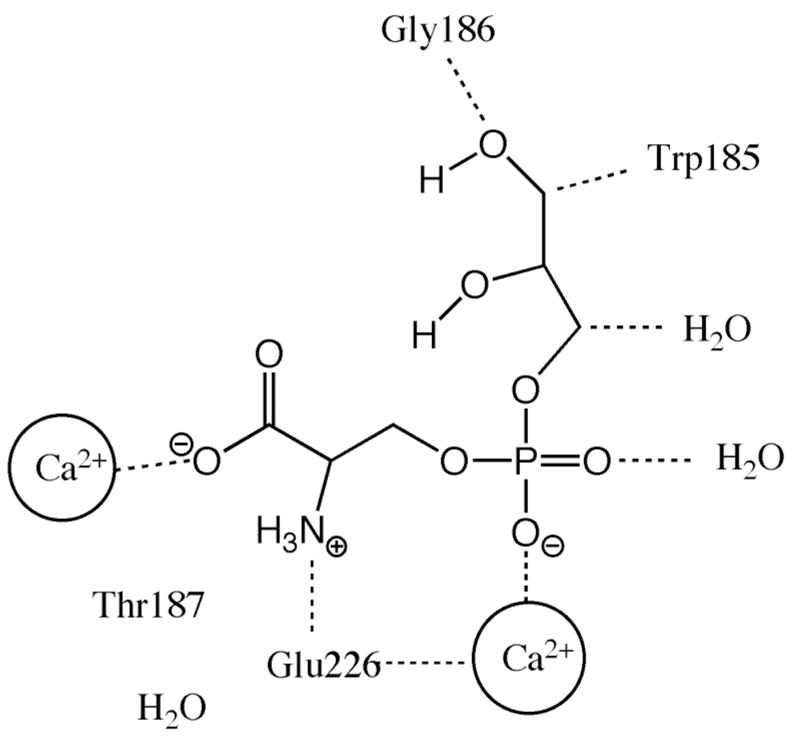
Three-component complex of Annexin V-Ca2+-glycerophosphoserine. The carboxylate and phosphate groups of the phosphatidylserine head group are bound via electrostatic attraction to the Ca2+ ions coordinated by the Annexin V protein. Additional stabilizing interactions occur by hydrogen bonding between the glycerol backbone of the glycerophosphoserine and amino acid residues of Annexin V.
A recent example of an artificial membrane surface targeting system, that employs ion-ion attraction as the keystone interaction, is the demonstration of synthetic metal coordination complexes as functional mimics of Annexin V. Annexin V is used extensively as a reagent for detecting the PS-rich membranes of dying and dead cells, but the protein has a number of practical limitations and there is a need for synthetic alternatives that are cheaper, more robust, and exhibit faster binding.25 Studies with zinc-dipicolylamine (Zn-DPA) complexes have shown that they can also associate selectively with PS-rich biomembranes using the three component assembly mechanism in Figure 4. In this case, Zn2+ ions act as the bridge between the dipicolylamine ligand and the anionic PS.26 Although the membrane binding process substitutes Zn2+ instead of Ca2+, the synthetic Zn-DPA coordination complexes have the same cell recognition properties as Annexin V and they are effective probes for in vitro assays of cell death. 27
Keystone Interactions at the Midpolar Membrane Interfacial Region (Ion-Dipole and Ionic Hydrogen Bonding)
Sequence analysis of TM proteins reveals that the amino acids in the membrane-spanning portion of the protein do not occur randomly. The interfacial region is rich with the aromatic amino acids, Trp and Tyr, which appear to act as “interfacial anchors” for the TM proteins.28 In constrast, Phe exhibits no preference for the interfacial region, instead occurring most often in the non-polar hydrocarbon region of the bilayer.13 There is evidence that the Trp and Tyr engage in cation-π interactions with the ammonium groups of the membrane phospholipids. The stabilizing effects of cation-π interactions are well known in protein stucture,29 but their importance in TM protein-lipid and TM protein-protein structure is still emerging.30 Recent computational and experimental studies suggest that model peptides containing Trp interact favorably with the head group of phosphatidylcholine (PC) by forming cation-π interactions between the indole ring of Trp and the quaternary ammonium of PC, as well as hydrogen bonds between the indole N-H and the phosphate oxygens.31 Furthermore, hydrogen bonding between the PC phosphate and the Tyr hydroxyl was more favorable than the alternative cation-π interaction with the PC quaternary ammonium.
