

Abstract
Autophagy plays a critical protective role maintaining energy homeostasis and protein and organelle quality control. These functions are particularly important in times of metabolic stress and in cells with high energy demand such as cancer cells. In emerging cancer cells, autophagy defect may cause failure of energy homeostasis and protein and organelle quality control, leading to the accumulation of cellular damage in metabolic stress. Some manifestations of this damage, such as activation of the DNA damage response and generation of genome instability may promote tumor initiation and drive cell-autonomous tumor progression. In addition, in solid tumors, autophagy localizes to regions that are metabolically stressed. Defects in autophagy impair the survival of tumor cells in these areas, which is associated with increased cell death and inflammation. The cytokine response from inflammation may promote tumor growth and accelerate cell non-autonomous tumor progression. The overreaching theme is that autophagy protects cells from damage accumulation under conditions of metabolic stress allowing efficient tolerance and recovery from stress, and that this is a critical and novel tumor suppression mechanism. The challenge now is to define the precise aspects of autophagy, including energy homeostasis and protein and organelle turnover, that are required for the proper management of metabolic stress that suppress tumorigenesis. Furthermore, we need to be able to identify human tumors with deficient autophagy, and to develop rational cancer therapies that take advantage of the altered metabolic state and stress responses inherent to this autophagy defect.
Keywords: autophagy, beclin1, cancer
Autophagy Activation Under Metabolic Stress in Normal Cells
The survival, growth and proliferation of normal mammalian cells require the presence of growth factor signals from their environment as well as the absence of stress signals which may activate checkpoints or cell death programs. In addition, nutrients and in most cases oxygen, are essential to sustain the normal physiological functions of cells by supporting metabolism while suppressing starvation, damage and checkpoint responses.1–9 These three types of signals crosstalk to each other and together dictate the survival status of mammalian cells.10,11 Mammals have evolved sophisticated organs and systems that work coordinately to provide a homeostatic environment where the concentrations of various growth factors, nutrients and oxygen fluctuate within a manageable range. As a result, the cells constituting a mammal experience only limited and transient metabolic stress such as suboptimal concentrations of growth factors, nutrients and oxygen. Moreover, mammalian cells have intricate adaptive responses to metabolic stress that activate specific cellular processes to cope with stress conditions. One central signaling pathway that coordinates these cellular responses is the insulin-like growth factor I (IGF-I)-AKT-mTOR (mammalian target of rapamycin) pathway.10,11 Growth and maintenance factors activate the PI3-dependent kinase 1-AKT kinase cascade, leading to the activation of mTOR. Nutrient or energy deprivation can signal to AMPK (AMP activated kinase), which negatively regulates mTOR. In turn, mTOR re-tunes the activities of the cellular catabolic and anabolic processes to sustain cell survival, growth and proliferation allowing adaptation to new conditions. Under non-stress conditions, for example, mTOR suppresses autophagy, whereas deprivation represses mTOR signaling thereby activating autophagy. Thus, the catabolic process of autophagy is a key component of this adaptive response to metabolic stress, which is regulated by mTOR and other signaling events.12–14
Autophagy is part of the complex cellular process that responds to metabolic stress through which intracellular components, including ribosomes, endoplasmic reticulum, mitochondria, protein aggregates and cytoplasm are engulfed and targeted for lysosomal degradation.12–14 This process not only allows for efficient mobilization and recycling of cell-internal resources, but also reorganizes cell metabolism to achieve the new balance between energy consumption and production. This process of autophagy-mediated lysosomal degradation of cellular constituents also serves the important function to prevent the accumulation of damaged proteins and organelles, particularly during stress. Indeed, the most fundamental and conserved function of autophagy is to respond to and mitigate metabolic stress, such as that triggered by growth factor, nutrient and oxygen deprivation.2,14,15
The important role of autophagy in normal cellular adaptation to metabolic stress in mammals has been elegantly demonstrated by the work of the Mizushima group through the generation of transgenic mice that expresses the GFP-tagged autophagy marker MAP-LC3. They were able to show that autophagy is rapidly induced under starvation conditions in the majority of mouse tissues.16,17 In addition, mutant mice deficient for the essential autophagy gene atg5 that have a profound defect in autophagy, fail to survive the neonatal starvation period.18 These atg5−/− mice exhibit severe nutrient and energy insufficiency within 10 hours of birth, as evidenced by the significantly lower amino-acid concentrations in plasma and tissues, and die within 24 hours.18 This provides an elegant example of the protective role for autophagy afforded by sustaining cellular metabolism during physiological nutrient deprivation in vivo.
