

Abstract
Accumulating evidence suggests dysfunction of the gamma-aminobutyric acid (GABA) system in major depressive disorder (MDD). Neuroimaging studies consistently report reductions of cortical GABA in depressed patients. Our post-mortem analyses demonstrate a reduction in the density and size of GABAergic interneurons in the dorsolateral prefrontal cortex (PFC) in MDD. The goal of this study was to test whether the level of glutamic acid decarboxylase (GAD), the GABA synthesizing enzyme, will also be reduced in the same cortical region in MDD. Levels of GAD-65 and GAD-67 proteins were investigated by Western blotting in samples from the dorsolateral PFC (BA9) in 13 medication-free subjects with MDD, and 13 psychiatrically healthy controls. The overall amount of GAD-67 was significantly reduced (−34 %) in depressed subjects as compared to matched controls. Since recent neuroimaging studies demonstrate that antidepressants modulate GABA levels, additional experiments were performed to examine the levels of GAD in 8 depressed subjects treated with antidepressant medications. Levels of GAD-67 were unchanged in these depressed subjects as compared to their respective controls (n=8). The overall amounts of GAD-65 were similar in depressed subjects compared to matched controls, regardless of antidepressant medication. Reduced levels of GAD-67, which is localized to somata of GABA neurons, further support our observation of a decreased density of GABAergic neurons in the PFC in depression. It is likely that a decrease in GAD-67 accounts for the reduction in GABA levels revealed by neuroimaging studies. Moreover, our data support previous neuroimaging observations that antidepressant medication normalizes GABA deficits in depression.
Keywords: Post-mortem, GAD, GABA, antidepressants, major depressive disorder, dorsolateral prefrontal cortex
Introduction
Several lines of evidence indicate that major depressive disorder (MDD) is associated with abnormalities in the gamma-aminobutyric acid (GABA) system (for review see Sanacora and Saricicek, 2007). Recent neuroimaging studies report reductions in GABA levels in the prefrontal and occipital cortex in depressed patients (Hasler et al., 2007; Sanacora et al., 1999; Sanacora et al., 2004). Reduced GABA concentrations were also demonstrated in the plasma and cerebrospinal fluid in depression (Brambilla et al., 2003; Gerner and Hare, 1981; Kasa et al., 1982; Petty et al., 1992). Moreover, a metabolomic analysis demonstrates reductions in the level of GABA as well as several fatty acids and glycerol in blood plasma of older depressed patients (Paige et al., 2007). Recent post-mortem morphometric analyses in MDD demonstrate a reduction in the density and size of GABAergic interneurons immunoreactive for calbindin protein in the dorsolateral prefrontal cortex (PFC; Rajkowska et al., 2007) suggesting GABAergic system dysfunction in depression.
GABA is synthesized from glutamate in GABAergic neurons by glutamic acid decarboxylase (GAD), the pyridoxal phosphate (PLP)-dependent enzyme (Martin et al., 1991). GAD exists in two isoforms, GAD-65 and GAD-67, which are the products of two independent genes (Erlander et al., 1991; Kaufman et al., 1991). Gene knockout studies in mice have helped define distinct roles for each isoform. Mice lacking GAD-67 have significantly reduced GABA levels and die at birth of a severe cleft palate (Asada et al., 1997). In contrast, GAD-65 knockout mice have normal basal levels of GABA and appear normal at birth, but develop fatal seizures and anxiety phenotypes (Asada et al., 1996). It has been observed that GAD-65 is more abundant in the nerve terminals, whereas GAD-67 is more concentrated in the neuronal cell bodies (Erlander et al., 1991; Erlander and Tobin, 1991; Kaufman et al., 1991). Thus, based on the different neuronal distributions of GAD isoforms, GAD-67 may be involved in the synthesis of GABA for general metabolic activity, whereas GAD-65 may be predominantly involved in synthesizing GABA for neuronal transmission (Martin and Rimvall, 1993).
Interestingly, it has been demonstrated that antidepressant therapies induce marked changes in GABAergic function. For example, GABA levels in the occipital cortex were increased in depressed patients after antidepressant treatments such as electroconvulsive therapy (ECT) or selective serotonin reuptake inhibitors (SSRIs; Sanacora et al., 2002; Sanacora et al., 2003) but not after cognitive behavioral therapy (Sanacora et al., 2006). Moreover, a number of earlier animal studies reveal that administration of tricyclic antidepressant drugs, inhibitors of monoamine oxidase, or electroconvulsive shock elevates GABA levels or increases its release (Bowdler et al., 1983; Korf and Venema, 1983; Patel et al., 1975; Perry and Hansen, 1973; Popov and Matthies, 1969). Collectively, these data clearly indicate a relationship between antidepressant medication and regulation of GABAergic transmission.
