

Abstract
The transforming growth factor (TGF) β signaling pathway is involved in many cellular processes including proliferation, differentiation, adhesion, motility and apoptosis. The loss of TGFβ signaling occurs early in carcinogenesis and its loss contributes to tumor progression. The loss of TGFβ responsiveness frequently occurs at the level of the TGFβ type II receptor (TGFβRII) which has been identified as a tumor suppressor gene (TSG). In keeping with its TSG role, the loss of TGFβRII expression is frequently associated with high tumor grade and poor patient prognosis. Reintroduction of TGFβRII into tumor cell lines results in growth suppression. Mutational loss of TGFβRII has been characterized, particularly in a subset of colon cancers with DNA repair enzyme defects. However, the most frequent cause of TGFβRII silencing is through epigenetic mechanisms. Therefore, re-expression of TGFβRII by use of epigenetic therapies represents a potential therapeutic approach to utilizing the growth suppressive effects of the TGFβ signaling pathway. However, the restoration of TGFβ signaling in cancer treatment is challenging because in late stage disease, TGFβ is a pro-metastatic factor. This effect is associated with increased expression of the TGFβ ligand. In this Review, we discuss the mechanisms associated with TGFβRII silencing in cancer and the potential usefulness of histone deacetylase (HDAC) inhibitors in reversing this effect. The use of HDAC inhibitors may provide a unique opportunity to restore TGFβRII expression in tumors as their pleiotropic effects antagonize many of the cellular processes, which mediate the pro-metastatic effects associated with increased TGFβ expression.
Keywords: TGFβ RII, Epigenetics, Histone modifications, Histone deacetylases, HDAC inhibitors, Therapy
Introduction
The transforming growth factor (TGF) β signaling pathway is involved in many cellular processes including proliferation, differentiation, adhesion, motility and apoptosis (1). These functions are frequently disrupted in malignant cells and the TGFβ type II receptor (TGFβRII) was subsequently demonstrated to be a tumor suppressor gene (TSG). The TGFβ signaling pathway mediates potent growth inhibition in normal cells and in line with its role as a TSG, cancer cells use both genetic and epigenetic mechanisms to inactivate TGFβRII. However, the relationship between the TGFβ signaling pathway and cancer progression is complex. The abrogation of TGFβ signaling provides a growth advantage to early stage malignant tumors but TGFβ assumes a pro-metastatic role in progressed tumors (2-4). Therefore, the loss of TGFβRII is associated with poor clinical outcome and is a predictor of poor prognosis in early stage breast cancer but over expression of the TGFβ ligand is associated with the metastatic phenotype in many tumors (5, 6). This dual nature of TGFβ presents a challenge with respect to restoring TGFβ signaling to take advantage of the growth suppressive effects. In this review, we will outline the epigenetic mechanisms involved in TGFβRII silencing and discuss whether epigenetic therapy with histone deacetylase (HDAC) inhibitors presents a unique opportunity to restore TGFβ signaling by restoring low levels of TGFβRII expression while minimizing the adverse effects associated with over expression of the TGFβ ligand.
Overview of TGFβ signaling
In the mammalian system, TGFβ's are classified as TGFβ1, TGFβ2, and TGFβ3 which are encoded by different genes but they all function through the same receptor signaling system (1). TGFβ is secreted as a latent complex bound to other extracellular proteins like latent TGFβ binding proteins that tether the TGFβ in the extracellular matrix (7). TGFβ binds to TGFβRIII at the cell surface which presents the ligand to the TGFβRII receptors. The intracellular signaling is initiated upon the selective binding of the active cytokine to the TGFβRII homodimer which has constitutively active Ser/Thr kinase activity (Fig. 1). Upon TGFβ binding, TGFβRII forms a heterotetramer with TGFβRI comprising of two identical TGFβRI/TGFβRII receptor heterodimers bound to dimeric TGFβ. Once the receptor complex is formed, TGFβRII transphosphorylates and activates the TGFβRI Ser/Thr kinase. Activation of TGFβRI propagates downstream signaling via the Smad family of proteins (8-10). The TGFβRI receptor directly interacts with and phosphorylates Smad 2 and Smad 3 (also termed as receptor activated Smads or R-Smads). These Smads bind to Smad 4 (also termed Co-Smad) which results in the translocation of this complex to the nucleus where the Smads regulate TGFβ-responsive gene expression (10-12).
Figure 1.
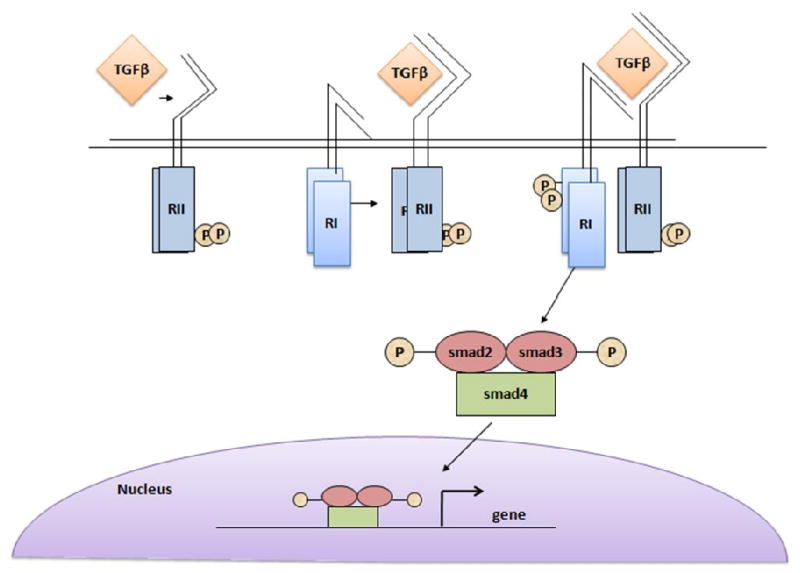
Schematic of the TGFβ signaling pathway. (RI-TGFβRI; RII-TGFβRII).
Cancer Epigenetics
The field of epigenetics in cancer research developed in 1983 from the finding of altered DNA methylation in cancer cells compared to normal tissue (11). Cancer cells show regions of global genome hypomethylation with specific promoters showing local hypermethylation. Unlike mutational alterations in gene activation, epigenetic changes affect gene expression (and so phenotype) without altering the DNA sequence (genotype). DNA hypomethylation is associated with gene (potentially oncogene) activation while DNA hypermethylation is associated with gene silencing as in X-chromosome inactivation and in cancer cells, leads to the silencing of many TSG's. The genome contains many CpG dinucleotides and methylation is associated with the 5′ carbon on cytosines adjacent and upstream of a guanosine (12, 13). This CpG methylation frequently occurs in the 5′ regulatory region of genes where it interferes with transcription factor binding.
As well as DNA methylation induced changes in gene transcription, the status of gene expression is also influenced by alterations in post translational modifications of histones (14). These modifications include acetylation, methylation, phosphorylation, sumoylation and ubiquination but acetylation and methylation are the most documented modifications with respect to the status of gene transcription. Aberrant patterns of histone methylation or acetylation/deacelylation contribute to TSG silencing in cancer.