Metabolic Stress During Tumorigenesis
Tumorigenesis is a process by which normal cells are progressively transformed into cancerous cells through the accumulation of genetic mutations and epigenetic modifications. During this process, especially in the development of solid tumors, cells may experience severe and prolonged metabolic stress conditions that are significantly different from the transient and mild events that are typically experienced by normal cells and tissues. For example, when a tumor reaches to the size of 0.2–2 mm in diameter, oxygen, growth factors and nutrients, including glucose and other small molecules such as amino acids, cannot diffuse efficiently to the cells at the center of the tumor mass due to inadequate vascularization.19 Similarly, in large, established tumors, the vasculature is profoundly abnormal and subject to intermittent collapse that similarly creates metabolic stress.20 As a result, the cells at the center of the microscopic tumors and in sectors of established tumors live in a metabolic stressed microenvironment characterized by hypoxia, low pH and nutrient deprivation. We found that these metabolically stressed tumor regions is where autophagy is localized and where it functions to support tumor cell survival.3,6,7,15
Metabolic stress in tumors elicits a number of important and distinct cellular responses that dramatically impact tumor progression including cell death by apoptosis or necrosis,9,21 cell survival through autophagy,3,6,7,15 and induction of the hypoxia-inducible factors and activation of transcription programs to promote angiogenesis and ameliorate stress.22 Substantial evidence exists that this intricate interaction between the emerging tumor and its unique microenvironment is an important phase for tumor progression.23,24
Autophagy Promotes Survival to Metabolic Stress and Suppresses Genome Damage
Autophagy, a critical program in alleviating metabolic stress in normal cells, is compromised in 45% to 75% human prostate, breast and ovarian cancers.25 Indeed, autophagy defect in the mouse as a result of targeted deletion of the beclin1 gene, promotes tumorigenesis.26,27 It has become clear that autophagy defect has significant impact on cell survival and tumorigenesis in response to metabolic stress. Using immortalized baby mouse kidney epithelial cells (iBMKs) from the wild type beclin1+/+ and isogenic beclin1+/− mice, and from the atg5+/+ and the atg5−/− mice, we demonstrated that the autophagy-compromised immortalized cells are more sensitive to metabolic stress both in vitro and in tumors in vivo.7,15 Similar sensitivity to metabolic stress in vitro and in vivo was observed with autophagy-defective beclin1+/− immortalized mouse mammary epithelial cells (iMMECs) when compared to those from wild type mice.3,28 Thus, as is the case with normal cells, tumor cells utilize the autophagy pathway predominantly as a survival mechanism to sustain viability during periods of metabolic stress.
Interestingly, when injected into nude mice, despite the fact that the autophagy-defective iBMK cells and iMMECs are more prone to cell death by apoptosis or necrosis in response to metabolic stress, these cells paradoxically exhibit much increased tumorigenicity.3,7,15 This interesting paradox of how loss of a survival pathway could enhance tumor growth was resolved by our further characterization of these immortalized cells with impaired autophagy. Strikingly, in comparison to their isogenic wild type counterparts, the autophagy-compromised cells display upregulation of the DNA damage response under conditions of metabolic stress, and an elevated rate of chromosome instability and accelerated progression to aneuploidy.3,7,15 It is noteworthy that metabolic stress and chromosome instability are the hallmarks of solid tumors, but the interrelationship has previously been unknown. Our findings suggest that deficient autophagy in association with inherent metabolic stress in the tumor microenvironment may be a cause of genome damage that enables and facilitates tumorigenesis.