GABA is a major component of neuronal circuitry in the PFC and cortical GABAergic interneurons can be divided into nonoverlapping subpopulations based on the calcium binding protein, calbindin, parvalbumin or calretinin they co-express (Conde et al., 1994; Lund and Lewis, 1993). Since a population of calbindin-immunoreactive interneurons was selectively reduced in the dorsolateral PFC in depression (Rajkowska et al., 2007), the aim of the present study was to investigate whether GAD-65 or GAD-67 protein would also be reduced in the dorsolateral PFC in depression. The levels of GAD-65 and GAD-67 were measured in medication-free subjects with MDD and their individually matched control cases. Additionally, the levels of GAD-65 and GAD-67 were also determined in medicated subjects with MDD and their corresponding controls. Medication-free refers to subjects in which antidepressant drugs were not detected in post-mortem blood samples; medicated subjects are those for which antidepressant drugs were detected in post-mortem blood samples. The levels of GAD-67 and GAD-65 were measured in homogenates from the gray matter of left dorsolateral PFC (Brodmann's Area 9) using Western blot method.
Material and methods
Human subjects
Post-mortem brain samples were collected at autopsy from the total of 34 subjects at the Cuyahoga County Coroner's Office in Cleveland, OH. Informed written consent was collected from the legal next-of-kin of all subjects. Next-of-kin were interviewed and retrospective psychiatric assessments were conducted in accordance with Institutional Review Board policies at University Hospitals of Cleveland and the University of Mississippi Medical Center. As previously described, a trained interviewer administered the Schedule for Affective Disorders and Schizophrenia: lifetime version (SADS-L; Endicott and Spitzer, 1978) and/or the Structured Clinical Interview for DSM-IV Psychiatric Disorders (SCID-IV) to knowledgeable next-of-kin of subjects about three months after the death to determine current and lifetime Axis I psychopathology. Diagnoses for Axis I major mental disorders were independently assessed by a clinical psychologist and a psychiatrist, and consensus diagnosis was reached in conference using information from the knowledgeable informants, the coroner's office, and available inpatient and outpatient records. Twenty one subjects met the DSM-IV criteria for major depressive disorder (MDD, American Psychiatric Association, 1994). None of the 21 control subjects met criteria for depression (see Tables 1 and 2). Among 21 depressed individuals, 16 died by suicide. Eleven out of 21 depressed subjects were hospitalized (1-35 times) due to their depression. Blood and urine samples from all subjects were examined by the coroner's office for psychotropic medications and substances of abuse, including antidepressants and ethanol (Tables 1, 2). Thirteen subjects did not have antidepressants present in their post-mortem toxicology screening, thus they are referred to as MDD “medication-free. In fact, nine out of these 13 “medication-free” MDD subjects did not have prescription for antidepressants in the last month of life (for further details see Table 1).
Table 1.
Demographic characteristics of medication-free MDD subjects and controls
| Parameter |
Controls (n=13) |
MDD (n=13) |
|---|---|---|
| Age (mean ± SEM) |
51 ± 5 y |
51 ± 4 y |
| Age range |
30 - 80 y |
30 - 78 y |
| PMI (mean ± SEM) |
21 ± 2 h |
24 ± 2 h |
| PMI range |
9 - 32 h |
11 - 44 h |
| pH (mean ± SEM) |
6.72 ± 0.07 |
6.58 ± 0.08 |
| pH range |
6.3 – 7.14 |
6.06 - 6.97 |
| Time in freezer (mean ± SEM) |
105 ± 8 mo |
114 ± 10 mo |
| Time in freezer range |
42 - 146 mo |
36 - 152 mo |
| Gender (F:M) |
6:7 |
5:8 |
| Medication history* |
none |
n=4 |
| Toxicology: | ||
| • Clean | n=9 | n=7 |
| • Antidepressant drugs | none | none |
| • Other |
n=4 (brompheniramine, n=1; CO, n=1; brompheniramine, orphenadrine, acetaminophen, n=1; caffeine, n=1) |
n=6 (CO, alprazolam, n=1; ethanol, n=1; propoxyphene, acetaminophen, n=1; CO, phenobarbital, phenytoin, n=1; diazepam, acetaminophen, n=1; diphenhydramine, n=1) |
| Cause of death |
Cardiovascular disease, n=12; Acute hemorrhagic pancreatitis, n=1 |
Suicide, n=9 (shot gun, n=3; CO poisoning, n=2; hanging, n=2; drug overdose, n=1; jumper, n= 1), Other causes, n=4 (all cardiovascular disease) |
| Diagnosis |
None, n=10; History of alcohol abuse, n=2; Specific phobia-situational type (heights), n=1 |
MDD, n=10; MDD and history of alcohol abuse, n=1; MDD and history of alcohol dependence, n=1; MDD, bulimia nervosa and drug abuse, n=1 |
| Smoking | Smokers, n=6; History of smoking, n=2 |
Smokers, n=6; History of smoking, n=2 |
PMI, Post-mortem interval; CO, carbon monoxide; MDD, major depressive disorder; mo, month.