Chromatin is packaged into nucleosomes which consist of 147 base pairs of DNA wound around a core histone octamer consisting of two copies each of histone (H) 2A, H2B, H3 and H4 (15). These units are stabilized by a linker H1 histone. Initially, the nucleosomes were thought to represent a stable scaffold on which gene transcription occurred. However, it is now known that chromatin is a dynamic structure. Nucleosomes can be repositioned and the tightness of the DNA winding around the core histone octamer can be varied making the associated gene promoters more or less accessible to the transcription machinery.
The alterations in chromatin structure affecting gene promoter accessibility are mediated by post-translational modifications of histones, most of which occur on lysines in the N-terminal tails which protrude through the DNA helix wrapped around the globular cores of the histones (Fig. 2). These histone modifications can affect gene transcription in two ways. Firstly, modifications can affect the interaction between the DNA and histones to open up the nucleosome structure making associated genes more accessible to the transcription machinery. Acetylation of lysines by histone acetyltransferases (HAT's) in the N-terminal tail of histones neutralizes the positive charge decreasing the electrostatic attraction for the negative DNA. Histone deacetylases (HDAC's) remove the acetyl groups resulting in compaction of the structure. However, other modifications, such as methylation, do not affect ionic interactions but still influence gene transcription. These methyl groups provide platforms for specific methyl binding proteins. This led Strahl and Allis in 2000 to propose that histone modifications formed a “histone code” (16). They proposed that these modifications acted in combinational or sequential fashion on one or more histone tails to specify unique downstream functions. Allis and co-workers subsequently proposed a “binary switch model” in which it was proposed that binding of effector molecules is not just a function of recognition of their specific histone marks but is also affected by modifications on adjacent residues (17, 18).
Figure 2.
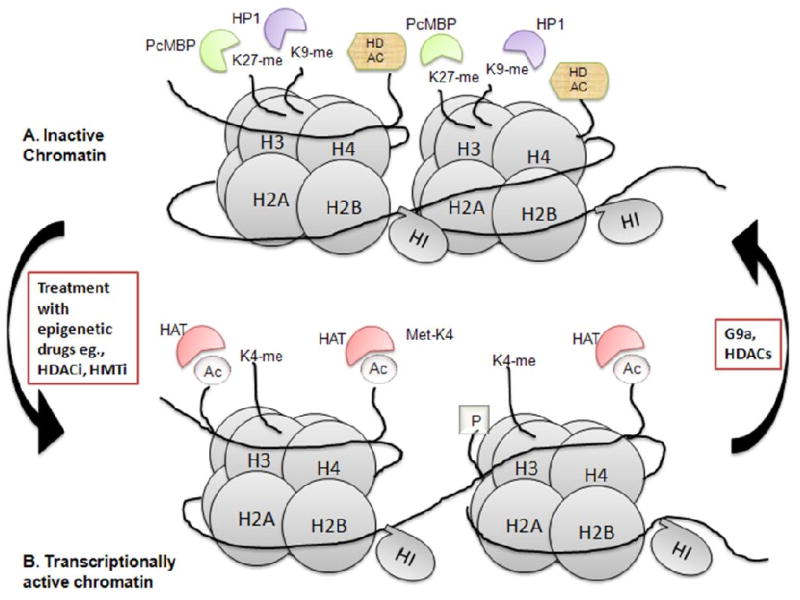
A model of chromatin depicting the epigenetic modifications associated with transcriptionally active and inactive chromatin. A. Inactive chromatin is associated with H3K9 and H3K7 methylation. The polycomb repressor methyl binding protein (PcMBP) binds the methyl group associated with H3K27 and HP1 binds the H3K9 methyl group. HDAC's are associated with the repressed chromatin which is tightly associated with the histones. B. The transcriptionally active chromatin is associated with acetylated H3 and H4 and H3K4 methylation. HAT's associate with the active, open chromatin.
The excitement generated by the study of epigenetics and cancer stems from the reversible nature of these epigenetic modifications. The identification of histone modifications associated with TSG silencing has important implications for cancer therapy. Both DNA demethylating and HDAC inhibitors have been developed. The use of these small molecule enzyme inhibitors, while challenging in terms of specificity, are proving more amenable to patient therapy than gene replacement therapy approaches.
Histone Modifications
Histone hyperacetylation is associated with active transcription whereas hypoacetylation is associated with gene repression (19, 20). The acetylation status is determined by the antagonistic HAT's and HDAC's. The acetyl group is added to the έ-amino of lysines on histones 3 and 4. Some of these same histone tails are substrates for methylation. Unlike acetylation, histone methylation can be associated with both gene activation and silencing (21-24). Histone methylation also occurs on histones 3 and 4, and six lysine targets have been identified. The H3-K9 methylation (H3K9me) is associated with heterochromatin regions (25, 26). This K9 methylation is catalyzed by the histone methyltransferase (hMTase) SUV39H1 which preferentially trimethylates lysine 9. This enzyme also recruits HP1 to the silenced gene promoter. Methylation of H4K20 is also characteristic of heterochromatic regions (27). It was subsequently shown that H3K9 methylation is involved in euchromatic gene silencing (28). However, the major H3K9 hMTase in euchromatin is G9a which preferentially mono- and di-methylates H3K9 in a processive manner (29, 30). The G9a protein contains the methyltransferase catalytic region (SET domain) and six contiguous ankyrin repeats which are involved in protein-protein interactions (31). There are two isoforms of G9a, a long and short form. The latter is a splice variant which lacks exon 10 (32). This short form contains the methyltransferase region and the ankyrin repeats. It is not known whether the long and short isoforms have identical function within the cell.
One known interaction partner of G9a is the G9a-like protein known as euHMTase1 (33, 34). This HMTase forms heteromeric complexes with G9a, and like G9a, methylates H3K9. However, in mouse embryo stem cells, knockout of G9a alone is sufficient to reduce H3K9 mono- and di-methylation. The complex formation with euHMTase 1 may function to stabilize the G9a protein, as the level of G9a was significantly decreased in euhMTase 1 knockout mouse stem cells. Unlike SUV39H1, G9a cannot itself bind and recruit HP1, but trimethylation by SUVH391 again recruits HP1 to euchromatically silenced genes. The H3K27 methylation is also associated with gene silencing and is catalyzed by the hMTase EZH2 which is part of the polycomb repressive complex and the methyl group recruits polycomb methyl binding proteins (35).
While H3K9 methylation is a major histone mark associated with silenced genes, H3K4 di- and tri-methylation are major histone methylation marks associated with transcriptionally active genes (24). The hMTase SET7 is responsible for both di- and tri-methylation of H3K4 and associates with the actively transcribing RNA polymerase II (36). Significantly, in Saccharomyces cerevisiae, methylated H3K4 interacts with the mediator complexes which in mammals and yeast contain HAT's (35, 37). The discovery of demethylase enzymes lagged behind their couterparts. Until their discovery, histone replacement within the nucleosome was thought to mediate the changes in histone modifications associated with gene transcription status. However specific demethylases have now been identified (reviewed in 38).
As implied in the binary switch mechanism, the silencing post-translational modifications are not totally independent of each other. For example, H3K9 methylation decreases p300 HAT activity without disrupting its association with chromatin (28). Methylation repressor complexes recruit HDAC's (35). These effects presumably serve to reinforce the silencing mechanism. These findings also illustrate the extreme plasticity of epigenetic silencing mechanisms which lends them to therapeutic manipulation.