Based on these results a clearer picture emerges that can explain the role of autophagy in suppressing progression of solid tumors (Fig. 1). While autophagy activation is important to mitigate the chronic and severe metabolic stress experienced by emerging and evolving tumor cells, autophagy inactivation further exacerbates the damaging impact of metabolic stress.6 The latter destabilizes cellular homeostasis and promotes cell death while elevating genome instability. Although defective autophagy and enhanced susceptibility to metabolic stress promote cell death, the resulting necrosis, and perhaps also apoptosis, provokes an inflammatory response associated with cytokine production and acceleration of tumor growth.15 Analogous to a wound-healing response, the chronic inflammation caused by cell death of autophagy-defective tumor cells under metabolic stress may provide a non-cell autonomous mechanism of tumor progression.6 Independently, increased genome instability expedites tumor evolution, progression and invasion by increasing the mutation rate and acquisition of additional functions that favor tumor growth in a cell-autonomous fashion.6
Figure 1.
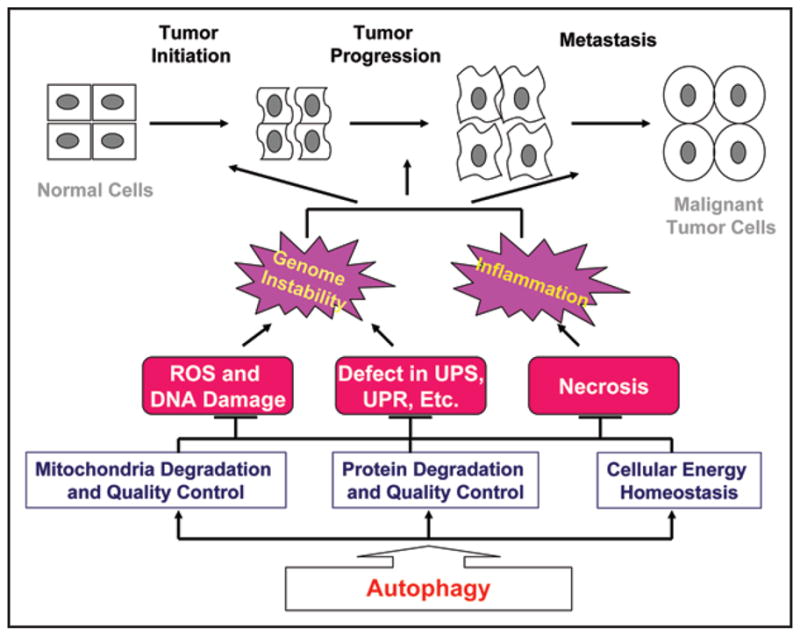
Autophagy suppresses tumorigenesis at different stages and by multiple mechanisms. Autophagy is cellular protective process that is critical for mitochondria degradation and quality control, protein quality control, and cellular energy homeostasis. Autophagy functions are particularly important under metabolic stress conditions. Autophagy defect may lead to various cellular damaging events and cell death by necrosis or apoptosis, ultimately causing genome instability and inflammation which in turn drive the cell autonomous (by genome instability) and cell non-autonomous (by necrosis and inflammation) evolution of tumors. Arrows indicate promotion, while T indicates inhibition.
It is intriguing to speculate how autophagy defect destabilizes cellular homeostasis in a manner that is manifested by elevated cell death and genome instability. We would like to discuss here the various aspects of autophagy function and how their loss may contribute to cell death and genome instability. We should keep in mind, however, that these activities are likely highly interrelated and may work synergistically in promoting cancer development.
The Role of Autophagy in Maintaining Cellular Energy Homeostasis
Metabolic stress such as hypoxia and nutrient starvation often leads to reduction of intracellular ATP levels, which are indicated by the activation of the AMP-activated kinase. Nutrient deprivation activates apoptosis in cultured cells if the cellular apoptotic machinery is intact.4,15 However, when apoptosis is inhibited by expression of anti-apoptotic Bcl-2 or by deficiency in proapoptotic Bax and Bak, nutrient deprivation leads to activation of autophagy enabling prolonged survival. Restoration of nutrients then allows recovery of normal cellular function.6 If metabolic stress persists and autophagy progresses unabated, this can lead ultimately to cell death.6 Inactivation of autophagy in these cells greatly exacerbates this nutrient deprivation-induced necrosis and inflammation.15 Interestingly, supplying a cell-permeable metabolic substrate, methyl pyruvate, can effectively reverse the deleterious consequences of autophagy inactivation.4 This strongly argues that one mechanism by which autophagy prolongs cell survival and limits cell death occurs mainly through providing metabolic substrates by recycling intracellular proteins and other components to maintain cellular energy homeostasis (Fig. 1).