Treatment with antidepressants within 4 weeks of time of death. The average ages, PMI, pH and time in freezer of depressed and control subjects were not statistically different.
Table 2.
Demographic characteristics of medicated MDD subjects and controls
| Parameter |
Controls (n=8) |
MDD (n=8) |
|---|---|---|
| Age (mean ± SEM) |
51 ± 6 y |
51 ± 7 y |
| Age range |
27 - 78 y |
20 - 77 y |
| PMI (mean ± SEM) |
21 ± 2 h |
21 ± 3 h |
| PMI range |
10 - 29 h |
10 - 29 h |
| pH (mean ± SEM) |
6.83 ± 0.04 |
6.57 ± 0.07 |
| pH range |
6.71 – 7.09 |
6.21 - 6.79 |
| Time in freezer (mean ± SEM) |
100 ± 17 mo |
105 ± 15 mo |
| Time in freezer range |
29 - 149 mo |
28 - 149 mo |
| Gender (F:M) |
2:6 |
2:6 |
| Medication history* |
none |
n=8 |
| Toxicology: | ||
| • Clean | n=7 | none |
| • Antidepressant drugs | none | n=8 (amitriptyline, n=1; sertraline, norsertraline, n=2; bupropion, venlafaxine, n=1; venlafaxine, n=1; nortriptyline, n=1; paroxetine, n=1; sertraline, n=1) |
| • Other |
n=1 (CO) |
n=5 (chlorpromazine, amantadine, lidocaine, n=1; diphenhydramine, n=1; diphenhydramine, hydrocodone, n=1; cannabinoids, n=1; lidocaine,n=1) |
| Cause of death |
Cardiovascular disease, n=7; Asphyxiation due to CO, n=1 |
Suicide, n=7; (shot gun, n=2; CO poisoning, n=1 ; hanging, n=2; drowning, n=1; overdose-hyperkalemia, n=1); Other causes, n=1 (pulmonary thromboembolism) |
| Diagnosis |
None, n=6; History of alcohol abuse, n=1; History of alcohol dependence (32 y prior to death), n=1 |
MDD, n=6; MDD, hypochondriasis, polysubstance dependence, and alcohol abuse, n=1; MDD, alcohol abuse and Marijuana abuse, n=1 |
| Smoking | Smokers, n=5; History of smoking, n=1 |
Smokers, n=3; History of smoking, n=1 |
PMI, Post-mortem interval; CO, carbon monoxide; MDD, major depressive disorder; mo, month.
Treatment with antidepressants within 4 weeks of time of death. The average ages, PMI, pH and time in freezer of depressed and control subjects were not statistically different.
Eight depressed subjects tested positively for different antidepressants in their toxicology screening, and thus they are referred to as MDD “medicated” (Table 2). All of these “medicated” MDD subjects had prescriptions for antidepressants in the last month of life. Depressed subjects and psychiatrically healthy controls were matched as close as possible for age, gender, post-mortem interval (PMI), tissue pH, and storage time in freezer. The duration of depression for antidepressant-free depressed subjects ranged from 0.2 to 50 years (13.8±4.1 y) and for medicated MDD subjects ranged from 0.25 to 30 years (7.4± 3.5 y).
Immunoblotting
Frozen sections from the blocks of tissue containing dorsolateral PFC were cut on a cryostat at 50 μm. The first two sections collected were stained for Nissl substance to distinguish Brodmann's area 9 (Rajkowska and Goldmann-Rakic, 1995). The immediately adjacent sections were used to collect punches of tissue. The diameter of the punches was adjusted to include all six cortical layers of the gray matter of area 9, and the underlying white matter was excluded.
