

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune syndrome marked by autoantibody production. Innate immunity is essential to transform humoral autoimmunity into the clinical lupus phenotype. Nitric oxide (NO) is a membrane-permeable signaling molecule involved in a broad array of biologic processes through its ability to modify proteins, lipids, and DNA and alter their function and immunogenicity. The literature regarding mechanisms through which NO regulates inflammation and cell survival is filled with contradictory findings. However, the effects of NO on cellular processes depend on its concentration and its interaction with reactive oxygen. Understanding this interaction will be essential to determine mechanisms through which reactive intermediates induce cellular autoimmunity and contribute to a sustained innate immune response and organ damage in SLE.
Keywords: Nitric oxide, systemic lupus erythematosus, reactive oxygen species, free radicals, oxidative stress
Introduction
The broad phenotype of systemic lupus erythematosus (SLE) is unified by autoantibody production, a function of the acquired immune response. However, an inappropriately active and sustained innate immune response is implicated in both the initiation and pathogenic consequences of autoantibody production in SLE. An important arm of the innate immune response is the production of reactive intermediates (RIs). The role of these RIs and subsequent oxidative products in the initiation and exacerbation of the lupus phenotype is discussed below.
Reactive intermediates
RIs are short-lived molecules formed by chemical reactions that are capable of rapidly modifying other molecules. Through these modifications, they act as signaling molecules for a broad array of cellular functions. RI’s important in biology include the reactive oxygen intermediates (ROIs such as super-oxide (SO) and hydrogen peroxide (H2O2)), reactive nitrogen intermediates (RNIs such as nitric oxide (NO) and peroxynitrite (ONOO−)), and hypochlorous acid (HOCl). The pathogenic potential of NO is partly dependent upon its concentration and whether its production occurs in proximity to the formation of ROI such as SO. Catalysis of RIs to less reactive molecules occurs through enzymes such as SO dismutase (which catalyzes the reaction of SO to H2O2). H2O2, not a free radical, is further catalyzed in some cell types to HOCl (bleach) by myeloperoxidase with antimicrobial effect.
H2O2 can also be converted to hydroxyl radicals (OH) in the presence of Fe(II) via the Fenton reaction or to water and oxygen by catalase [1]. NO is produced by one of the three known isoforms of NO synthase (NOS). NOS dimerizes to form the active enzymes. Uncoupled monomers or low arginine substrate conditions result in NOS producing SO [2]. NO and SO rapidly combine to form ONOO−. Thus, RI production depends on catalytic enzyme activity and substrate availability as well as the amount and activity of detoxifying enzymes (Figure 1) [3].
Figure 1.
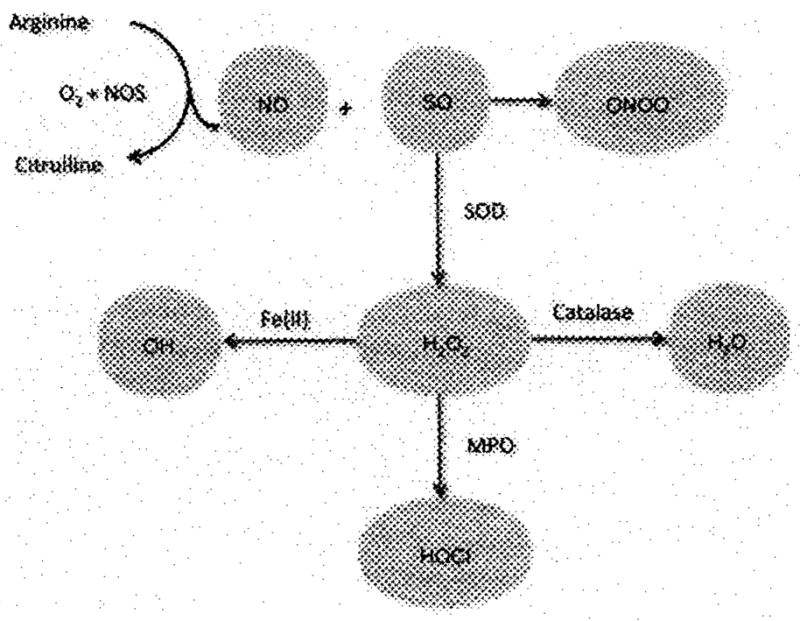
The fate of NO and SO is dependent on the production of other RI and detoxifying pathways. NO is produced by NOS, which also produces SO when arginine is in short supply or if NOS is uncoupled. SO is also produced by inflammatory cells or cells under stress. NO reacts rapidly and preferentially with SO to form ONOO−. SO can be catalyzed to hydrogen peroxide (H2O2) by SO dismutase (SOD). H2O2 can undergo the Fenton reaction with Fe(II) to form OH which is converted into water by catalase. Myeloperoxidase found in neutrophils and other cells converts H2O2 to HOCl.
NO biology
NO is a membrane-permeable free radical that is formed by one of the three NOS using arginine and oxygen as substrates (Figure 1). NO has the potential to induce both physiologic and pathologic effects, a seemingly contradictory notion that complicates interpretation of the literature. The effect of NO production on cellular processes is largely dependent on its concentration and the local presence of other free radicals. Lower concentrations of NO have a direct effect on processes such as proliferation and cell survival, while higher concentrations have an indirect effect through both oxidative stress (via a first-order, diffusion-limited reaction with SO to form ONOO−) and nitrosative stress (through a second-order reaction with NO to form N2O3). Because NO is freely diffusible across cell membranes and SO is not, the reaction of SO and NO occurs within cells/organelles producing SO [activated leukocytes, endothelial cells, and mitochondria] in proximity to diffusible NO from the target cell or a neighboring cell [4].