Epigenetic Drugs and Reactivation of TSG Expression
The extensive changes in DNA methylation and histone modifications involved in silencing of TSG's led to studies on the effects of epigenetic drugs on TSG expression. Early studies focused on decitabine (5-azacytidine; 5-Aza-C) a DNA demethylating agent and one of the first HDAC inhibitors discovered, trichostatin A (TSA). The DNA demethylating inhibitor 5-Aza-C was found to inhibit DNA methylases at high concentrations but to inhibit hMTase's at lower concentrations and so not only affect CpG methylation but also the post-translational modifications of histones. Treatment with the DNA methyltransferase (DNMT) inhibitor reverses H3K9 methylation (39). The re-expression of the TSG's desmocollin and MAPSIN by 5-Aza-C is associated with both reduced H3K9 methylation and reduced expression of the hMTase G9a in breast tumor cells (40). In head and neck squamous carcinoma cell line Tu159 which does not express cyclin dependent kinase inhibitor p16 (CDKN2A), the promoter DNA is methylated and shows high association with the H3K9 methylation mark (41). Consequently, p16 expression is repressed in this cell line and induced by 5-Aza-C. In contrast, the HDAC inhibitor TSA did not induce expression. However, studies on the histone modifications showed that TSA did induce H3 acetylation and decreased the extent of H3K9 methylation associated with the p16 promoter. The decreased H3K9me was not due to TSA mediated repression of the expression of G9a or SUV39H1. Rather, the hMTases possess chromodomains which recognize and bind to H3K9. The DNMT's also interact with HDAC 1 and 2 and so inhibition of the HDAC's may affect the activity and/or interaction of these enzymes.
Some of the new generation HDAC inhibitors (based on TSA structure/activity) possess the ability to affect DNA methylation by disrupting DNMT interaction with the chromatin (42, 43). Desipeptide was able to activate expression of the TSG's p16, SALL3 and GATA4 in lung, colon and pancreatic cancer cell lines. The activation was associated with decreased G9a and SUV39H1 expression and decreased H3K9 methylation associated with the genes' promoters. Apicidin was also able to activate expression of these TSG's while TSA was not. Therefore, epigenetically silenced TSG's can be reactivated by epigenetic drugs including HDAC inhibitors. Pertinent to this finding is the observation that epigenetic drugs do not activate epigenetically silenced promoters to a fully euchromatic state (44). The re activated promoters retain some of the transcriptional repressive marks, again pointing to the plasticity of epigenetic mechanisms.
Genetic Silencing of TGβRII in Cancer
As discussed in the Introduction, the TGFβ signaling pathway, in keeping with its tumor suppressive function in early stage carcinogenesis, is inactivated in many tumor types. The loss of negative growth regulation by TGFβ affords tumor cells a selective growth advantage associated with increased resistance to apoptosis in response to growth factor deprivation and hypoxic stress resulting in increased tumorigenicity. Frequently, inhibition of TGFβ signaling occurs by abrogation of TGFβRII or TGFβRI function emphasizing their tumor suppressor role (45-50). Mutations of Smad 4 are also common events contributing to loss of TGFβ signaling in cancer progression but the main focus of this review is on TGFβRII receptor silencing.
The mechanism of silencing of TGFβ receptors covers the spectrum of genetic and epigenetic changes (reviewed in 51). The mutations in the TGFβ receptors occur independently of each other and do not directly affect the functioning of the other receptors. The first genetic mutation of TGFβRII was described in two gastric cancer cell lines. These cell lines contained TGFβRII mRNA truncated at exons 2 and 3, respectively. The exon 2 mutant lacks most of the receptor including the TGFβ binding region. The exon 3 truncation contains the TGFβ binding domain but lacking the c-terminus including the Ser/Thr kinase. This protein is potentially a dominant negative receptor (52).
The most common mutational loss of TGFβRII occurs in a subset of human colon carcinomas termed hereditary non-polyposis colon cancer (HNPCC; 53-57). These tumors display the replication error repair positive (RER+) phenotype which is characterized by microsatellite instability (MSI). The TGFβRII sequence contains a tract of 10 adenine residues at nucleotides 709-718 which is subject to frame-shift mutation due to replication error in these repeated base pairs. Deletion or insertion of an adenine within this region leads to formation of an early stop codon resulting in expression of a truncated TGFβRII of 129 amino acids (compared to 565 amino acids in the wild type). Subsequent studies showed that mutations in the TGFβRII gene were not seen until human colon adenomas transitioned to overt carcinoma indicating that RII mutation is a late event in the high MSI colon cancer phenotype. The status of TGFβRII has been studied in other types of high MSI tumors. While gastric cancers which show mutations in BAT sequences (assessed by PCR using primers which span microsatellite sequences with multiple base pair polyadenine tracts) have corresponding frame shift mutations in TGFβRII (58), MSI associated TGFβRII mutations were uncommon in pancreatic, pituitary and breast carcinomas (51, 59).
Epigenetic Silencing of TGFβRII in Cancer
The loss of TGFβ signaling also occurs independently of TGFβRII mutation involving changes in the DNA sequence. The loss of TGFβ signaling has been reported in many tumor types due to dramatic down regulation of TGFβRII protein expression, even in cancers where mutational inactivation occurs. Studies on the incidence of CpG island (CGI) methylation indicate that TGFβRII promoter DNA methylation is an infrequent event. The defective expression of TGFβRII in primary NSCLC showed that promoter methylation correlated with loss of TGFβRII expression when carcinoma and “normal” paracarcinoma tissue was examined (60). Interestingly, increased DNMT expression is not an indication that all TSG's will undergo DNA methylation. Increased DNMT expression has been observed in colon cancer (61). However, the CpG Island methylation phenotype (CIMP) which is associated with a high incidence of MSI is associated with TGFβRII mutation due to silencing of DNA repair enzymes (62, 63).
Examination of the status of CpG island methylation in many other tumor types has also revealed that DNA methylation was not the underlying cause of epigenetic regulation of TGFβRII in human pancreatic, lung and prostate cancer tumors and lymphoma (64-67). The reduced expression of TGFβRII in agnogenic myeloid metaplasia was not associated with mutation or CpG methylation (68). Yamashita and co-workers (67) studied the CpG methylation status of the TGFβRII promoter in human prostate tumors and in a rat model of prostate carcinoma. Neither DNA methylation nor mutation of the TGFβRII promoter was detected in 27 patient tumors analyzed. Moreover, only 3 out of 7 invasive dorsolateral lobe and 0 out of 5 ventral lobe prostate adenocarcinomas induced in F344 rats by 3,2′-dimethyl-4-aminobiphenyl and testosterone treatment showed TGFβRII methylation. This group went on to study TGFβRII CpG methylation in 33 human cancer cell lines comprising 5 prostate, 6 ovarian, 6 lung, 7 pancreas and 9 stomach cancer derived lines. Only one ovarian and one pancreatic cell line showed DNA methylation. However, chromatin immunoprecipitation (ChIP) analysis revealed that these cell lines which did not express TGFβRII had decreased H3 acetylation and increased H3K9 trimethylation associated with the TGFβRII promoter. Similar results were found in breast cancer cell lines with silenced TGFβRII (65).