The Role of Autophagy in Mitochondria Quality Control and Degradation
Mitochondria are organelles where oxidative phosphorylation takes place. Under normal conditions mitochondria consume glucose metabolites and oxygen and produce the bulk of ATP required for cellular functions. A byproduct of oxidative phosphorylation is the production of reactive oxygen species (ROS) that can leak from mitochondria, particularly, damaged mitochondria. Chronic ROS exposure can result in oxidative damage to cellular proteins, lipids and nucleic acids.29 Mitochondria are also important regulators of apoptotic programmed cell death. Efflux of mitochondrial components such as cytochrome c and SMAC from the mitochondrial inter-membrane space triggers effector caspase activation and apoptosis.30 Mitochondrial dysfunction resulting from autophagy defect may thereby contribute in multiple ways to tumorigenesis. Moreover, ATP reduction and ROS induction are also major causes of necrosis that can promote cellular damage and inflammation, potentially influencing tumorigenesis (Fig. 1).31
In response to transient nutrient and oxygen fluctuations, mitochondrial function and number are adjusted accordingly to accommodate environmental changes. Autophagy may play a critical role in maintaining a pool of functional mitochondria through degration of defective mitochondria constantly at a low rate.32 Under chronic metabolic stress, particularly amino acid deficiency, mitochondria degradation accelerates. It appears that autophagy is the only process by which mitochondria are degraded.32–34 Autophagy-deficient cells and organisms exhibit profound morphological as well as functional changes, including elevation of ROS production.18,33–37 The accumulation of stressed mitochondria likely contributes to the DNA damage observed in the autophagy deficient cells under metabolic stress conditions, which in turn would increase chromosome instability.3,7 In addition, failure to adapt cellular metabolism machinery through autophagic degradation of mitochondria and ribosomes to achieve the new balance between energy consumption and production may further disrupt the cellular energy homeostasis. The potential role for autophagy in controlling the turnover of other organelles such as endoplasmic reticulum and peroxisomes may also limit oxidative damage under metabolic stress conditions.
The Role of Autophagy in Protein Degradation
Cellular proteins can be degraded via two major pathways, ubiquitin-dependent proteasome degradation and autophagy-dependent lysosomal degradation. Experimental evidence suggests that these two pathways are functionally interrelated. One important phenotype in some tissues of mice with atg5 or atg7 gene disruption and defective autophagy is the accumulation of ubiquitinated proteins.38,39 There are at least several possibilities to explain why autophagy deficiency increases the accumulation of ubiquitinated proteins. First, ubiquitination may be an intrinsic signal for proteins targeted for autophagic degradation, therefore, autophagy deficiency leads to accumulation of these ubiquitinated proteins. Second, the autophagosome-bound proteins in autophagy-defective cells may be re-tagged with ubiquitin to reroute them to the proteasome-dependent degradation pathway to compensate for defective lysosome-mediated protein turnover. Third, failure of proper turnover of damaged proteins by autophagy leads to their accumulation that may antagonize proteasome-mediated protein degradation. In any case, it is predicted that the proteasome-dependent protein degradation might be affected in autophagy-deficient cells, in particular under metabolic stress such as amino acid deficiency, which requires protein degradation through autophagy.