A clear picture of cation-π interactions within a protein-phospholipid complex is provided by the anti-PC antibody McPC603 bound to the head group of PC.32 The cation-π interactions work in concert with other forces to facilitate formation of a tightly-bound protein-phospholipid complex.33 The quaternary ammonium of PC makes contact with Trp107 of the heavy chain of McPC603, interacting with the partially negative ring face of the indole (Figure 6). Additionally, the phosphate group of PC also participates in two important interactions with MCPC603, one to Tyr33 of the McPC603 heavy chain, and one to Tyr100 of the light chain. Interestingly, these Tyr rings associate with the phosphate not via their hydroxyl groups, but through ion-dipole interactions with the CH residues on the aromatic ring. These phosphate-CH edge contacts contribute a combined favorable interaction energy of approximately 1.25 kcal/mole.33
Figure 6.
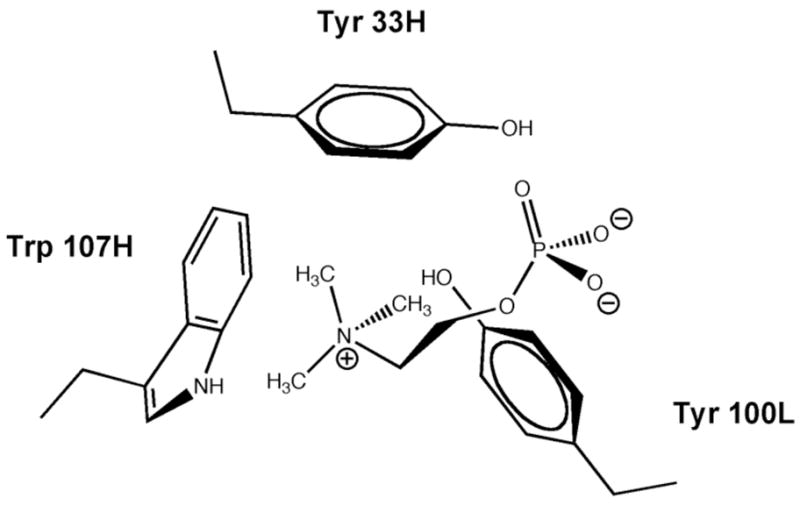
Chemical illustration of the PC head group in the McPC603 binding pocket. The quaternary ammonium can make ion-dipole interactions with Trp107, and to a lesser extent Tyr33.
Ionic hydrogen bonding is another keystone interaction in the membrane interfacial region, and a good example of its dominance is the selective association of phosphatidic acid (PA) with PA-binding proteins.34 PA is a minor component of cell membranes but it plays a key role in many cellular processes.35 Synthetic membrane-active peptides harboring lysine and arginine residues have been shown to induce deprotonation of PA and form very strong hydrogen bonds with the dianionic phosphomonoester head group (Figure 7). Thus, PA acts a preferred docking site for lysine and arginine residues through an electrostatic/hydrogen bond switch. The cone shape of the PA is thought to favor a deep location in the midpolar interfacial region, where the lower dielectric amplifies the strength of the ionic hydrogen bonds. Using this PA binding model, it should be possible to design new types of synthetic recognition molecules that reside in the membrane interfacial region and employ ionic hydrogen bonding as the keystone interaction for sensing the presence of PA, or selective disruption of undesired PA/protein association.