The finding that epigenetic silencing of the TGFβRII gene involves transcriptional repression due to changes in promoter associated proteins has led to detailed analysis of the underlying mechanisms. Given the importance of TGFβRII as a growth suppressor of early stage cancers, many of these studies have examined the effect of HDAC inhibitors on TGFβRII expression and the associated alterations in promoter associated proteins. Most of these detailed analyses have been perfomed in cancer cell lines. These studies have implicated not only histone modifications as important in determining transcriptional status of the TGFβRII gene but other transcription factors that are targets of HAT's and HDAC's.
Histone Modifications Associated with the Epigenetic Silencing of TGFβRII
Osada and co-workers (66, 69) have performed an extensive characterization of the histone modifications associated with the epigenetically silenced TGFβRII promoter in several lung cancer cell lines and documented the changes in histone modification status associated with HDAC inhibitor induced re expression. In studies on six lung cancer cell lines, they showed 3 distinct patterns of histone modification associated with the TGFβRII promoter indicative of progressive degrees of TGFβRII silencing. The expression of TGFβRII in the A549 cell line was associated with H3 acetylation and H3K4 methylation (pattern I). The remaining cell lines which did not express TGFβRII all lacked H3 acetylation but pattern II showed very low H3K4 methylation and no DNA methylation whereas pattern III was positive for DNA methylation and lacked H3K4 methylated histone association. The pattern II cell lines showed strong and persistent TGFβRII induction upon treatment with TSA whereas pattern III cell lines showed only a transient weak induction or no effect. The TSA mediated induction of TGFβRII in the pattern II cell lines was associated with transient induction of H3K9 acetylation which coincided with a decrease in H3K9 methylation but persisent methylation of H3K4.
The Role of the Sp1 Family and NF-Y in the Epigenetic Repression of TGFβRII
The promoter for TGFβRII is well characterized (70). It does not contain a TATA or CAAT box near the transcription initiation site. Like most TATA-less promoters, basal promoter activity is dependent upon an Sp1 which binds a consensus sequence which is located at -25bp upstream from the transcription start site. This Sp1 site is located within the basal core promoter (-47 to -1). There is a second Sp1 site at -143. In addition, the promoter contains two other positive regulatory sequences designated PRE1 (-219 to 172) and PRE2 (+1 to +35). The first PRE binds an AP1/CREB complex. The NF-Y transcription factor binds to an inverted CCAAT box in a negative regulatory element (NRE2) of TGFβRII promoter at position -83, which is in close proximity to the novel Sp1 site at position -102.
The Brattain laboratory explored the mechanisms underlying TGFβRII silencing in the MCF7lL breast carcinoma cell line. The MCF7L cell line is a late passage variant of MCF cells which has lost TGFβRII expression but retains low level TGFβRI expression. The early passage MCF-7E (Early) retains TGFβRII expression and so TGFβ responsiveness. Initial studies revealed that MCF-7L cells had low Sp1 protein level compared to MCF-7E cells. The Sp gene family consists of four members (71). While Sp1, 2 and 4 are activators of gene transcription, Sp3 can be an activator or a repressor. The low Sp1 level in MCF-7L accounts for the loss or reduced expression of TGFβRII. Treatment with 5′-Aza-C reversed the effect of low Sp1 thereby leading to restoration of TGFβ receptor expression and consequently TGFβ mediated signal transduction (72, 73). As well as a reduced level of Sp1, the MCF-7L cell line has higher expression of the transcriptional repressor Sp3 protein than the MCF-7E cell line (74). Treatment with 5′-aza-C decreased the expression of Sp3 (75). These studies illustrate that the Sp1/Sp3 ratio plays a pivotal role in determining the activation/repression status of the TGFβRII gene. The inhibitory effect of Sp3 on TGFβRII transcription is not just due to its intrinsic repressor activity but it also competes with Sp1 for binding to the GC box resulting in reduced Sp1 mediated gene activation. Transfection of Sp3 causes significant repression of both TGFβRI and TGFβRII expression in MCF-7E after binding to the promoters.
The HDAC inhibitor TSA was also able to induce TGFβRII expression in the MCF7L cell line (76). This induction required the GC box at -25 within the TGFβRII promoter. In the presence of TSA, ectopic Sp3 led to TGFβRII promoter activation. Transfection of the wild type HAT p300 with Sp3 stimulated TGFβRII promoter activity while co-transsfection of an acetyltransferase domain deleted p300 did not. A key lysine in the Sp3 inhibitory domain is the target of acetylation (77) which converts Sp3 from a transcriptional repressor to an activator of transcription. These findings are summarized in Fig. 3.
Figure 3.
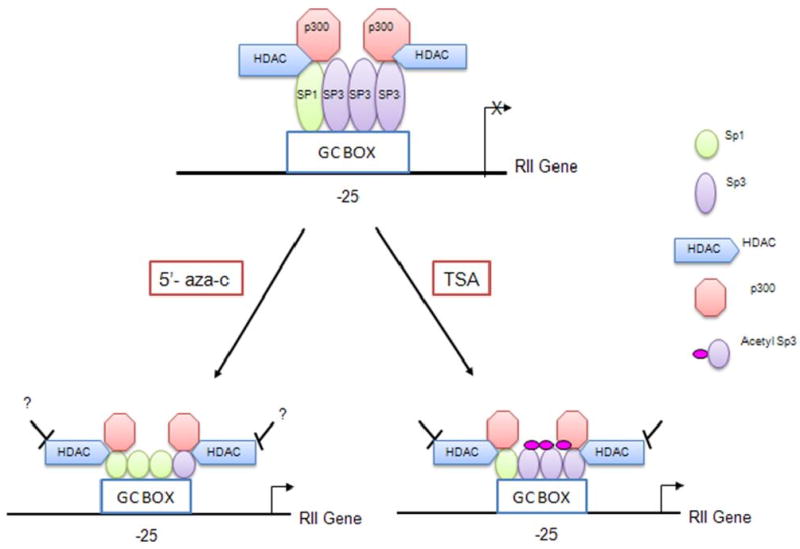
A model representing the role of Sp1 and Sp3 in the epigenetic silencing of TGFβRII. The MCF7L cell line has low Sp1 expression. Therefore, in the absence of drug treatment, more Sp3 is bound to the TGFβRII promoter than Sp1 resulting in repression. Following treatment with 5-Aza-C, Sp1 expression is increased resulting in displacement of Sp3 and re activation of TGFβRII expression. Following treatment with TSA, Sp3 becomes acetylated converting it from a transcriptional repressor to an activator resulting in re-activation of TGFβRII promoter activity. [Figure derived from the work of Ammanamanchi and Brattain, 2001a (74), 2001b (75) and 2003 (76)].