How defects in autophagy may specifically alter cellular function dependent on protein turnover by the proteasome pathway is not yet known. It is well established that the normal function of ubiquitin-dependent proteolysis system (UPS) is essential for normal cell growth, cell cycle progression and cell death.40,41 For example, cell cycle progression is driven by the periodical oscillation of cyclin-dependent kinase (CDK) activities, which is by and large caused by the ubiquitin-dependent proteasomal degradation of key regulators such as cyclins and CDK inhibitors. It is possible that the deregulation of UPS in autophagy-deficient cells could indirectly contribute to loss of cell cycle control and genome instability observed in the autophagy deficient cells under metabolic stress. Alternatively, autophagy defects and the excessive accumulation of damaged proteins can activate cellular stress responses including the unfolded protein response (UPR) and toll-like receptor signaling that can have far-reaching effects on cellular functions leading to tumor progression. Finally, the accumulation of damaged or misfolded proteins is associated with toxicity and this failure to “take out the (protein) garbage” in autophagy-defective cells may be in itself a source of oxidative stress resulting in DNA damage and cell death (Fig. 1).
The unique tumor suppression mode of autophagy that limits cellular damage and death thereby suppressing tumor initiation and tumor progression, introduces a dilemma in targeting autophagy for cancer therapy. Blocking autophagy in tumor cells either pharmacologically or genetically results in increased tumor cell death.3,4,7,15 Therefore, combining autophagy inhibitors with other cancer chemotherapeutics can enhance the efficacy of acute cell killing. However, autophagy inhibitors may increase genome instability in the surviving cancer cells and may also promote the cell non-autonomous tumor progression, which would together expedite cancer relapse. Again, the devil is in the details. By carefully choosing the tumor types and drug combinations and regimens, it is possible that autophagy inhibitors can enhance the efficacy of the existing chemotherapeutics to acutely reduce the tumor burden to a threshold that cancer recurrence from the surviving cells becomes less an issue. Given the fact that a number of relevant autophagy-compromised mouse models are available, it is likely we can determine the best ways to translate our knowledge in autophagy into cancer treatment practice. Moreover, based on the high prevalence of monoallelic deletion of autophagy gene deletion (e.g., beclin1) in human tumors, it is clearly possible that autophagy status can serve as a prognostic marker for some tumor chemotherapy regimens. Lastly, promoting autophagy as a means to limit cellular damage as a strategy for cancer prevention should be considered.
Going forward, identification of the key cellular damaging events in autophagy-defective cells that promote tumor progression and neuronal degeneration will also be critical. We must bear in mind that autophagy defects and the resulting damaging effects of stress may be distinct in different cell types, and may vary depending on the physiological context. For example, how stress from defects in autophagy is manifested in proliferating cancer cells may be very different from that manifested in post-mitotic, differentiated neuronal cells. Identification of the mechanisms by which autophagy serves to manage metabolic stress will be important for establishing the role of autophagy in tumor suppression, as well as in limiting neuronal degeneration and aging.
References
- 1.Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007;120:379–83. doi: 10.1242/jcs.03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karantza Wadsworth V, Patel S, Kravchuk O, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- 6.Mathew R, Karantza Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–81. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathew R, White E. Why sick cells produce tumors: the protective role of autophagy. Autophagy. 2007;3:502–5. doi: 10.4161/auto.4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson DA, White E. Exploiting different ways to die. Genes Dev. 2004;18:1223–6. doi: 10.1101/gad.1212404. [DOI] [PubMed] [Google Scholar]
- 10.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20:267–75. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 13.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–7. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 15.Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 17.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 19.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 20.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 21.Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 2004;18:2095–107. doi: 10.1101/gad.1204904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–7. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds and signaling: tissue architecture regulates development, homeostasis and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 26.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karantza Wadsworth V, White E. Role of autophagy in breast cancer. Autophagy. 2007;3:610–3. doi: 10.4161/auto.4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 2007;39:443–55. doi: 10.1080/03602530701468516. [DOI] [PubMed] [Google Scholar]
- 30.Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005;121:671–4. doi: 10.1016/j.cell.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 31.Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 32.Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. Embo J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy. 2006;2:80–4. doi: 10.4161/auto.2.2.2460. [DOI] [PubMed] [Google Scholar]
- 34.Kim I, Rodriguez Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Qi H, Taylor R, Xu W, Liu LF, Jin S. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3:337–46. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- 36.Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scherz Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–7. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 40.Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005;12:1178–90. doi: 10.1038/sj.cdd.4401692. [DOI] [PubMed] [Google Scholar]
- 41.Reinstein E, Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann Intern Med. 2006;145:676–84. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]