Tissue samples were prepared according to a method published previously (Karolewicz et al., 2008). Samples were homogenized in ice-cold TE buffer (10 mM Tris-HCl and 1 mM ethylene-diaminetetraacetate, EDTA) containing protease inhibitors (Protease Inhibitor Cocktail Tablets - Complete™, Boehringer Mannheim GmbH). The homogenized tissue was centrifuged at 900 × g for 10 min. Total protein concentration was determined in the resulting supernatant using the bicinchoninic acid (BCA) method (Pierce Biotechnology, Inc, Rockford, IL, USA). Samples were mixed with sample buffer (125 mM Tris base, 20% glycerol, 4% SDS, 10% mercaptoethanol, 0.05% bromophenol blue, pH 6.8) and heated at 95°C for 8 min. Solubilized protein (20 μg per lane) was subjected to 10 % Criterion Precast Tris-HCl gel electrophoresis (Bio-Rad Laboratories, Hercules, CA, USA) and transferred to nitrocellulose membranes (Hybond ECL; Amersham Biosciences, Piscataway, NJ, USA). After transfer, blots were blocked in 5% non-fat milk/TBS (20 mM Tris base and 0.5 M NaCl, pH 7.5 ) or 5% non-fat milk/PBS (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4 and KH2PO4, pH 7.4) for 2 h, then incubated (overnight at 4° C) with mouse anti-GAD-67 monoclonal antibodies (diluted 1:1000; Chemicon, Temecula, CA, USA; no. MAB5406). GAD-65 was labeled using rabbit polyclonal antibodies diluted 1:1000 (Chemicon, no. AB5082). As a control for transfer and loading beta-tubulin was detected on each blot using anti-tubulin (Abcam Inc., Cambridge, MA, USA; no. ab6046) antibody, diluted 1:10,000. Membranes were washed two times for 15 min in TBS buffer and incubated with secondary anti-mouse antibody for GAD-67 (diluted 1:2000; Amersham Biosciences, no. NA931) or anti-rabbit secondary antibody for GAD-65 and beta-tubulin (diluted 1:5000; Amersham Biosciences; no. NA934). After incubation, blots were washed 3 or 4 times for 15 min and developed using enhanced chemiluminescence detection (ECL; PerkinElmer Life Sciences, Inc., Boston, MA, USA) and immediately exposed to film (Hyperfilm-ECL, Amersham Biosciences).
Immunoreactivities of GAD-67 and GAD-65 were investigated in two different experiments in pairs of depressed subjects and matched controls. Pairs of subjects were matched as closely as possible for age, gender, post-mortem interval, tissue pH, and storage time in freezer. A maximum five pairs of control/depressed subjects were run on the same gel with duplicates run on separate gels. There was a linear relationship between optical density values and protein concentrations for all antibodies used (Figure 1). In order to minimize inter-blot variability and to aid in quantifying blots, each gel was loaded with 3 concentrations of the same cortical tissue standard (dissected from a healthy control subject) consisting of 10, 20, and 40 μg of total protein.
Figure 1.
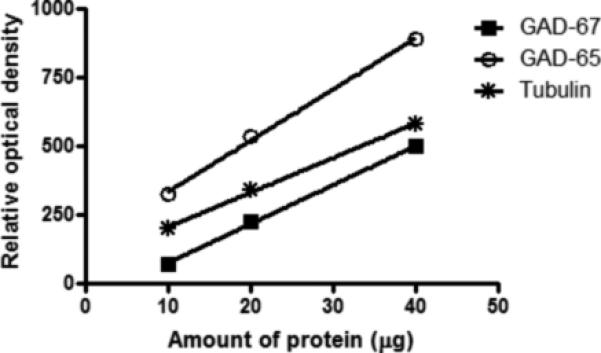
Relationship between the optical density values of Western-blotted protein immunoreactivities and protein amounts for GAD-67, GAD-65, and tubulin. Wells were loaded with three concentrations of cortical tissue consisting of 10, 20, and 40 μg total protein.
Data analysis
Band densities were analyzed using MCID Elite 7.0 (Imaging Research, St. Catherines, Ontario, Canada). Relative optical density values were converted to arbitrary protein units using the standards. Data were then normalized to beta-tubulin immunoreactivity detected on the same blot.
The levels of GAD-67 and GAD-65 immunoreactivity in depressed subjects were compared to their respective controls in two separate experiments using nonparametric Wilcoxon signed ranks tests (SPSS, version 16.0). In the first experiment, MDD subjects medication-free (n=13) were compared to their matched psychiatrically healthy control subjects (n=13), whereas, in the additional experiment, MDD subjects medicated at the time of death (n=8) were compared to their matched controls (n=8).
Pearson correlation analyses was used to assess the correlations between investigated proteins and potentially confounding factors such as age, pH, post-mortem interval (PMI), and storage time in freezer. In order to adjust for multiple comparisons (two GAD isoforms analyzed per brain region) and to avoid a Type 1 error, the p-value ≤ 0.025 was considered as a threshold for significance.