The mechanism of repression of TGFβRII gene was studied in MIA-PaCa2 cells. The pancreatic cell line MIAPaCa-2 is highly tumorigenic and resistant to TGFβ induced growth inhibition. This is due to loss of TGFβRII gene expression (78). Expression of TGFβRII was restored by the HDAC inhibitors MS275, TSA and sodium butyrate and the TGFβRII promoter activity of a -219/+36 TGFβRII luciferase construct was increased 60-fold following TSA (79). Deletion of the region -100 to -47 dramatically reduced the TGFβRII promoter activity. This region contains the inverted CAAT box which binds the NF-Y transcription factor. Mutation of this site revealed that it was required for stimulation of TGFβRII promoter activity by the HDAC inhibitors. Significantly, Osada et al. (2001; 66) in their studies on TSA stimulation of TGFβRII promoter activity in lung cancer cell lines showed that the inverted CAAT box was required for TSA responsiveness. The activity of NF-Y is modulated by association with HAT's, NF-YA with PCAF and NF-YB with p300. DNA affinity pull-down assay (DAPA) revealed that following TSA treatment, but not in untreated cells, NF-YA associated with PCAF at the TGFβRII promoter. Transfection of a PCAF expressing vector stimulated the TGFβRII promoter activity in the absence of HDAC inhibitor. The Sp1 site at -102 (adjacent to the inverted CAAT box) was also required for TSA mediated induction of TGFβRII expression in the pancreatic cell line (80). The Sp1 bound to this site interacts with NF-Y. TSA treatment interferes with HDAC 1 binding to this Sp1. A time course study of protein association by DAPA with the -112/-66 region of the TGFβRII promoter containing the NF-Y and Sp1 site revealed that binding of Sp1 and NF-Y did not change up to 24h following TSA treatment. However, p300 and PCAF association with the promoter increased while HDAC 1 binding decreased. These changes were associated with increased acetylation of H4 associated with the promoter.
All the above findings lead to a model of TGFβRII epigenetic silencing in which compact nucleosome structure is maintained by HDAC and methyltransferase association. Silencing is maintained by both histone modifications and the non-acetylated state of key transcription factors which are also associated with the promoter. Treatment with HDAC inhibitors alters chromatin structure by histone acetylation, loss of repressive methylation marks and gain of activation methylation signatures. The associated HDAC leaves the complex. The “opening” up of the chromatin structure allows access of HAT's resulting in p300 acetylation of Sp3 at the -25 GC box and PCAF which associates with NF-Y at the inverted CAAT box resulting in enhanced TGFβRII promoter activity. These studies are also indicative of the pivotal role of HDACs in maintaining the transcriptionally repressive chromatin state and in down regulating transcription factor activity.
Histone Deacetylases as Cancer Therapeutic Targets
The HDAC enzymes fall into 4 classes (20, 81). In addition to the three HDAC classes, there is a fourth group termed sirtuins. While inhibitors to sirtuins are being developed, they are not as well studied as the HDAC's and will not be considered further here. The enzyme activities of the HDAC family are zinc dependent and are classified based on size, structure and similarity to Saccharomyces cerevisiae enzymes. The class I enzymes are localized to the nucleus while class II enzymes undergo nucleo-cytoplasmic shuttling. While HAT's and HDAC's were named for their histone substrates, the first targets identified, these enzymes do not exclusively modify histones (82-86). The tumor suppressor p53 is acetylated at multiple sites which affects its activity. Many other transcription factors also undergo acetylation. This post translational modification can affect DNA binding activity and interaction with other co-activators or repressors. As the acetylation site lysine is frequently the target for other modifications such as ubiquination and sumoylation, stability and cellular localization can also be affected. The HDAC6 enzyme is cytoplasmic and is involved in tubulin and Hsp90 acetylation.
While mutation (truncation) of the HAT p300 is seen in MSI phenotype tumors, particularly colon cancer (86), mutations of HDAC's are relatively uncommon. However, studies on the expression of HDAC's in cancer have revealed that class I HDAC's are over expressed in many tumor types (87-91) and HDAC 2 over expression is associated with poor prognosis in gastric (92) and prostatic cancer (93). In contrast to the increased expression of Class I HDAC's observed in tumors, decreased class II HDAC expression is associated with poor prognosis in NSCLC (94)
Given the high HDAC activity of tumors compared to normal tissue, HDAC inhibitors have entered clinical trials. There are four major classes of HDAC inhibitors (95, 96). The hydroxamic acids include Vorinostat (SAHA), Belinostat and TSA. These HDAC inhibitors inhibit all classes of HDAC (I, IIa, IIb and IV). The short chain fatty acids include valproic acid and sodium butyrate and they inhibit class I and II HDAC's. The benzamide MS-275 selectively inhibits HDAC's 1, 2 and 3 while the cyclic tetrapeptide Desipeptide is selective for HDAC's 1 and 2.They result in differentiation, growth arrest and apoptosis of cancer cells. The notable feature of these first and second generation drugs is that most affect all Class I and II HDAC's. However, these drugs are generally well-tolerated as normal cells are resistant to the doses which are effective against cancer cells, possibly due to the increased HDAC expression associated with the malignant phenotype. Given the pleiotropic effects of these drugs there is the potential to affect most of the genome but in fact HDAC inhibitors affect the expression of only 8-20% of the total number of genes indicating these drugs do have some selectivity. Clearly, HDAC inhibitors affect most of the cellular processes important in transformation but as epigenetic drugs, perhaps one of their most important effects is activation of re expression of TSG's.
The Potential Use of HDAC inhibitors in Restoring TGFβRII Expression in Cancer
It is evident from the above discussion that as well as non-reversible genetic silencing, malignant cells employ reversible epigenetic mechanisms to silence TGFβRII. Reactivation of the repressed TGFβRII would lead to therapeutic benefit from the growth inhibitory and apoptotic effects of the TGFβ signaling pathway which includes down regulation of the inhibitor of apoptosis family member survivin (97). Even in cancers such as colon carcinoma in which mutational frame shift inactivation of TGFβRII is well documented (in the high MSI, RER+ sub population) there are tumor subsets in which TGFβRII is inactivated by epigenetic mechanisms. It has been demonstrated that expression of epigenetically silenced TSG's can be reactivated by epigenetic therapies. The HDAC's can restore expression of TSG's where transcriptional repression by histone (or transcription factor) modifications are involved. In certain lung, colon, breast and prostate tumors there is accumulating evidence that these modifications account for TGFβRII silencing. Therefore, these tumors would be amenable to re expression of TGFβRII by use of HDAC inhibitors. Several HDAC inhibitors have entered clinical trials and they are proving to be a reasonably well tolerated therapy (83-86).
However, there are several challenges to the utilization of reactivation of TGFβRII to take advantage of its tumor suppressive effect. Firstly, it will be necessary to identify the patient population which would be amenable to this approach. Characterizations of patient populations with respect to global epigenetic changes such as acetylation status are becoming increasingly used to predict patient therapy outcome and prognosis. Mutation can be analyzed by BAT- TGFβRII PCR and promoter methylation using methylation specific primers. TGFβRII expression would have to be evaluated in biopsies. However, immunohistochemstry is routinely used to determine the estrogen receptor status of breast tumors. The patient population meeting the criteria of non-mutational loss or low expression of TGFβRII could then be “targeted” to HDAC therapy. This approach might be useful in terms of the non-specificity of HDAC inhibitors. The design of HDAC inhibitors specific for cancer specific HDAC's is challenging, both because of HDAC structure and because research is ongoing to identify which specific HDAC's would represent the best target in different tumor types based on the specific TSG,s which they are involved in silencing. The identification of specific patient populations with specific gene defects for HDAC therapy could enable more effective use of the drugs. Such an approach may prove useful in breast cancer patients where HDAC inhibitors could potentially be used to induce ER expression in anti-estrogen resistant, ER negative tumors (98). The conversion to ER positive status could potentially make these tumors responsive to Tamoxifen.