NO itself can reversibly nitrosylate thiol groups on proteins in a fashion much akin to phosphorylation. This process may be indirectly mediated by carrier thiol peptides or proteins such as thioredoxin [5]. NO also modifies iron centers in proteins such as p450 enzymes and soluble guanylate cyclase, the latter being responsible for the vasodilating effect of NO. N2O3 can modify zinc finger domains and nitrosylate thiol groups. ONOO− can oxidize lipids such as those found in LDL or arachidonic acid [6]. ONOO− can also act as a peroxide substrate for peroxidases such as those found in cyclooxygenase (COX) [7]. Finally, ONOO− can nitrate DNA (Figure 2) [8].
Figure 2.
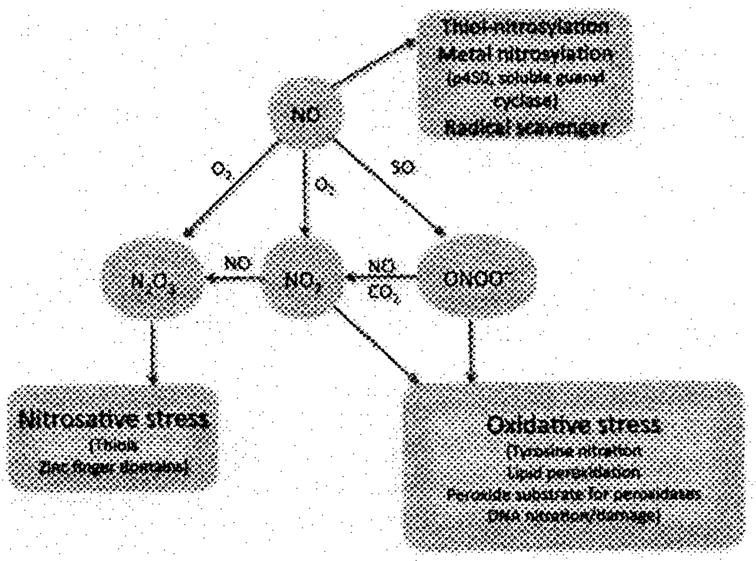
The actions of NO on biologically relevant molecules. NO can nitrosylate thiol amino acids in proteins or peptides to form nitrosothiols. It can nitrosylate iron in p450 enzymes and guanylate cyclase to alter enzyme activity. NO also acts as a radical scavenger of SO (SO or O2−). When this latter reaction occurs, oxidative stress occurs through the action of ONOO−, which itself can nitrate protein tyrosines and DNA, a process that can alter enzyme function and increase immunogenicity of DNA and proteins/peptides. ONOO− and NO2 can oxidize lipids to form mediators of inflammation. Finally, ONOO− can as act as a peroxide substrate to increase the activity of enzymes such as COX.
Primary and secondary effects of NO on cellular processes
Different cellular proteins have different concentration thresholds for modification by NO or its oxidative/nitrosative products. Below 30 nM, NO can have a protective and proliferative effect by activating c-GMP-mediated processes, the most recognized of this is c-GMP-mediated vasodilatation from endothelium-derived NO. In the 30–100nM concentration range, the Akt pathway is activated, leading to phosphorylation of BAD and caspase-6, both of which lead to cell survival [9]. S-nitrosylation of caspases by NO in the cytoplasm has an anti-apoptotic effect. On the other hand, mitochrondrial NO can combine with SO to form ONOO−, which nitrates cytochrome c, leading to mitochondria-mediated apoptosis [10]. Another example lies in the interaction between RNI and eicosanoid synthesis. NO produced in the presence of SO, as is often seen in inflamed tissues or in the presence of antiphospholipid antibodies [11], results in the rapid formation of ONOO−, which in turn nitrates and inhibits the activity of prostacyclin synthase with an IC50 of about 50 nM [12]. ONOO− also acts as a peroxide substrate to COX2 and increases its activity [7]. This results in increased availability of prostaglandin H2 (PGH2) substrate for thromboxane and PGE2 synthases in the vasculature and glomerulus.
The inhibition of prostacyclin synthase and activation of COX2 can reverse the vasodilatory and antithrombotic effects of PGI2 and increase the formation and, thus, inflammatory effects of TXA2 and PGE2. At 100 nM, hypoxia-induced factor 1α is stabilized, increasing proliferation and protection against cellular injury. Above 400 nM, p53 is phosphorylated and acetylated, leading to cell cycle arrest and apoptosis. Above 800 nM, nitrosative stress (with the formation of N2O3) occurs, which tends to result in nitrosation of thiol, lysine, and zinc fingers [8]. Specific to apoptotic signaling, N2O3 can lead to nitrosation of PARP and caspases [9].
NOS isoforms
The two isoforms [eNOS and neuronal (nNOS)] are generally constitutively expressed and are calcium dependent. In the vascular system, NO produced by eNOS acts as a vasodilator through activation of soluble guanylate cyclase.
The inducible NOS gene (NOS2) transcribes iNOS that is primarily expressed in immune cells including macrophages and macrophage-like cells such as mesangial cells. iNOS expression is stimulated in murine cells by several cytokines and toll-like receptor ligands such as lipopolysaccharide (LPS), interleukin-6 (IL6), interferon-γ (IFNγ), IL1β, and tumor necrosis factor-α (TNFα). In human monocytes and macrophages, induction in vitro can be attained with IFN γ and LPS, but the levels of NO produced are log-fold lower than those seen in murine macrophages.