Results
Medication-free subjects with MDD vs controls
Nine out of thirteen medication-free depressed subjects had reduced amount of GAD-67 as compared to their respective controls, two depressed subject had unchanged level of GAD, and two depressed subject had slightly higher level of GAD as compared to matched control. The overall amount of GAD-67 immunoreactivity from medication-free MDD subjects was significantly lower (−34%) compared to control subjects (Wilcoxon signed ranks test, Z= −2.341, p=0.019; Fig. 2). Figure 3 shows GAD-67 immunoreactivity from individual medication-free MDD subjects expressed as percentages of values from paired control subjects. The amount of GAD-65 immunoreactivity from depressed subjects was unchanged as compared to controls (Wilcoxon signed ranks test, Z= −1.153, p=0.249; Fig. 2). Pearson correlation analyses revealed no association between level of GAD-67 or GAD-65 proteins and potentially confounding variables such as: Age, PMI, brain pH, gender or storage time in freezer. The lack of influence of these variables on the level of GAD proteins was observed whether all subjects (MDD + controls) or each of the diagnostic groups were tested separately.
Figure 2.

Medication-free MDD subjects vs controls. Immunoblots of GAD-67 (A), GAD-65 (C) and tubulin from two representative pairs of control and medication-free MDD subjects used in the analysis; (B) significant decrease in the GAD-67 immunoreactivity (−34%) was observed in depressed subjects (n=13) as compared to controls (n=13); (D) amount of GAD-65 immunoreactivity from control (n=13) and depressed (n=13) subjects. Normalized optical density values for the individual subjects (circles) and mean values (horizontal lines) are presented.
Figure 3.

GAD-67 immunoreactivity from individual medication-free MDD subjects expressed as percentages of values from paired control subjects. Each bar is an average of duplicate comparisons.
Medicated subjects with MDD vs controls
In contrast to medication free depressed subjects, depressed subjects that were medicated had unchanged levels of GAD-67 (Wilcoxon signed ranks test, Z= −0.14, p=0.889) and GAD-65 (Wilcoxon signed ranks test, Z= −0.84, p=0.401) proteins as compared to their matched controls (Fig. 4).
Figure 4.

Medicated MDD subjects vs controls. Immunoblots of GAD-67 (A), GAD-65 (C) and tubulin from two representative pairs of control and medicated MDD subjects used in the analysis; (B) amount of GAD-67 immunoreactivity from control (n=8) and depressed (n=8) subjects; (D) amount of GAD-65 immunoreactivity from control (n=8) and depressed (n=8) subjects. Normalized optical density values for the individual subjects (circles) and mean values (horizontal lines) are presented.
The level of GAD-67 and GAD-65 proteins in medicated MDD subjects was not significantly influenced by age, PMI, brain pH, gender or storage time in freezer. Similarly, in control subjects the level of GAD proteins was not correlated with any of these variables.
Discussion
We have found that the level of GAD-67, but not GAD-65, was significantly reduced in medication-free depressed subjects as compared to their matched controls. In contrast, depressed subjects medicated with antidepressants at the time of death had unchanged levels of both GAD isoforms as compared to corresponding control subjects. Together, these data raise the intriguing possibility that GAD-67 is involved in the pathophysiology of depression and antidepressants are able to normalize a GAD deficit in MDD.
GAD-67 is found abundantly in cell bodies and proximal dendrites, and GAD-65 is predominantly present in axon terminals (Kaufman et al., 1991). Hence, a selective decrease in GAD-67 protein suggests that somata and dendrites of specific population of GABAergic neurons may be affected in depression. Low levels of GAD-67 protein immunoreactivity in depression may suggest either a loss of cell bodies and/or their dendritic processes or decreased rate of GAD-67 protein synthesis per unchanged number of neurons. In fact, we have previously reported a selective reduction in the density of calbindin-positive GABAergic interneurons in the dorsolateral PFC (Rajkowska et al., 2007). Therefore, it is likely that reduced GAD-67 immunoreactivity in the dorsolateral PFC is due to a reduction in the density and size of calbindin-positive somata of GABAergic interneurons. Other post-mortem studies investigating GAD in depression found reductions in GAD-65/67 immunopositive structures in the dorsolateral PFC (Gos et al., 2008) and in GAD protein level in the cerebellum (Fatemi et al., 2005).
A reduction in GAD-67 in post-mortem dorsolateral PFC of medication-free MDD subjects reported herein supports a recent MRI spectroscopy observation of a reduced GABA level in the PFC in unmedicated living depressed subjects (Hasler et al., 2007). This and other neuroimaging studies which consistently report reduced GABA levels in MDD (Sanacora et al., 1999; Sanacora et al., 2004) measure total amount of GABA, consisting of metabolic and neurotransmitter pools in cellular and extracellular compartments. Given that the present study demonstrates a reduction in the level of GAD-67 isoform which is predominantly involved in the synthesis of metabolic or cytosolic GABA, it could be suggested that reduced metabolic pool of GABA significantly contributes to the GABA deficits observed by neuroimaging studies in MDD.