Figure 7.
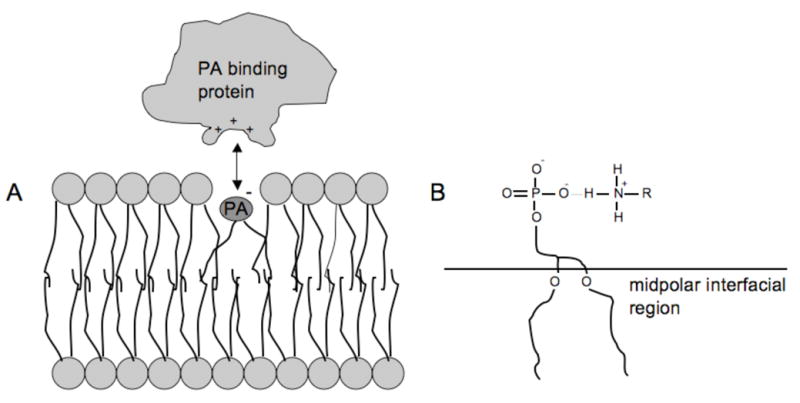
(A) Association of PA and PA-binding protein in the midpolar region induces, (B) deprotonation of the PA and formation of very stable ionic hydrogen bonds. Figure adapted from reference 36.
Keystone Interactions in the Non-polar Hydrocarbon Interior (van der Waals Forces and Neutral Hydrogen Bonding)
The non-polar hydrocarbon region of the bilayer consists of phospholipid acyl chains of varying lengths and degrees of unsaturation. In some cases, these lipids associate laterally to form discrete domains in the membrane. These “lipid rafts,” which are often rich in sphingomyelin (SM) and cholesterol,36 are stabilized by chain packing that maximizes van der Waals contacts in the membrane interior. Indeed, hydrogen bonding in the interfacial region of the membrane between the cholesterol hydroxyl and the SM head group is not a controlling factor.37 Another consequence of the variability in acyl chain length among the membrane phospholipids is the phenomenon of hydrophobic matching. This occurs when TM proteins of varying lengths cause localized thickening and thinning of the membrane bilayer. Proteins are approximately 400 times less compressible than the phospholipids in a membrane bilayer,38 which is a consequence of the increased capacity of phospholipids to undergo stretching and compression. While it is the phospholipids that usually undergo the structural remodeling to produce hydrophobic matching, the TM proteins can also adjust their thickness by lateral association and tilting of TM segments.39
One of the best characterized examples of lateral protein-protein aggregation in a membrane is dimerization of the TM protein glycophorin A.40 On first inspection, glycophorin A does not appear structurally prone to self-association. It lacks polar residues for hydrogen bonding in its TM domain, which is rich in Gly, Val, and Ile residues. Direct interhelical packing occurs between Gly residues 79 and 83 of each helix, consistent with TM interhelical interactions mediated by the commonly observed GxxxG motif.41, 42 In this case, the very small Gly side-chains allow a closely packed dimerization interface (Figure 8) that can produce strong van der Waals forces and promote an unusual hydrogen bonding effect, namely Cα-H· · · O hydrogen bonding.43 With an approximate pKa of 18–20, the glycine CHα proton is much less acidic than the more common hydrogen bond donors, but nevertheless CHα hydrogen bonds to opposing peptide backbone carbonyls appears to stabilize helix-helix dimers, especially those where the helices can form close contacts.44 Experimental studies of Cα-H· · · O bond strength in membrane bilayers have measured the interaction to be 0.88 kcal/mole.45 Thus, to influence helix-helix association in a meaningful way, several simultaneous interactions must be formed.
Figure 8.