The targeted induction of TGFβRII expression would also allow choice of targeted combinatorial therapies. There is considerable interest in the therapeutic use of vitamin D in treating cancer. Studies on the effectiveness of vitamin D against breast and colon cancer revealed that for maximum inhibition, vitamin D required a functional TGFβ signaling system (99, 100). Therefore, the combination of HDAC inhibitors with vitamin D in patients with epigenetically silenced TGFβRII could potentially enhance the therapeutic effectiveness of vitamin D. Of course, these approaches would all be subject to determination of optimal dosing and timing of the drug, and in the case of combination therapies, the order of administration.
However, the major challenge to overcome in utilizing re expression of TGFβRII as a cancer therapy is ensuring that the restoration of the TGFβ pathway will not lead to pro-metastatic signaling (2-4). In cell lines with silenced TGFβRII, the reintroduction of the TGFβRII receptor gene leads to growth inhibition and apoptosis pointing to the potential usefulness of the approach (45, 46, 101). Significantly, high expression of the TGFβ ligand is associated with the pro-metastatic effect of the TGFβ signaling pathway in advanced cancer. The mechanisms by which TGFβ functions as a pro-metastatic factor still remain to be fully elucidated, but the TGFβ appears to be involved in modifying the surrounding tumor microenvironment as well as functioning to promote the epithelial-to- mesenchymal (EMT) transition. This latter process is involved in tumor cell metastasis. The disruption of cell junction integrity through loss of E-cadherin is part of this process. The EMT may also involve Ras and PI-3 kinase signaling. Significantly, HDAC inhibitors may inhibit some of these effects. HDAC inhibitors, through their pan inhibition of class I and II HDAC's not only affect histone and nuclear protein acetylation but also have cytoplasmic effects, one of which is acetylation of the chaperone protein Hsp90 (102). Acetylation disrupts the interaction of Hsp90 with its client proteins which require this interaction for correct folding. Several of the downstream components of the Ras and PI-3 kinase signaling pathways are client proteins of Hsp90. The HDAC inhibitor TSA prevented the EMT in renal proximal tubular epithelial cells (103). The HDAC inhibitor SAHA profoundly inhibited the growth of several pancreatic cancer cell lines. The cells also underwent increased apoptosis. These effects were associated with effects on cell cycle components (increased p21, and decreased cyclin D and Myc) but the drug also induced increased expression of E-cadherin (104). Increased expression of E-cadherin was also observed in NSCLC cells treated with MS275 (105). Significantly, E-cadherin is a target of epigenetic silencing during tumor progression and reactivation of expression may contribute to the anti invasive effects of HDAC inhibitors (86, 106).
In conclusion, HDAC inhibitors could provide a unique opportunity to harness the tumor suppressive effects of TGFβ for therapeutic benefit. In targeted patient populations, HDAC inhibitors could be used to reactivate expression of the epigenetically silenced TGFβRII providing the therapeutic benefit of growth inhibitory and pro-apoptotic TGFβ signaling while pro-actively affecting the processes such as the EMT which contribute to the pro-metastatic effects associated with tumor TGFβ ligand over expression in late malignancy.
Acknowledgments
The authors wish to thank Dr. Michael G. Brattain for helpful discussions and advise in the preparation of this article. We thank Kelly Houston for secretarial help. Support for the work came from National Institutes of Health Grant CA 72001 to Dr. Brattain.
References
- 1.Massague J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 3.Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 4.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Gobbi H, Dupont WD, Simpson JF, et al. Transforming growth factor-beta and breast cancer risk in women with mammary epithelial hyperplasia. J Natl Cancer Inst. 1999;91:2096–101. doi: 10.1093/jnci/91.24.2096. [DOI] [PubMed] [Google Scholar]
- 6.Gobbi H, Arteaga CL, Jensen RA, et al. Loss of expression of transforming growth factor beta type II receptor correlates with high tumour grade in human breast in-situ and invasive carcinomas. Histopathology. 2000;36:168–77. doi: 10.1046/j.1365-2559.2000.00841.x. [DOI] [PubMed] [Google Scholar]
- 7.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–7. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 8.Miyazono K. TGF-beta signaling by Smad proteins. Cytokine Growth Factor Rev. 2000;11:15–22. doi: 10.1016/s1359-6101(99)00025-8. [DOI] [PubMed] [Google Scholar]
- 9.Liu F. Receptor-regulated Smads in TGF-beta signaling. Front Biosci. 2003;8:s1280–303. doi: 10.2741/1149. [DOI] [PubMed] [Google Scholar]
- 10.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–93. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 11.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 12.Baylin SB. Mechanisms underlying epigenetically mediated gene silencing in cancer. Semin Cancer Biol. 2002;12:331–7. doi: 10.1016/s1044-579x(02)00053-6. [DOI] [PubMed] [Google Scholar]
- 13.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. New Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 14.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci USA. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luger K, Richmond TJ. The histone tails of the nucleosome. Curr Opin Genet Dev. 1998;8:140–6. doi: 10.1016/s0959-437x(98)80134-2. [DOI] [PubMed] [Google Scholar]
- 16.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 17.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–9. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Fischle W, Cheung W, Jacobs S, Khorasanizadeh S, Allis CD. Beyond the double helix: writing and reading the histone code. Novartis Found Symp. 2004;259:3–17. discussion -21, 163-9. [PubMed] [Google Scholar]
- 19.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 20.Mai A, Massa S, Rotili D, et al. Histone deacetylation in epigenetics: an attractive target for anticancer therapy. Med Res Rev. 2005;25:261–309. doi: 10.1002/med.20024. [DOI] [PubMed] [Google Scholar]
- 21.Jenuwein T, Laible G, Dorn R, Reuter G. SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell Mol Life Sci. 1998;54:80–93. doi: 10.1007/s000180050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 23.Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends in cell biology. 2001;11:266–73. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- 24.Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–98. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 25.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 26.Peters AH, Mermoud JE, O'Carroll D, et al. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Gen. 2002;30:77–80. doi: 10.1038/ng789. [DOI] [PubMed] [Google Scholar]
- 27.Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Gen. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 28.Stewart MD, Li J, Wong J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol. 2005;25:2525–38. doi: 10.1128/MCB.25.7.2525-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tachibana M, Nozaki M, Takeda N, Shinkai Y. Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J. 2007;26:3346–59. doi: 10.1038/sj.emboj.7601767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patnaik D, Chin HG, Esteve PO, Benner J, Jacobsen SE, Pradhan S. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. J Biol Chem. 2004;279:53248–58. doi: 10.1074/jbc.M409604200. [DOI] [PubMed] [Google Scholar]