Our findings of a reduction in GAD-67 levels in untreated depressed subjects and a lack of a reduction in depressed subjects treated with antidepressants raise the possibility that antidepressants may normalize GAD-67 level in depression. In the present study eight depressed subjects had antidepressants (sertraline, citalopram, paroxetine, venlafaxine, amitriptyline, and nortriptyline) present in their toxicology screenings (Table 2). Among these drugs are selective serotonin reuptake inhibitors (SSRIs), and inhibitors of reuptake of both serotonin and norepinephrine. Thus, the observed protective effect of antidepressants on the cortical GAD-67 level could be explained, to some extent, by the action of serotonin on GABAergic neurons.
More direct evidence for the influence of SSRIs on GABA system is provided by Sanacora et al (2002; 2003) demonstrating that chronic treatment with fluoxetine and citalopram as well as electroconvulsive therapy results in higher levels of GABA as compared to levels recorded before treatment. Moreover, acute administration of the SSRI citalopram increased total occipital GABA levels in healthy subjects (Bhagwagar et al., 2004) with the magnitude similar to that observed after chronic (on average 8 weeks) treatment with SSRIs. The ability of serotoninergic antidepressant drugs to normalize GABA levels in depression and even increase it in healthy subjects indicates close interactions between these two systems and suggests that GABAergic mechanism may indirectly contribute to the mechanism of action of clinically active antidepressants.
The evidence exists that facilitation of GABAergic transmission produces antidepressant effects in humans (Nielsen et al, 1990; Smith et al, 1998; Petty et al, 1995). Interestingly, agents acting at GABA-A or GABA-B receptors exhibit antidepressant-like activity in animal screening procedures (Lloyd et al., 1983; Nowak et al., 2006). If there is a deficit in GABA neurotransmission in depression, then adaptive changes in the density of GABA receptors might be predicted. Studies of GABA-A receptor binding in post-mortem tissues from depressed and suicide subjects have not demonstrated consistent alterations as increases, decreases or no change were found in the benzodiazepine binding sites (Pandey et al., 1997; Cheetham et al., 1988; Rochet et al., 1992; Kugaya et al., 2003; Zhu et al., 2006). Moreover, gene expression studies report reduced (Merali et al, 2004) or up-regulated (Choudary et al., 2005) transcripts encoding specific subunits of the GABA-A receptor in depression and suicide. Collectively, these post-mortem studies of GABA-A receptors do not provide consistent evidence for GABA receptor dysregulation as a pathological marker of depression. Differences between experimental techniques and subject characteristics (e.g. medication exposure, post-mortem interval, brain tissue pH) are key factors that may be associated with discrepancies between post-mortem studies.
One of the shortcomings of this study is that GAD protein immunoreactivity was measured in post-mortem tissue homogenates as opposed to measuring GAD immunoreactivity localized to specific subpopulations of cortical GABA interneurons. However, we have previously reported that calbindin-positive GABAergic interneurons were selectively reduced in the same brain region in MDD (Rajkowska et al., 2007); thus it was plausible to speculate that lower GAD-67 protein levels could reflect a reduction in interneurons expressing calbindin. However, among 21 MDD subjects examined in the present study, only 7 subjects who were antidepressant-free were the same as those analyzed in our study on reductions in GABA-calbindin immunoreactive neurons (Rajkowska et al., 2007). There was no significant correlation between the amount of GAD-67 and the density or size of calbindin immunoreactive neurons. This discrepancy could be explained by the fact, that GAD-67 was measured in homogenates collected from the entire extend of available area 9 and across all six cortical layers. In contrast, calbindin immunoreactive neurons were counted only in a very narrow strip of cortical area 9 and only within one cortical layer (layer II).
Another possible weakness of our study is that we classify the MDD subjects as “medication-free” based on clean post-mortem toxicology screening. However, upon examination of the medical records we can determine that nine out of these 13 “medication-free MDD subjects did not have a prescription for antidepressants in the last month of life (Table 1). Therefore, it is likely that a majority of our “medication-free” MDD subjects were antidepressants naïve for at least 4 weeks before their death. Further studies are needed to test the influence of antidepressants on the GAD protein level in the brain.
Next limitation of the present study is a relatively small sample size (n=8) of “medicated” MDD subjects. Thus, our observation on unchanged level of GAD-67 and GAD-65 in these subjects needs to be tested on a larger number of subjects treated with antidepressants.
In summary, to our knowledge, this is the first observation of selective reduction in the GAD-67 immunoreactivity in the dorsolateral PFC of antidepressant-free MDD subjects and a lack of reduction in medicated MDD subjects as compared to matched controls. The present findings are consistent with studies using other approaches to implicate the GABA system in the pathophysiology of depression. Further indirect evidence is provided here of a regulatory effect of antidepressants on GABAergic system. Additional studies of the GABAergic markers will lead to a better understanding of the role of GABA in the pathology of depression and may lead to the development of more effective approaches to the treatment of depressive symptoms.