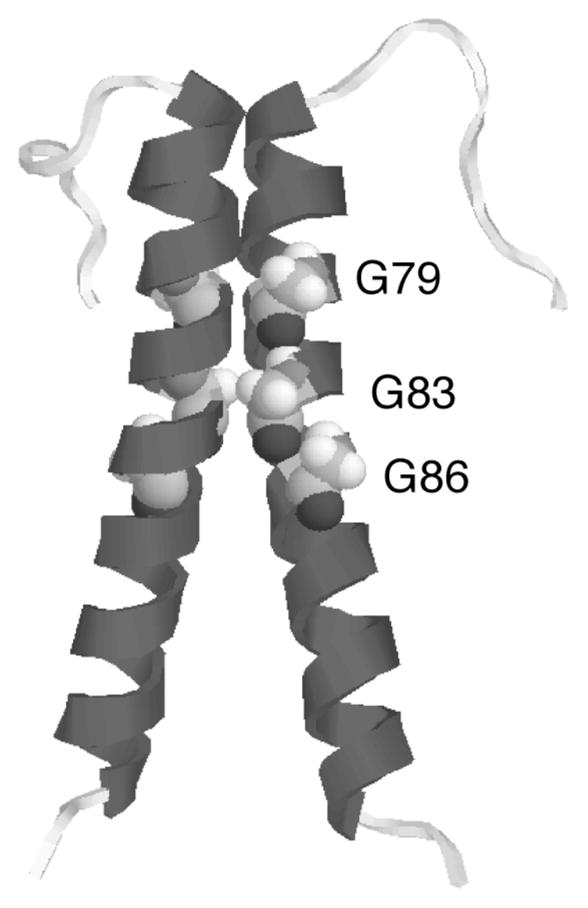
Crystal structure (PDB 1AFO) of a dimer of the transmembrane protein glycophorin A. The helix-helix contact region is formed by a series of cofacial glycine residues that allow the transmembrane helices to pack tightly together, maximizing van der Waals contacts between the helices.
Like glycophorin A, most TM proteins have a non-polar helical TM segment that matches the hydrocarbon interior of the bilayer. Many TM proteins are receptors for hormones that exert their effect by inducing receptor dimerization or conformational change. In instances where mutations introduce polar amino acid side-chains into the TM region of these receptor proteins, the stability gained by side-chain hydrogen bonding can often drive ligand-independent receptor activation, a situation that can lead to numerous cellular malfunctions (Figure 9).46 This phenomenon is often observed with Receptor Tyrosine Kinases (RTKs) which elicit biochemical signals at the plasma membrane via lateral dimerization, in part due to the interaction of their TM domains. Two ligand activation models have been proposed for RTKs.47 The most popular is that RTKs exist in a monomer-dimer equilibrium, and it is the active dimer which achieves stability upon ligand binding. A modified model envisions the ligand binding to an inactive form of the TM dimer and switching it to an active conformation. In line with their crucial role in the regulation of cell growth, mutations in TM domains of RTKs can induce improper signaling events leading to pathological phenotypes, especially cancer.48 A classic example is the Val664Glu mutation in the TM domain of the rat oncogenic form of the Neu RTK, which causes ligand-independent receptor dimerization and upregulation of constitutive kinase activity.49 The structural basis for this transformation remains the subject of ongoing experimental,50 and theoretical investigation.51 There is evidence that the Glu664 forms interhelical hydrogen bonds (either two Glu664 residues on separate helices self-associate, or Glu664 forms a hydrogen bond with the backbone carbonyl of Ala661 on the partner helix), however, other explanations focus on changes in the relative orientation of the two TM helices.52 In any case, there is interest in discovering synthetic TM peptides and peptidomimetics that can disrupt these pathogenic hydrogen-bonding events in the non-polar membrane interior.53, 54
Figure 9.