- 31.Milner CM, Campbell RD. The G9a gene in the human major histocompatibility complex encodes a novel protein containing ankyrin-like repeats. Biochem J. 1993;290(Pt 3):811–8. doi: 10.1042/bj2900811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown SE, Campbell RD, Sanderson CM. Novel NG36/G9a gene products encoded within the human and mouse MHC class III regions. Mamm Genome. 2001;12:916–24. doi: 10.1007/s00335-001-3029-3. [DOI] [PubMed] [Google Scholar]
- 33.Tachibana M, Ueda J, Fukuda M, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19:815–26. doi: 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008;27:2681–90. doi: 10.1038/emboj.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Cao R, Xia L, et al. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8:1207–17. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 37.Malik S, Roeder RG. Epigenetics? Mediator does that too! Mol Cell. 2008;31:305–6. doi: 10.1016/j.molcel.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–40. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen CT, Weisenberger DJ, Velicescu M, et al. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2′-deoxycytidine. Cancer Res. 2002;62:6456–61. [PubMed] [Google Scholar]
- 40.Wozniak RJ, Klimecki WT, Lau SS, Feinstein Y, Futscher BW. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene. 2007;26:77–90. doi: 10.1038/sj.onc.1209763. [DOI] [PubMed] [Google Scholar]
- 41.Coombes MM, Briggs KL, Bone JR, Clayman GL, El-Naggar AK, Dent SY. Resetting the histone code at CDKN2A in HNSCC by inhibition of DNA methylation. Oncogene. 2003;22:8902–11. doi: 10.1038/sj.onc.1207050. [DOI] [PubMed] [Google Scholar]
- 42.Bartova E, Pachernik J, Harnicarova A, et al. Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. Journal Cell Sci. 2005;118(Pt 21):5035–46. doi: 10.1242/jcs.02621. [DOI] [PubMed] [Google Scholar]
- 43.Wu LP, Wang X, Li L, et al. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol Cell Biol. 2008;28:3219–35. doi: 10.1128/MCB.01516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–9. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 45.Sun L, Wu G, Willson JK, et al. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J Biol Chem. 1994;269:26449–55. [PubMed] [Google Scholar]
- 46.Brattain MG, Markowitz SD, Willson JK. The type II transforming growth factor-beta receptor as a tumor-suppressor gene. Curr Opin Oncol. 1996;8:49–53. doi: 10.1097/00001622-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 47.Markowitz SD, Roberts AB. Tumor suppressor activity of the TGF-beta pathway in human cancers. Cytokine Growth Factor Rev. 1996;7:93–102. doi: 10.1016/1359-6101(96)00001-9. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Han W, Zborowska E, et al. Reduced expression of transforming growth factor beta type I receptor contributes to the malignancy of human colon carcinoma cells. J Biol Chem. 1996;271:17366–71. doi: 10.1074/jbc.271.29.17366. [DOI] [PubMed] [Google Scholar]
- 49.Ko Y, Koli KM, Banerji SS, et al. A kinase-defective transforming growth factor-beta receptor type II is a dominant-negative regulator for human breast carcinoma MCF-7 cells. Int J Oncol. 1998;12:87–94. doi: 10.3892/ijo.12.1.87. [DOI] [PubMed] [Google Scholar]
- 50.Biswas S, Chytil A, Washington K, et al. Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res. 2004;64:4687–92. doi: 10.1158/0008-5472.CAN-03-3255. [DOI] [PubMed] [Google Scholar]
- 51.Kim SJ, Im YH, Markowitz SD, Bang YJ. Molecular mechanisms of inactivation of TGF-beta receptors during carcinogenesis. Cytokine Growth Factor Rev. 2000;11:159–68. doi: 10.1016/s1359-6101(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 52.Park K, Kim SJ, Bang YJ, et al. Genetic changes in the transforming growth factor beta (TGF-beta) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-beta. Proc Natl Acad Sci USA. 1994;91:8772–6. doi: 10.1073/pnas.91.19.8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–8. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 54.Parsons R, Myeroff LL, Liu B, et al. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55:5548–50. [PubMed] [Google Scholar]
- 55.Wang J, Sun L, Myeroff L, et al. Demonstration that mutation of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J Biol Chem. 1995;270:22044–9. doi: 10.1074/jbc.270.37.22044. [DOI] [PubMed] [Google Scholar]
- 56.Grady WM, Myeroff LL, Swinler SE, et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59:320–4. [PubMed] [Google Scholar]
- 57.Markowitz S. TGF-beta receptors and DNA repair genes, coupled targets in a pathway of human colon carcinogenesis. Biochim Biophys Acta. 2000;1470:M13–20. doi: 10.1016/s0304-419x(99)00031-1. [DOI] [PubMed] [Google Scholar]
- 58.Pinto M, Oliveira C, Cirnes L, et al. Promoter methylation of TGFbeta receptor I and mutation of TGFbeta receptor II are frequent events in MSI sporadic gastric carcinomas. J Pathol. 2003;200:32–8. doi: 10.1002/path.1327. [DOI] [PubMed] [Google Scholar]
- 59.Schmitt FC, Soares R, Gobbi H, et al. Microsatellite instability in medullary breast carcinomas. Int J Cancer. 1999;82:644–7. doi: 10.1002/(sici)1097-0215(19990827)82:5<644::aid-ijc5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 60.Hougaard S, Norgaard P, Abrahamsen N, Moses HL, Spang-Thomsen M, Skovgaard Poulsen H. Inactivation of the transforming growth factor beta type II receptor in human small cell lung cancer cell lines. Br J Cancer. 1999;79:1005–11. doi: 10.1038/sj.bjc.6690161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt WM, Sedivy R, Forstner B, Steger GG, Zochbauer-Muller S, Mader RM. Progressive up-regulation of genes encoding DNA methyltransferases in the colorectal adenoma-carcinoma sequence. Mol Carcinog. 2007;46:766–72. doi: 10.1002/mc.20307. [DOI] [PubMed] [Google Scholar]
- 62.Ogino S, Kawasaki T, Ogawa A, Kirkner GJ, Loda M, Fuchs CS. TGFBR2 mutation is correlated with CpG island methylator phenotype in microsatellite instability-high colorectal cancer. Hum Pathol. 2007;38(4):614–20. doi: 10.1016/j.humpath.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 63.Teodoridis JM, Hardie C, Brown R. CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett. 2008;268:177–86. doi: 10.1016/j.canlet.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 64.Di Bartolo DL, Cannon M, Liu YF, et al. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood. 2008;111:4731-. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hinshelwood RA, Huschtscha LI, Melki J, et al. Concordant epigenetic silencing of transforming growth factor-beta signaling pathway genes occurs early in breast carcinogenesis. Cancer Res. 2007;67:11517–27. doi: 10.1158/0008-5472.CAN-07-1284. [DOI] [PubMed] [Google Scholar]
- 66.Osada H, Tatematsu Y, Sugito N, Horio Y, Takahashi T. Histone modification in the TGFbetaRII gene promoter and its significance for responsiveness to HDAC inhibitor in lung cancer cell lines. Mol Carcinog. 2005;44:233–41. doi: 10.1002/mc.20135. [DOI] [PubMed] [Google Scholar]
- 67.Yamashita S, Takahashi S, McDonell N, et al. Methylation silencing of transforming growth factor-beta receptor type II in rat prostate cancers. Cancer Res. 2008;68:2112–21. doi: 10.1158/0008-5472.CAN-07-5282. [DOI] [PubMed] [Google Scholar]