Acknowledgements
We gratefully acknowledge the work of Drs James C. Overholser, Herbert Y. Meltzer, Bryan L. Roth, George Jurjus, Lisa Konick, Lesa Dieter and Nicole Herbst in the retrospective psychiatric diagnoses and tissue collection. The excellent assistance of the Cuyahoga County Coroner's Office, Cleveland, OH is greatly appreciated. This publication was supported by MH60451 (GR), MH67996 (CAS); RR017701 (BK, DM, CAS, GR); National Alliance for Research on Schizophrenia and Depression (BK), and American Foundation for Suicide Prevention (BK).
Footnotes
Statement of interest
There are no financial or non-financial interests to disclose for any of the authors related to this submission. Dr. Rajkowska has served in 2008 as a consultant to Lilly Research Laboratories, a division of Eli Lilly and Company.
References
- American Psychiatric Association . Diagnostic and statistical manual of mental disorder. 4th edn. American Psychiatry Press; Washington DC: 1994. [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochemical and Biophysical Research Communications. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwagar Z, Wylezinska M, Taylor M, Jezzard P, Matthews PM, Cowen PJ. Increased brain GABA concentrations following acute administration of a selective serotonin reuptake inhibitor. American Journal of Psychiatry. 2004;161:368–370. doi: 10.1176/appi.ajp.161.2.368. [DOI] [PubMed] [Google Scholar]
- Bowdler JM, Green AR, Minchin MC, Nutt DJ. Regional GABA concentration and [3H]-diazepam binding in rat brain following repeated electroconvulsive shock. Journal of Neural Transmission. 1983;56:3–12. doi: 10.1007/BF01243369. [DOI] [PubMed] [Google Scholar]
- Brambilla P, Perez J, Barale F, Schettini G, Soares JC. GABAergic dysfunction in mood disorders. Molecular Psychiatry. 2003;8:721–737. doi: 10.1038/sj.mp.4001362. [DOI] [PubMed] [Google Scholar]
- Cheetham SC, Crompton MR, Katona CL, Parker SJ, Horton RW. Brain GABAA/benzodiazepine binding sites and glutamic acid decarboxylase activity in depressed suicide victims. Brain Research. 1988;460:114–123. doi: 10.1016/0006-8993(88)91211-5. [DOI] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, Myers RM, Bunney WE, Jr, Akil H, Watson SJ, Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA. Local circuit neurons immunoreactive for calretinin, calbindin D-28k or parvalbumin in monkey prefrontal cortex: distribution and morphology. Journal of Comparative Neurology. 1994;341:95–116. doi: 10.1002/cne.903410109. [DOI] [PubMed] [Google Scholar]
- Endicott J, Spitzer RL. A diagnostic interview: the schedule for affective disorders and schizophrenia. Archives of General Psychiatry. 1978;35:837–844. doi: 10.1001/archpsyc.1978.01770310043002. [DOI] [PubMed] [Google Scholar]
- Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- Erlander MG, Tobin AJ. The structural and functional heterogeneity of glutamic acid decarboxylase: a review. Neurochemical Research. 1991;16:215–226. doi: 10.1007/BF00966084. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Earle JA, raghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophrenia Research. 2005;72:109–122. doi: 10.1016/j.schres.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Gerner RH, Hare TA. CSF GABA in normal subjects and patients with depression, schizophrenia, mania, and anorexia nervosa. American Journal of Psychiatry. 1981;138:1098–1101. doi: 10.1176/ajp.138.8.1098. [DOI] [PubMed] [Google Scholar]
- Gos T, Günther K, Bielau H, Dobrowolny H, Mawrin C, Trübner K, Brisch R, Steiner J, Bernstein HG, Jankowski Z, Bogerts B. Suicide and depression in the quantitative analysis of glutamic acid decarboxylase-immunorective neuropil. Journal of Affective Disorders. 2008 doi: 10.1016/j.jad.2008.04.021. Published Online: 5 Jun 2008. DOI: 10.1016/j.jad.2008.04.021
- Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Archives of General Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- Karolewicz B, Szebeni K, Gilmore T, Maciag D, Stockmeier CA, Ordway GA. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. International Journal of Neuropsychopharmacology. 2008 doi: 10.1017/S1461145708008985. Published Online: 23 Jun 2008. DOI:10.1017/S1461145708008985