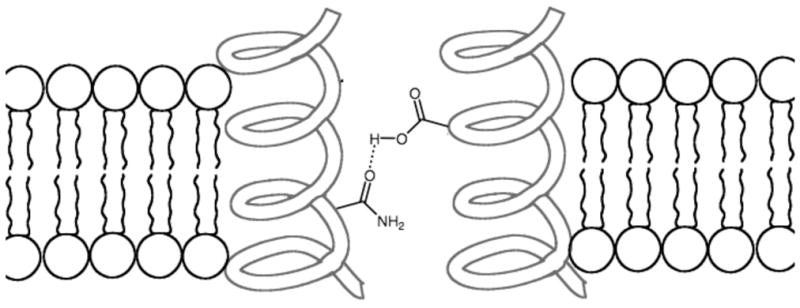
Mutations that introduce polar side-chains into the TM segment of a protein can promote lateral helix-helix association in the non-polar membrane interior.
Synthetic association systems that employ the keystone interactions of van der Waals contacts and neutral hydrogen bonding to selectively recognize TM helices and modulate lateral protein-protein association would have many useful applications as reagents for cell biology research and as potential therapeutics. Recently, a computational method for designing peptides that target (TM) helices of integrins in micelles and cell membranes has been developed.55 Computed helical anti-membrane protein (CHAMP) is a computational approach to design peptides that will target the TM helices of proteins. The TM domains of two platelet integrins, αIIbβ3 and ανβ3, were targeted in the initial study. A peptide with high geometric complementarity to the TM target was selected by CHAMP, and the membrane insertion ability was facilitated by the inclusion of solubility enhancing groups on the N and C termini. Experimental analysis in micelles and also bacterial and mammalian membranes provided evidence for high affinity homodimerization of the synthetic peptides. There was also strong heterodimerization with the TM domain of the target integrins. The ability to specifically target TM helices in a sequence specific manner depended on the geometric complementarity of the target-host complex. In the future CHAMP, or similarly designed peptides, will provide molecular reagents that can assess the consequences of inhibiting lateral protein-protein association within membranes.
Conclusion
Designing peptides or small molecule mimetics of lipid-binding proteins or molecules that associate with TM proteins is very challenging, in part because the driving forces for protein-lipid or TM protein-protein association are a composite of many non-covalent interactions that act simultaneously. Electrostatic forces, cation-π interactions, van der Waals forces, and hydrogen bonding all play a role in maintaining proper membrane structure and function. The simplifying concept of different keystone interactions within the three regions of a biological membrane provides a tractable intellectual basis for designing molecular recognition systems. Eventually this simplifying concept may not be necessary because increasing computing power will enable high throughput calculations of proposed molecular designs, including high quality Molecular Dynamics simulations of complete bilayer assemblies.56
Acknowledgments
This work was partially supported by the NIH (B.D.S.), the American Heart Association (SDG #0735350N) and an Indiana University Biomedical Research Grant (R.V.S.).
Footnotes
Bridges and aqueducts constructed by the Roman Empire still stand today even though the stones of which many are made are not joined by cement or any other adhesives. Despite the size and architectural complexity of these structures, only a few key contacts between specific “keystone components” provide the stabilizing interactions that hold these structures together. If these keystone interactions are disrupted, the entire structure will collapse.