In most cells, signaling pathways for iNOS induction converge on the janus kinase/signal transducer and activator of transcription and/or the nuclear factor kappaB (NF-κB) pathways. iNOS is expressed during pathological states in human eNOS cells, synovial fibroblasts, polymorphonuclear cells, lymphocytes, and natural killer cells. In normal human tissue, expression is strong in myocytes, skeletal muscle, and Purkinje cells.
iNOS produces log-fold higher amounts of NO than the constitutively expressed isoforms. When iNOS is uncoupled or in a low arginine environment, SO is produced with NO, resulting in ONOO− formation. ONOO− produced by immune cells is capable of killing intracellular pathogens and tumor cells. Glutathione peroxidase, catalase, SO dismutase, heme oxygenase, and antioxidants serve to protect host cells during inflammatory states by reducing the total ROI burden that can contribute to ONOO− production [3].
Increased markers of NO production with lupus disease in humans
Given that no selective inhibitors of iNOS have been approved or successfully completed the early development for human use, it is difficult to directly study the pathogenic potential of iNOS in human lupus. However, several independent studies have demonstrated a significant correlation between markers of systemic NO production and global lupus disease activity [3]. One of these studies demonstrated more prominent increases in markers of NO production among African-Americans with lupus disease activity [13].
This predisposition to produce increased NO in response to disease activity may be inherited, as two NOS2 polymorphisms were significantly more prevalent in female African-American SLE subjects than matched controls. Supporting a functional role for the polymorphisms in combating infection are studies reporting increased markers of systemic NO production and improved malaria survival in some African populations with these polymorphisms [14].
Tissue- and cell-specific NOS expression
Skin
The skin often reflects disease activity in SLE, and iNOS expression in this organ appears to parallel that activity. Immunostaining for iNOS protein and mRNA was elevated in 33% of epidermal tissue samples from cutaneous lupus subjects before exposure to ultraviolet B (UVB) irradiation, but expression was increased in all samples after UVB exposure [15]. Among subjects with systemic disease, skin biopsy specimens from the buttocks revealed higher iNOS expression in endothelial cells and keratinocytes than in controls. Endothelial expression correlated with lupus disease activity [16]. The presence of iNOS in unaffected skin eNOS cells suggests systemic expression, while its induction with UV exposure offers one mechanism for increased expression during disease activity.
eNOS cells
Lupus patients often display a phenotype of defective eNOS function, as subjects with SLE have reduced endothelium-dependent vasodilation [17]. The mechanism behind this defect is unclear, but the increased levels of circulating endothelial cells seen in lupus subjects may be a maker of damage to the endothelium. The level of circulating endothelial cells in lupus subjects correlated inversely with complement levels, and these cells stained for nitrotyrosine. This observation, combined with the observation that eNOS cells stain for iNOS even in non-lesional skin, suggests an immune complex-mediated production of ONOO− by iNOS in endothelial cells [16,18].
Renal cortex
More recent longitudinal observational studies demonstrate increased markers of systemic NO production (serum NOX) in lupus patients with proliferative lupus nephritis when compared with those with non-proliferative renal disease or those with lupus, but without nephritis. In the same nephritis patients, those who did not achieve renal response to therapy had significantly higher serum NOX levels in the first 3 months of therapy than those who achieved a renal response [19]. This provides the rationale for the hypothesis that sustained RNI production leads to renal damage in lupus nephritis.
Several laboratories have described increased iNOS expression in the glomeruli of subjects with proliferative lupus nephritis [13,20,21]. In biopsies from those with class IV disease, citrulline staining increased with iNOS staining, suggesting that the iNOS was functionally active [22]. In one study, glomerular iNOS staining co-localized with markers of apoptosis and staining for p53, a proapoptotic signaling molecule [21]. This is consistent with the known effect of higher levels of NO on p53 phosphorylation [9]. In another study, in patients with class IV LN, iNOS expression was increased in glomerular, tubular, and interstitial cells. iNOS expression in the tubulointerstitium correlated significantly with total lesion index on biopsy and the extent of proteinuria and creatinine clearance at the time of biopsy. iNOS and NFκB co-localized with apoptotic cells in the glomerulus [23]. These data suggest that two mechanisms for iNOS-mediated glomerular damage in proliferative nephritis are increased by signaling for apoptosis via increased p53 activity and through activation of NFκB.
Potential pathogenic mechanisms of increased RNI production suggested by studies of human SLE subjects
One mechanism through which NO can be pathogenic, in the setting of SLE, is through the creation of neoepitopes by ONOO−-mediated nitration of nucleophilic domains on self-antigens. In one study, serum from SLE patients bound more avidly to ONOO− nitrated versus native poly-L-tyrosine. Binding of serum from patients with high dsDNA antibody titers was inhibited by nitrated poly-L-tyrosine, nitrated BSA, nitrated DNA, and nitrated chromatin much more effectively than native forms of these antigens. DNA, modified in this fashion, is also a better immunogen for inducing anti-dsDNA antibody production in experimental animals [24]. Similarly, peroxynitrite-treated DNA is more immunogenic than native DNA as the antigen for dsDNA antibody testing of serum from patients with SLE [25,26]. These combined studies suggest that ONOO− modifications of self-antigens can create neoepitopes with increased binding affinity over native antigens. Whether the increased immunogenicity of nitrated DNA stems from cross-reactivity of these epitopes with native DNA or later leads to epitope spreading to unmodified epitopes has not been determined.