- 68.Hemavathy KC, Chang TH, Zhang H, et al. Reduced expression of TGF beta1RII in agnogenic myeloid metaplasia is not due to mutation or methylation. Leuk Res. 2006;30:47–53. doi: 10.1016/j.leukres.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 69.Osada H, Tatematsu Y, Sugito N, Horio Y, Takahashi T. Histone modification in the TGFbetaRII gene promoter and its significance for responsiveness to HDAC inhibitor in lung cancer cell lines. Mol Carcinog. 2005;44:233–41. doi: 10.1002/mc.20135. [DOI] [PubMed] [Google Scholar]
- 70.Bae HW, Geiser AG, Kim DH, et al. Characterization of the promoter region of the human transforming growth factor-beta type II receptor gene. J Biol Chem. 1995;270:29460–8. doi: 10.1074/jbc.270.49.29460. [DOI] [PubMed] [Google Scholar]
- 71.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 72.Ammanamanchi S, Kim SJ, Sun LZ, Brattain MG. Induction of transforming growth factor-beta receptor type II expression in estrogen receptor-positive breast cancer cells through SP1 activation by 5-aza-2′-deoxycytidine. J Biol Chem. 1998;273:16527–34. doi: 10.1074/jbc.273.26.16527. [DOI] [PubMed] [Google Scholar]
- 73.Periyasamy S, Ammanamanchi S, Tillekeratne MP, Brattain MG. Repression of transforming growth factor-beta receptor type I promoter expression by Sp1 deficiency. Oncogene. 2000;19:4660–7. doi: 10.1038/sj.onc.1203822. [DOI] [PubMed] [Google Scholar]
- 74.Ammanamanchi S, Brattain MG. Sp3 is a transcriptional repressor of transforming growth factor-beta receptors. J Biol Chem. 2001a;276:3348–52. doi: 10.1074/jbc.M002462200. [DOI] [PubMed] [Google Scholar]
- 75.Ammanamanchi S, Brattain MG. 5-azaC treatment enhances expression of transforming growth factor-beta receptors through down-regulation of Sp3. The Journal of biological chemistry. 2001b;276:32854–9. doi: 10.1074/jbc.M103951200. [DOI] [PubMed] [Google Scholar]
- 76.Ammanamanchi S, Freeman JW, Brattain MG. Acetylated sp3 is a transcriptional activator. J Biol Chem. 2003;278:35775–80. doi: 10.1074/jbc.M305961200. [DOI] [PubMed] [Google Scholar]
- 77.Spengler ML, Brattain MG. Sumoylation inhibits cleavage of Sp1 N-terminal negative regulatory domain and inhibits Sp1-dependent transcription. J Biol Chem. 2006;281:5567–74. doi: 10.1074/jbc.M600035200. [DOI] [PubMed] [Google Scholar]
- 78.Venkatasubbarao K, Ammanamanchi S, Brattain MG, Mimari D, Freeman JW. Reversion of transcriptional repression of Sp1 by 5 aza-2′ deoxycytidine restores TGF-beta type II receptor expression in the pancreatic cancer cell line MIA PaCa-2. Cancer Res. 2001;61:6239–47. [PubMed] [Google Scholar]
- 79.Huang W, Zhao S, Ammanamanchi S, Brattain M, Venkatasubbarao K, Freeman JW. Trichostatin A induces transforming growth factor beta type II receptor promoter activity and acetylation of Sp1 by recruitment of PCAF/p300 to a Sp1. NF-Y complex J Biol Chem. 2005;280:10047–54. doi: 10.1074/jbc.M408680200. [DOI] [PubMed] [Google Scholar]
- 80.Zhao S, Venkatasubbarao K, Li S, Freeman JW. Requirement of a specific Sp1 site for histone deacetylase-mediated repression of transforming growth factor beta Type II receptor expression in human pancreatic cancer cells. Cancer Res. 2003;63:2624–30. [PubMed] [Google Scholar]
- 81.Thiagalingam S, Cheng KH, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 82.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 83.Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol. 2007;74:659–71. doi: 10.1016/j.bcp.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 84.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 85.Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007;13:23–9. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- 86.Sigalotti L, Fratta E, Coral S, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol. 2007;212:330–44. doi: 10.1002/jcp.21066. [DOI] [PubMed] [Google Scholar]
- 87.Ishihama K, Yamakawa M, Semba S, et al. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol. 2007;60:1205–10. doi: 10.1136/jcp.2005.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3:28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 89.Weichert W, Denkert C, Noske A, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia. 2008;10:1021–7. doi: 10.1593/neo.08474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weichert W, Denkert C, Noske A, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia. 2008;10:1021–7. doi: 10.1593/neo.08474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weichert W, Roske A, Niesporek S, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14:1669–77. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 92.Song J, Noh JH, Lee JH, et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. Apmis. 2005;113:264–8. doi: 10.1111/j.1600-0463.2005.apm_04.x. [DOI] [PubMed] [Google Scholar]
- 93.Weichert W, Roske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98:604–10. doi: 10.1038/sj.bjc.6604199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Osada H, Tatematsu Y, Saito H, Yatabe Y, Mitsudomi T, Takahashi T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int J Cancer. 2004;112:26–32. doi: 10.1002/ijc.20395. [DOI] [PubMed] [Google Scholar]
- 95.Dejligbjerg M, Grauslund M, Litman T, et al. Differential effects of class I isoform histone deacetylase depletion and enzymatic inhibition by belinostat or valproic acid in HeLa cells. Mol Cancer. 2008;7:70. doi: 10.1186/1476-4598-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Khan N, Jeffers M, Kumar S, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–9. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 97.Wang J, Yang L, Yang J, et al. Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 2008;68:3152–60. doi: 10.1158/0008-5472.CAN-07-5348. [DOI] [PubMed] [Google Scholar]
- 98.Hostetter CL, Licata LA, Keen JC. Timing is everything: Order of administration of 5-aza 2′ deoxycytidine, trichostatin A and tamoxifen changes estrogen receptor mRNA expression and cell sensitivity. Cancer Lett. 2008 doi: 10.1016/j.canlet.2008.10.005. Article in press. [DOI] [PubMed] [Google Scholar]
- 99.Li F, Ling X, Huang H, et al. Differential regulation of survivin expression and apoptosis by vitamin D3 compounds in two isogenic MCF-7 breast cancer cell sublines. Oncogene. 2005;24:1385–95. doi: 10.1038/sj.onc.1208330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang L, Yang J, Venkateswarlu S, Ko T, Brattain MG. Autocrine TGFbeta signaling mediates vitamin D3 analog-induced growth inhibition in breast cells. J Cell Physiol. 2001;188:383–93. doi: 10.1002/jcp.1125. [DOI] [PubMed] [Google Scholar]
- 101.Anumanthan G, Halder SK, Osada H, et al. Restoration of TGF-beta signalling reduces tumorigenicity in human lung cancer cells. Br J Cancer. 2005;93:1157–67. doi: 10.1038/sj.bjc.6602831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chinnaiyan P, Allen GW, Harari PM. Radiation and new molecular agents, part II: targeting HDAC, HSP90, IGF-1R, PI3K, and Ras. Semin Radiat Oncol. 2006;16:59–64. doi: 10.1016/j.semradonc.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 103.Yoshikawa M, Hishikawa K, Marumo T, Fujita T. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-beta1 in human renal epithelial cells. J Am Soc Nephrol. 2007;18:58–65. doi: 10.1681/ASN.2005111187. [DOI] [PubMed] [Google Scholar]
- 104.Kumagai T, Wakimoto N, Yin D, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121:656–65. doi: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 105.Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944–50. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 106.Digel W, Lubbert M. DNA methylation disturbances as novel therapeutic target in lung cancer: preclinical and clinical results. Crit Rev Oncol Hematol. 2005;55:1–11. doi: 10.1016/j.critrevonc.2005.02.002. [DOI] [PubMed] [Google Scholar]