References
- 1.Singer SJ, Nicolson GL. Science. 1972;175:720–731. doi: 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- 2.Carpten JD, et al. Nature. 2007;448:439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 3.Partridge AW, Therien AG, Deber CM. Biopolymer. 2002;66:350–358. doi: 10.1002/bip.10313. [DOI] [PubMed] [Google Scholar]
- 4.de Hoog CL, Mann M. Annu Rev Genomics Hum Genet. 2004;5:267–293. doi: 10.1146/annurev.genom.4.070802.110305. [DOI] [PubMed] [Google Scholar]
- 5.Fahy E, et al. J Lipid Res. 2005;46:839–861. doi: 10.1194/jlr.E400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Drews J. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith MEA, Wang D-N. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho W, Stahelin RV. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 9.Wimley WC, White SH. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 10.Northrup SH, Erickson HP. Proc Natl Acad Sci USA. 1992;89:3338–3342. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunningham BC, Wells JA. J Mol Biol. 1993;234:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- 12.Scott DL, Mande AM, Sigler PB, Honig B. Biophys J. 1994;67:493–504. doi: 10.1016/S0006-3495(94)80546-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stahelin RV, Cho W. Biochemistry. 2001;40:4672–4678. doi: 10.1021/bi0020325. [DOI] [PubMed] [Google Scholar]
- 14.Diraviyam K, Murray D. Biochemistry. 2006;45:2584–2598. doi: 10.1021/bi051901t. [DOI] [PubMed] [Google Scholar]
- 15.Snitko Y, Koduri R, Han S-K, Othman R, Baker SF, Molini BJ, Wilton DC, Gelb MH, Cho W. Biochemistry. 1997;36:14325–14333. doi: 10.1021/bi971200z. [DOI] [PubMed] [Google Scholar]
- 16.McLaughlin S, Murray D. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 17.Arbuzova A, Murray D, McLaughlin S. Biochim Biophys Acta. 1998;1376:369–379. doi: 10.1016/s0304-4157(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Crocker E, McLaughlin S, Smith SO. J Biol Chem. 2003;278:21459–21466. doi: 10.1074/jbc.M301652200. [DOI] [PubMed] [Google Scholar]
- 19.Victor K, Jacob J, Cafiso DS. Biochemistry. 1999;38:12527–12536. doi: 10.1021/bi990847b. [DOI] [PubMed] [Google Scholar]
- 20.Cho W, Stahelin RV. Biochim Biophys Acta. 2006;1761:838–49. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Shao X, Davletov BA, Sutton RB, Sudhof TC, Rizo J. Science. 1996;273:248–251. doi: 10.1126/science.273.5272.248. [DOI] [PubMed] [Google Scholar]
- 22.Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gomez-Fernandez JC. EMBO J. 1999;18:6329–6338. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards AS, Newton AC. Biochemistry. 1997;36:15615–15623. doi: 10.1021/bi9718752. [DOI] [PubMed] [Google Scholar]
- 24.Swairjo MA, Concha NO, Kaetzel MA, Dedman JR, Seaton BA. Nat Struct Biol. 1995;2:968–974. doi: 10.1038/nsb1195-968. [DOI] [PubMed] [Google Scholar]
- 25.Hanshaw RG, Smith BD. Bioorg Med Chem. 2005;13:5035–5042. doi: 10.1016/j.bmc.2005.04.071. [DOI] [PubMed] [Google Scholar]
- 26.O’Neil EJ, Smith BD. Coord Chem Rev. 2006;250:3068–3080. [Google Scholar]
- 27.Hanshaw RG, Lakshmi C, Lambert TN, Smith BD. ChemBiochem. 2005;12:2214–2220. doi: 10.1002/cbic.200500149. [DOI] [PubMed] [Google Scholar]
- 28.White SH, Wimley WC. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 29.Dougherty DA, Stauffer DA. Science. 1990;250:1558–1560. doi: 10.1126/science.2274786. [DOI] [PubMed] [Google Scholar]
- 30.Sanderson JM. Org Biomol Chem. 2005;3:201–212. doi: 10.1039/b415499a. [DOI] [PubMed] [Google Scholar]