ONOO− can also modify lipids. Peroxidation of arachidonate by ONOO− can lead to formation of isoprostanes [6] that stimulate monocyte adhesion to endothelial cells [27] and induce vasoconstriction in smooth muscles [28]. ONOO− can oxidize arachidonic acid or lipids such as those found in LDL. Oxidized, but not native, LDL complexes with β2-glycroprotein I. Antibodies to this complex were elevated in subjects with SLE and antiphospholipid syndrome, and this antibody/antigen complex enhances influx of oxidized LDL into foamy macro-phages, providing a plausible mechanism for accelerated atherosclerosis in SLE [29]. Some phospholipids within oxidized LDL have platelet-activating factor-like activity and can stimulate growth of smooth muscle cells [30].
ONOO− derived from myeloperoxidase and not iNOS can lead to nitration of tyrosine 166 in apolipoprotein A–I within HDL. Nitration of this amino acid leads to a loss of cholesterol efflux capacity of HDL [31], and this modification along with chlorination of this site confers a 6- to 16-fold risk for cardiovascular disease that is independent of Framingham risk factors [32]. The extent to which this phenomenon occurs in SLE subjects is unclear. Thus, the complete clinical effect of ONOO− formation on lupus disease phenotype and cardiovascular disease associated with SLE is unknown.
Perl and colleagues identified that normal T cells express eNOS and nNOS (but not iNOS) and that expression of these NOS isoforms increased with CD3/CD28 costimulation. In addition, they demonstrated that NO induced an increase of mitochondrial hyperpolarization (MHP) in normal human T cells[33]. In contrast, they found that T lymphocytes of SLE patients exhibited persistent MHP and mitochondrial mass, accounting for increased production of ROI. These data suggest that mitochondrial dysfunction leading to ATP depletion is ultimately responsible for diminished activation-induced apoptosis and sensitizes lupus T cells to necrosis [34–36]. More recent studies have identified that NO-induced MHP in SLE T cells leads to activation of mTOR, a sensor of mitochondrial potential and target of the drug rapamycin [37].
Observational murine studies implicating iNOS activity in lupus disease progression
Although iNOS activity can suppress parasitemia or tumor growth, its over-expression in the setting of lupus disease activity is associated with not only organ damage but also more subtle changes in cellular phenotype and survival. Several studies involving murine models of lupus support this hypothesis. Both MRLMpJ-Faslpr/J (MRL/lpr) and (New Zealand Black × New Zealand White)F1 (NZB/W) mice develop spontaneous proliferative lupus nephritis. MRL/lpr mice developed increasing levels of urine NO metabolites (nitrate + nitrite or NOX) in parallel with the onset of glomerulonephritis [38].
This increase in iNOS activity was associated with formation of nitrotyrosine (NTyr), a product of ONOO−-mediated nitration of tyrosine (Tyr). Such modifications reduced the activity of catalase in the MRL/lpr kidney. Because catalase removes SO, its inactivation may have exposed cells to increased oxidative stress and could, thus, lead to redox signaling and transcriptional events [39]. iNOS expression is increased in the brain tissue of NZB/W mice compared with BALB/c mice and is associated with an increase in p53. The pro-apoptotic role of p53 activated by iNOS may lead to brain injury in SLE [40].
Potential mechanisms of increased iNOS expression in MRL/lpr mice
Immune complex formation and tissue deposition are not dependent on iNOS activity in murine lupus, as iNOS inhibitor therapy had no effect on glomerular immune complex deposition in MRL/lpr mice [38]. However, nephritogenic autoantibodies, when injected into young, disease susceptible MRL/lpr mice, induced NOS2 gene transcription in both an Fc-receptor-dependent and an Fc-receptor-independent manner. Similarly, MRL/lpr mesangial cells exposed to nephritogenic anti-dsDNA antibodies had increased levels of NOS2 message compared with control antibodies [41]. The histone deacetylase inhibitor trichostatin A, which attenuated renal disease in MRL/lpr mice, also inhibited NO production in cultured MRL/lpr mesangial cells [42]. This suggests that epigenetic regulation has a direct and/or indirect effect on iNOS expression. LPS/IFNγ-induced iNOS expression in MRL/lpr mesangial cells can be abrogated through inactivation of the interferon regulatory factor-1 (IRF1) gene.
It is difficult to conclude whether this mechanism directly reduces the iNOS expression seen in MRL/lprIRF1 −/− mice, as this genetic manipulation also reduces anti-dsDNA antibody production and glomerular immune complex deposition in these mice[43]. In NZB/W mesangial cells, IL20 and IL20 receptors are upregulated, and addition of IL20 induces expression of iNOS along with IL6, RANTES, IP 10, and MCP-1 [44]. Some interventions that do not directly inhibit iNOS enzyme also reduce expression of iNOS. For instance, chemical induction of heme oxygenase-1 and oral administration of mycophenolate mofetil both effectively treated glomerulonephritis in MRL/lpr mice while concurrently reducing iNOS expression in the kidney[45–47]. These data suggest that glomerular iNOS expression in murine models can be induced by nephritogenic autoantibodies and innate immune factors such as IL20 through several independent mechanisms.
Effects of manipulation of iNOS in murine lupus
Several studies utilizing competitive inhibitors of iNOS suggest that iNOS activity is pathogenic in murine lupus. Inhibiting iNOS activity in MRL/lpr mice before disease onset with the non-specific arginine analog LNG-monomethyl-L-arginine (L-NMMA) reduced 3NTyr formation in the kidney, partially restored renal catalase activity, and inhibited cellular proliferation and necrosis within the glomerulus[38,39,48].
This effect occurred without a change in immunoglobulin or complement deposition in the glomerulus, suggesting that increased iNOS expression occurred distal to immune complex deposition and complement activation [38]. The partially selective iNOS inhibitor L-N6-(1-iminoethyl)lysine (L-NIL) had a similar effect when used to treat these mice prior to disease onset. L-NIL-treated mice had lower glomerular pathology scores compared with controls [48]. L-NMMA used to treat NZB/W mice with nephritis had a similar but less profound effect on proteinuria and renal histopathology than did L-NMMA given before disease onset. L-NMMA monotherapy was less effective in treating the more aggressive, rapidly progressive nephritis seen in MRL/lpr mice [49].
In contrast to the effectiveness of pharmacologic iNOS inhibition in murine lupus was the observation that iNOS −/− MRL/lpr mice, while having reduced signs of vasculitis and IgG rheumatoid factor production, had similar glomerular pathology to their MRL/lpr wild-type litter-mates [50]. To address whether the beneficial effect of L-NMMA in lupus was unrelated to iNOS, MRL/lpr NOS2−/− mice were administered an iNOS-selective inhibitor prior to and throughout the progression of disease. NOS2−/− mice had elevated anti-dsDNA antibody levels and had, as observed in the past, no significant reductions in glomerular pathology or proteinuria. However, iNOS inhibitor therapy significantly reduced proteinuria and podocyte flattening/eNOS cell swelling by electron microscopy [51]. This suggests that iNOS inhibitor therapy reduces pathologic changes in podocyte and endothelial cell pathology in this model in an iNOS-independent (and possibly eNOS-dependent) fashion.
Potential mechanisms for pathogenicity of RNI suggested by studies in murine models of lupus
The mechanisms through which iNOS activity may bepathogenic in SLE have been studied in animal models and in vitro (Table I). As mentioned above, ONOO−, a by-product of iNOS activity, can nitrate protein amino acids and change the catalytic activity of enzymes. One such enzyme, catalase, serves to protect host tissues from free radical attack [39]. In vascular tissue, prostacyclin synthase [52] and eNOS [53] are inactivated by ONOO−, leading to vasoconstriction. These observations suggest that one mechanism through which iNOS activity is pathogenic is via deactivation of tissue protective enzymes.
Table I.
Mechanisms through which reactive nitrogen species can affect cell cycle/apoptosis and inflammation.
| Reactive Species | Target | Process | Effect | Reference(s) |
|---|---|---|---|---|
| NOX | Caspases | Nitrosation | Reduced enzyme activity | [9,10] |
| NOX | p53 | NO-induced DNA strand breaks? | Phosphorylation of p53 | [9,59] |
| NOX | HIF1α | Disruption of Hsp90–client protein interactions | HIF1α stabilized, half-life increased | [9,59] |
| NOX | Nitrosylation | Cytochrome c oxidase complex IV S2 | Inhibition of cytochrome c and mitochondrial membrane hyperpolarization | [60] |
| ONOO− | eNOS | Thiol oxidation | eNOS uncoupling/SO production | [52] |
| ONOO− | PGI2 synthase | Nitration | Reduced enzyme activity | [52] |
| ONOO− | Catalase | Nitration | Reduced enzyme activity | [39] |
| ONOO− | Cytochrome c | Nitration | Mitochrondia-induced apoptosis | [10] |
| ONOO− | Protein Tyr | Nitration | Neoepitope formation/increased antigenicity | [24] |
| ONOO− | DNA | Nitration | Neoepitope formation/increased antigenicity | [26] |
| ONOO− | Arachidonic acid | Peroxidation | Formation of isoprostanes | [6] |
| ONOO− | LDL | Oxidation | Increased uptake by macrophages, increased binding of β2-glycoprotein I | [29] |
| ONOO− | COX2 | Peroxide substrate | Increases enzyme activity | [7] |
NOX = NO or N2O3
Because nuclear antigens are presented in late apoptotic blebs [54], regulation of apoptosis and clearance of apoptotic cells is an important area of investigation. NO and ONOO− are both integral in regulating non-receptor-mediated apoptosis in many cellular systems [55]. To investigate the role of iNOS activity in apoptosis, MRL/lpr mice with active disease were treated with L-NMMA, an iNOS inhibitor. Compared with controls, treated mice exhibited reduced levels of splenocyte apoptosis. Treatment of cultured splenocytes isolated from mice with active disease with an NO donor resulted in increased levels of apoptosis [56]. NO or other RNI appeared to increase non-receptor-mediated apoptosis despite the well-described defect in receptor-mediated apoptosis in this murine model of lupus [57].
iNOS activity can lead to ONOO− production only if iNOS activity is accompanied by equimolar levels of SO. One mechanism for simultaneous production of SO and NO is through the parallel production of SO by the reductase domain of iNOS itself. Support for this mechanism in lupus comes from experiments involving pharmacological inhibition of iNOS in the MRL/lpr and NZB/W models. Mice given iNOS–specific and non-specific inhibitors demonstrated significant reductions in markers of systemic oxidant stress (urine F2-isoprostanes) compared with mice treated with distilled water [58]. This observation raises the possibility that some of the pathogenic effects of iNOS activity in SLE arise from its ability to produce ROI in proximity to NO.
Conclusions
The effects of NO production in vivo are dependent on focal cellular levels of NO production and its proximity to other reactive species. This makes it impossible to declare NO as either a pathogenic or protective molecule in lupus. It also complicates attempts to study the effect of RI on pathogenic lupus cellular processes in vitro. Future work investigating the effect of NO and reactive oxygen production in lupus will require determination of the concentration and cellular location of NO and reactive oxygen production in affected tissues in vivo. Newer live imaging and ex vivo and in vivo spin trap techniques are potential tools for this inquiry. This knowledge will provide a rational basis for mechanistic studies in vitro and the development of targeted therapies. Drugs that modulate the effects of reactive intermediate production in lupus hold promise but have yet to be tested in human SLE.