- 31.Sanderson JM, Whelan EJ. Phys Chem Chem Phys. 2004;6:1012–1017. [Google Scholar]
- 32.Satow Y, Cohen GH, Padlan EA, Davies DP. J Mol Biol. 1986;190:593–604. doi: 10.1016/0022-2836(86)90245-7. [DOI] [PubMed] [Google Scholar]
- 33.Novotny J, Bruccoleri RE, Saul FA. Biochemistry. 1989;28:4735–4749. doi: 10.1021/bi00437a034. [DOI] [PubMed] [Google Scholar]
- 34.Kooijman EE, Tielman DP, Testerink C, Munnik T, Rijkers DTS, Burger KNJ, de Kruijff B. J Biol Chem. 2007;282:11356–64. doi: 10.1074/jbc.M609737200. [DOI] [PubMed] [Google Scholar]
- 35.Testerink C, Munnik T. Trends Plant Sci. 2005;10:368–375. doi: 10.1016/j.tplants.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Ohanian J, Ohanian V. Cell Mol Life Sci. 2001;58:2053–2068. doi: 10.1007/PL00000836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kan C-C, Ruan Z, Bittman R. Biochemistry. 1996;30:7759–7766. doi: 10.1021/bi00245a013. [DOI] [PubMed] [Google Scholar]
- 38.Harroun TA, Heller WT, Weiss TM, Yang L, Huang HW. Biophys J. 1999;76:937–945. doi: 10.1016/S0006-3495(99)77257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nyholm TKM, Özdirekcan S, Killian JA. Biochemistry. 2007;46:1457–1465. doi: 10.1021/bi061941c. [DOI] [PubMed] [Google Scholar]
- 40.Smith SO, Song D, Shekar S, Groesbeek M, Ziliox M, Aimoto S. Biochemisry. 2001;40:6553–6558. doi: 10.1021/bi010357v. [DOI] [PubMed] [Google Scholar]
- 41.Kleiger G, Grothe R, Mallick P, Eisenberg D. Biochemistry. 2002;41:5990–5997. doi: 10.1021/bi0200763. [DOI] [PubMed] [Google Scholar]
- 42.Russ WP, Engelman DM. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 43.Shi Z, Olson CA, Bell AJ, Kallenbach NR. Biophys Chem. 2002;101:267–279. doi: 10.1016/s0301-4622(02)00171-0. [DOI] [PubMed] [Google Scholar]
- 44.Senes A, Ubarretxena-Belandia I, Engelman DM. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arbely E, Arkin IT. J Am Chem Soc. 2004;126:5362–5363. doi: 10.1021/ja049826h. [DOI] [PubMed] [Google Scholar]
- 46.Partridge AW, Therien AG, Deber CM. Proteins. 2004;54:648–656. doi: 10.1002/prot.10611. [DOI] [PubMed] [Google Scholar]
- 47.Li E, Hristova K. Biochemistry. 2006;45:6241–51. doi: 10.1021/bi060609y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ménard S, Casalini P, Campiglio M, Pupa SM, Tagliabue E. Cell Mol Life Sci. 2004;61:2965–2978. doi: 10.1007/s00018-004-4277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brennan PJ, Kumogai T, Berezov A, Murali R, Greene MI. Oncogene. 2000;19:6093–6101. doi: 10.1038/sj.onc.1203967. [DOI] [PubMed] [Google Scholar]
- 50.Beevers AJ, Kukol A. J Mol Biol. 2006;361:945–953. doi: 10.1016/j.jmb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Aller P, Garnier N, Genest M. J Biomol Struct Dynam. 2006;24:209–228. doi: 10.1080/07391102.2006.10507114. [DOI] [PubMed] [Google Scholar]
- 52.Aller P, Voiry L, Garnier N, Genest M. Biopolymers. 2005;77:184–197. doi: 10.1002/bip.20176. [DOI] [PubMed] [Google Scholar]
- 53.Bennasroune A, Fickova M, Gardin A, Dirrig-Grosch S, Aunis D, Crémal G, Hubert P. Mol Biol Cell. 2004;15:3464–3474. doi: 10.1091/mbc.E03-10-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berezov A, Chen J, Liu Q, Zhang HT, Greene MI, Murali R. J Biol Chem. 2002;277:28330–28339. doi: 10.1074/jbc.M202880200. [DOI] [PubMed] [Google Scholar]
- 55.Yin H, Slusky S, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, de Grado WF. Science. 2007;315:1817–22. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 56.Periole X, Huber T, Marrink SJ, Sakmar TP. J Am Chem Soc. 2007;129:10126–10132. doi: 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]