Acknowledgments
Special thanks go to Gary Gilkeson, MD, for reviewing the manuscript before submission.
Footnotes
Declaration of interest: This publication was made possible by Grant AR045476 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, GCRC Grant M01RR001070, an award from the VA Research Enhancement Award Program, a VA Merit Award funding from the Arthritis Foundation, and the Alliance for Lupus Research. Special thanks go to Gary Gilkeson, MD, for reviewing the manuscript before submission. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. Special thanks go to Gary Gilkeson, MD, for reviewing the manuscript before submission. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Jezek P, Zackova M, Ruzicka M, Skobisova E, Jaburek M. Mitochondrial uncoupling proteins—Facts and fantasies. Physiol Res. 2004;53(Suppl 1):S199–S211. [PubMed] [Google Scholar]
- 2.Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, Yokoyama M, Kawashima S, Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97(9):864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 3.Oates JC, Gilkeson GS. The biology of nitric oxide and other reactive intermediates in systemic lupus erythematosus. Clin Immunol. 2006;121(3):243–250. doi: 10.1016/j.clim.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Espey MG, Thomas DD, Miranda KM, Wink DA. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc Natl Acad Sci USA. 2002;99(17):11127–11132. doi: 10.1073/pnas.152157599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tannenbaum SR, White FM. Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem Biol. 2006;1(10):615–618. doi: 10.1021/cb600439h. [DOI] [PubMed] [Google Scholar]
- 6.Moore KP, Darley-Usmar V, Morrow J, Roberts LJ., II Formation of F2-isoprostanes during oxidation of human low-density lipoprotein and plasma by peroxynitrite. Circ Res. 1995;77(2):335–341. doi: 10.1161/01.res.77.2.335. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin DC, Landino LM, Marnett LJ. Reactions of prostaglandin endoperoxide synthase with nitric oxide and peroxynitrite. Drug Metab Rev. 1999;31(1):273–294. doi: 10.1081/dmr-100101918. [DOI] [PubMed] [Google Scholar]
- 8.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. 2004;385(1):1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- 9.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic Biol Med. 2008;45(1):18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leon L, Jeannin JF, Bettaieb A. Post-translational modifications induced by nitric oxide (NO): Implication in cancer cells apoptosis. Nitric Oxide. 2008;19(2):77–83. doi: 10.1016/j.niox.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 11.Delgado Alves J, Mason LJ, Ames PR, Chen PP, Rauch J, Levine JS, Subang R, Isenberg DA. Antiphospholipid antibodies are associated with enhanced oxidative stress, decreased plasma nitric oxide and paraoxonase activity in an experimental mouse model. Rheumatology (Oxford) 2005;44(10):1238–1244. doi: 10.1093/rheumatology/keh722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou MH, Yesilkaya A, Ullrich V. Peroxynitrite inactivates prostacyclin synthase by heme-thiolate-catalyzed tyrosine nitration. Drug Metab Rev. 1999;31(2):343–349. doi: 10.1081/dmr-100101922. [DOI] [PubMed] [Google Scholar]
- 13.Oates JC, Christensen EF, Reilly CM, Self SE, Gilkeson GS. Prospective measure of serum 3-nitrotyrosine levels in systemic lupus erythematosus: Correlation with disease activity. Proc Assoc Am Physicians. 1999;111(6):611–621. doi: 10.1046/j.1525-1381.1999.99110.x. [DOI] [PubMed] [Google Scholar]
- 14.Oates JC, Levesque MC, Hobbs MR, Smith EG, Molano ID, Page GP, Hill BS, Weinberg JB, Cooper GS, Gilkeson GS. Nitric oxide synthase 2 promoter polymorphisms and systemic lupus erythematosus in African-Americans. J Rheumatol. 2003;30(1):60–67. [PubMed] [Google Scholar]
- 15.Kuhn A, Fehsel K, Lehmann P, Krutmann J, Ruzicka T, Kolbbachofen V. Aberrant timing in epidermal expression of inducible nitric oxide synthase after UV irradiation in cutaneous lupus erythematosus. J Invest Dermatol. 1998;111(1):149–153. doi: 10.1046/j.1523-1747.1998.00253.x. [DOI] [PubMed] [Google Scholar]
- 16.Belmont HM, Levartovsky D, Goel A, Amin A, Giorno R, Rediske J, Skovron ML, Abramson SB. Increased nitric oxide production accompanied by the up-regulation of inducible nitric oxide synthase in vascular endothelium from patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40(10):1810–1816. doi: 10.1002/art.1780401013. [DOI] [PubMed] [Google Scholar]
- 17.El-Magadmi M, Bodill H, Ahmad Y, Durrington PN, Mackness M, Walker M, Bernstein RM, Bruce IN. Systemic lupus erythematosus: An independent risk factor for endothelial dysfunction in women. Circulation. 2004;110(4):399–404. doi: 10.1161/01.CIR.0000136807.78534.50. [DOI] [PubMed] [Google Scholar]
- 18.Clancy R, Marder G, Martin V, Belmont HM, Abramson SB, Buyon J. Circulating activated endothelial cells in systemic lupus erythematosus: Further evidence for diffuse vasculopathy. Arthritis Rheum. 2001;44(5):1203–1208. doi: 10.1002/1529-0131(200105)44:5<1203::AID-ANR204>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 19.Oates JC, Shaftman SR, Self SE, Gilkeson GS. Association of serum nitrate and nitrite levels with longitudinal assessments of disease activity and damage in systemic lupus erythematosus and lupus nephritis. Arthritis Rheum. 2008;58(1):263–272. doi: 10.1002/art.23153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furusu A, Miyazaki M, Abe K, Tsukasaki S, Shioshita K, Sasaki O, Miyazaki K, Ozono Y, Koji T, Harada T, et al. Expression of endothelial and inducible nitric oxide synthase in human glomerulonephritis. Kidney Int. 1998;53(6):1760–1768. doi: 10.1046/j.1523-1755.1998.00907.x. [DOI] [PubMed] [Google Scholar]
- 21.Wang JS, Tseng HH, Shih DF, Jou HS, Ger LP. Expression of inducible nitric oxide synthase and apoptosis in human lupus nephritis. Nephron. 1997;77(4):404–411. doi: 10.1159/000190316. [DOI] [PubMed] [Google Scholar]
- 22.Bollain YGJJ, Ramirez-Sandoval R, Daza L, Esparza E, Barbosa O, Ramirez D, Pacheco-Tovar G, Avalos-Diaz E, Rodriguez-Padilla C, Herrera-Esparza R. Widespread expression of inducible NOS and citrulline in lupus nephritis tissues. Inflamm Res. 2009;58(2):61–66. doi: 10.1007/s00011-009-7215-1. [DOI] [PubMed] [Google Scholar]
- 23.Zheng L, Sinniah R, Hong Hsu SI. Renal cell apoptosis and proliferation may be linked to nuclear factor-kappaB activation and expression of inducible nitric oxide synthase in patients with lupus nephritis. Hum Pathol. 2006;37(6):637–647. doi: 10.1016/j.humpath.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Khan F, Ali R. Antibodies against nitric oxide damaged poly L-tyrosine and 3-nitrotyrosine levels in systemic lupus erythematosus. J Biochem Mol Biol. 2006;39(2):189–196. doi: 10.5483/bmbrep.2006.39.2.189. [DOI] [PubMed] [Google Scholar]
- 25.Dixit K, Ali R. Role of nitric oxide modified DNA in the etiopathogenesis of systemic lupus erythematosus. Lupus. 2004;13(2):95–100. doi: 10.1191/0961203304lu492oa. [DOI] [PubMed] [Google Scholar]
- 26.Habib S, Moinuddin Ali R. Peroxynitrite-modined DNA: A better antigen for systemic lupus erythematosus anti-DNA autoantibodies. Biotechnol Appl Biochem. 2006;43(Pt 2):65–70. doi: 10.1042/BA20050156. [DOI] [PubMed] [Google Scholar]
- 27.Huber J, Bochkov VN, Binder BR, Leitinger N. The isoprostane 8-iso-PGE2 stimulates endothelial cells to bind monocytes via cyclic AMP- and p38 MAP kinase-dependent signaling pathways. Antioxid Redox Signal. 2003;5(2):163–169. doi: 10.1089/152308603764816523. [DOI] [PubMed] [Google Scholar]
- 28.Fukunaga M, Makita N, Roberts LJ, II, Morrow JD, Takahashi K, Badr KF. Evidence for the existence of F2-isoprostane receptors on rat vascular smooth muscle cells. Am J Physiol. 1993;246(6):C1619–C1624. doi: 10.1152/ajpcell.1993.264.6.C1619. [DOI] [PubMed] [Google Scholar]
- 29.Lopez LR, Kobayashi K, Matsunami Y, Matsuura E. Immunogenic oxidized low-density lipoprotein/beta2-glycoprotein I complexes in the diagnostic management of atherosclerosis. Clin Rev Allergy Immunol. 2009;37(1):12–19. doi: 10.1007/s12016-008-8096-8. [DOI] [PubMed] [Google Scholar]
- 30.Heery JM, Kozak M, Stafforini DM, Jones DA, Zimmerman GA, McIntyre TM, Prescott SM. Oxidatively modified LDL contains phospholipids with platelet-activating factor-like activity and stimulates the growth of smooth muscle cells. J Clin Invest. 1995;96(5):2322–2330. doi: 10.1172/JCI118288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng DQ, Brubaker G, Wu Z, Zheng L, Willard B, Kinter M, Hazen SL, Smith JD. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol. 2008;28 (11):2063–2070. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J Lipid Res. 2009;50 (Suppl):S346–S351. doi: 10.1194/jlr.R800086-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagy G, Koncz A, Perl A. T cell activation-induced mitochondrial hyperpolarization is mediated by Ca2+- and redox-dependent production of nitric oxide. J Immunol. 2003;171(10):5188–5197. doi: 10.4049/jimmunol.171.10.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gergely P, Jr, Niland B, Gonchoroff N, Pullmann R, Jr, Phillips PE, Perl A. Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol. 2002;169(2):1092–1101. doi: 10.4049/jimmunol.169.2.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagy G, Barcza M, Gonchoroff N, Phillips PE, Perl A. Nitric oxide-dependent mitochondrial biogenesis generates Ca2+ signaling profile of lupus T cells. J Immunol. 2004;173(6):3676–3683. doi: 10.4049/jimmunol.173.6.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gergely P, Jr, Grossman C, Niland B, Puskas F, Neupane H, Allam F, Banki K, Phillips PE, Perl A. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):175–190. doi: 10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, Phillips PE, Crow MK, Oess S, Muller-Esterl W, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182(4):2063–2073. doi: 10.4049/jimmunol.0803600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinberg JB, Granger DL, Pisetsky DS, Seldin MF, Misukonis MA, Mason SN, Pippen AM, Ruiz P, Wood ER, Gilkeson GS. The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: Increased nitric oxide production and nitric oxide synthase expression in MRL-lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-monomethyl-L-arginine. J Exp Med. 1994;179(2):651–660. doi: 10.1084/jem.179.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keng T, Privalle CT, Gilkeson GS, Weinberg JB. Peroxynitrite formation and decreased catalase activity in autoimmune MRL-lpr/lpr mice. Mol Med. 2000;6(9):779–792. [PMC free article] [PubMed] [Google Scholar]
- 40.Wang HP, Hsu TC, Hsu GJ, Li SL, Tzang BS. Cystamine attenuates the expressions of NOS- and TLR-associated molecules in the brain of NZB/W F1 mice. Eur J Pharmacol. 2009;607(1–3):102–106. doi: 10.1016/j.ejphar.2009.02.039. [DOI] [PubMed] [Google Scholar]
- 41.Qing X, Zavadil J, Crosby MB, Hogarth MP, Hahn BH, Mohan C, Gilkeson GS, Bottinger EP, Putterman C. Nephritogenic anti-DNA antibodies regulate gene expression in MRL/lpr mouse glomerular mesangial cells. Arthritis Rheum. 2006;54(7):2198–2210. doi: 10.1002/art.21934. [DOI] [PubMed] [Google Scholar]
- 42.Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111(4):539–552. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reilly CM, Olgun S, Goodwin D, Gogal RM, Jr, Santo A, Romesburg JW, Ahmed SA, Gilkeson GS. Interferon regulatory factor-1 gene deletion decreases glomerulonephritis in MRL/lpr mice. Eur J Immunol. 2006;36(5):1296–1308. doi: 10.1002/eji.200535245. [DOI] [PubMed] [Google Scholar]
- 44.Li HH, Cheng HH, Sun KH, Wei CC, Li CF, Chen WC, Wu WM, Chang MS. Interleukin-20 targets renal mesangial cells and is associated with lupus nephritis. Clin Immunol. 2008;129(2):277–285. doi: 10.1016/j.clim.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Takeda Y, Takeno M, Iwasaki M, Kobayashi H, Kirino Y, Ueda A, Nagahama K, Aoki I, Ishigatsubo Y. Chemical induction of HO-1 suppresses lupus nephritis by reducing local iNOS expression and synthesis of anti-dsDNA antibody. Clin Exp Immunol. 2004;138(2):237–244. doi: 10.1111/j.1365-2249.2004.02594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lui SL, Tsang R, Wong D, Chan KW, Chan TM, Fung PC, Lai KN. Effect of mycophenolate mofetil on severity of nephritis and nitric oxide production in lupus-prone MRL/lpr mice. Lupus. 2002;11(7):411–418. doi: 10.1191/0961203302lu214oa. [DOI] [PubMed] [Google Scholar]
- 47.Yu CC, Yang CW, Wu MS, Ko YC, Huang CT, Hong JJ, Huang CC. Mycophenolate mofetil reduces renal cortical inducible nitric oxide synthase mRNA expression and diminishes glomerulosclerosis in MRL/lpr mice. J Lab Clin Med. 2001;138(1):69–77. doi: 10.1067/mlc.2001.115647. [DOI] [PubMed] [Google Scholar]
- 48.Reilly CM, Farrelly LW, Viti D, Redmond ST, Hutchison F, Ruiz P, Manning P, Connor J, Gilkeson GS. Modulation of renal disease in MRL/lpr mice by pharmacologic inhibition of inducible nitric oxide synthase. Kidney Int. 2002;61(3):839–846. doi: 10.1046/j.1523-1755.2002.00230.x. [DOI] [PubMed] [Google Scholar]
- 49.Oates JC, Ruiz P, Alexander A, Pippen AM, Gilkeson GS. Effect of late modulation of nitric oxide production on murine lupus. Clin Immunol Immunopathol. 1997;83(1):86–92. doi: 10.1006/clin.1997.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilkeson GS, Mudgett JS, Seldin MF, Ruiz P, Alexander AA, Misukonis MA, Pisetsky DS, Weinberg JB. Clinical and serologic manifestations of autoimmune disease in MRL-lpr/lpr mice lacking nitric oxide synthase type 2. J Exp Med. 1997;186(3):365–373. doi: 10.1084/jem.186.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Njoku C, Self SE, Ruiz P, Hofbauer AF, Gilkeson GS, Oates JC. Inducible nitric oxide synthase inhibitor SD-3651 reduces proteinuria in MRL/lpr mice deficient in the NOS2 gene. J Investig Med. 2008;56(7):911–919. doi: 10.231/JIM.0b013e3181889e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zou MH, Cohen R, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11(2):89–97. doi: 10.1080/10623320490482619. [DOI] [PubMed] [Google Scholar]
- 53.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109(6):817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boyd CS, Cadenas E. Nitric oxide and cell signaling pathways in mitochondrial-dependent apoptosis. Biol Chem. 2002;383(3–4):411–423. doi: 10.1515/BC.2002.045. [DOI] [PubMed] [Google Scholar]
- 56.Oates JC, Gilkeson GS. Nitric oxide induces apoptosis in spleen lymphocytes from MRL/lpr mice. J Investig Med. 2004;52(1):62–71. doi: 10.1136/jim-52-01-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh AK. Lupus in the Fas lane? J Royal Coll Phys Lond. 1995;29(6):475–478. [PMC free article] [PubMed] [Google Scholar]
- 58.Njoku CJ, Patrick KS, Ruiz P, Jr, Oates JC. Inducible nitric oxide synthase inhibitors reduce urinary markers of systemic oxidant stress in murine proliferative lupus nephritis. J Investig Med. 2005;53(7):347–352. doi: 10.2310/6650.2005.53705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Hypoxic inducible factor 1 alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci USA. 2004;101(24):8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Jin B, Li L, Block ER, Patel JM. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288(4):C840–C849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]