- Kasa K, Otsuki S, Yamamoto M, Sato M, Kuroda H, Ogawa N. Cerebrospinal fluid gamma-aminobutyric acid and homovanillic acid in depressive disorders. Biological Psychiatry. 1982;17:877–883. [PubMed] [Google Scholar]
- Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. Journal of Neurochemistry. 1991;56:720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf J, Venema K. Desmethylimipramine enhances the release of endogenous GABA and other neurotransmitter amino acids from the rat thalamus. Journal of Neurochemistry. 1983;40:946–950. doi: 10.1111/j.1471-4159.1983.tb08078.x. [DOI] [PubMed] [Google Scholar]
- Kugaya A, Sanacora G, Verhoeff NP, Fujita M, Mason GF, Seneca NM, Bozkurt A, Khan SA, Anand A, Degen K, Charney DS, Zoghbi SS, Baldwin RM, Seibyl JP, Innis RB. Cerebral benzodiazepine receptors in depressed patients measured with [123I]iomazenil SPECT. Biological Psychiatry. 2003;54:792–799. doi: 10.1016/s0006-3223(02)01788-2. [DOI] [PubMed] [Google Scholar]
- Lloyd KG, Morselli PL, Depoortere H, Fournier V, Zivkovic B, Scatton B, Broekkamp C, Worms P, Bartholini G. The potential use of GABA agonists in psychiatric disorders: evidence from studies with progabide in animal models and clinical trials. Pharmacology, Biochemistry, and Behavior. 1983;18:957–966. doi: 10.1016/s0091-3057(83)80021-5. [DOI] [PubMed] [Google Scholar]
- Lund JS, Lewis DA. Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. Journal of Comparative Neurology. 1993;328:282–312. doi: 10.1002/cne.903280209. [DOI] [PubMed] [Google Scholar]
- Martin DL, Martin SB, Wu SJ, Espina N. Cofactor interactions and the regulation of glutamate decarboxylase activity. Neurochemical Research. 1991;16:243–249. doi: 10.1007/BF00966087. [DOI] [PubMed] [Google Scholar]
- Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. Journal of Neurochemistry. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- Merali Z, Du L, Hrdina P, Palkovits M, Faludi G, Poulter MO, Anisman H. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. Journal of Neuroscience. 2004;24:1478–1485. doi: 10.1523/JNEUROSCI.4734-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen NP, Cesana B, Zizolfi S, Ascalone V, Priore P, Morselli PL. Therapeutic effects of fengabine, a new GABAergic agent, in depressed outpatients: a double-blind study versus clomipramine. Acta Psychiatrica Scandinavica. 1990;82:366–371. doi: 10.1111/j.1600-0447.1990.tb01402.x. [DOI] [PubMed] [Google Scholar]
- Nowak G, Partyka A, Pałucha A, Szewczyk B, Wierońska JM, Dybała M, Metz M, Librowski T, Froestl W, Papp M, Pilc A. Antidepressant-like activity of CGP 36742 and CGP 51176, selective GABAB receptor antagonists, in rodents. British Journal of Pharmacology. 2006;149:581–590. doi: 10.1038/sj.bjp.0706845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige LA, Mitchell MW, Krishnan KR, Kaddurah-Daouk R, Steffens DC. A preliminary metabolomic analysis of older adults with and without depression. International Journal of Geriatric Psychiatry. 2007;22:418–423. doi: 10.1002/gps.1690. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Conley RR, Pandey SC, Goel S, Roberts RC, Tamminga CA, Chute D, Smialek J. Benzodiazepine receptors in the post-mortem brain of suicide victims and schizophrenic subjects. Psychiatry Research. 1997;71:137–149. doi: 10.1016/s0165-1781(97)00060-7. [DOI] [PubMed] [Google Scholar]
- Patel GJ, Schatz RP, Constantinides SM, Lal H. Effect of desipramine and pargyline on brain gamma-aminobutyric acid. Biochemical Pharmacology. 1975;24:57–60. doi: 10.1016/0006-2952(75)90313-5. [DOI] [PubMed] [Google Scholar]
- Perry TL, Hansen S. Sustained drug-induced elevation of brain GABA in the rat. Journal of Neurochemistry. 1973;21:1167–1175. doi: 10.1111/j.1471-4159.1973.tb07572.x. [DOI] [PubMed] [Google Scholar]
- Petty F, Kramer GL, Gullion CM, Rush AJ. Low plasma gamma-aminobutyric acid levels in male patients with depression. Biological Psychiatry. 1992;32:354–363. doi: 10.1016/0006-3223(92)90039-3. [DOI] [PubMed] [Google Scholar]
- Petty F, Trivedi MH, Fulton M, Rush AJ. Benzodiazepines as antidepressants: does GABA play a role in depression? Biological Psychiatry. 1995;38:578–591. doi: 10.1016/0006-3223(95)00049-7. [DOI] [PubMed] [Google Scholar]
- Popov N, Matthies H. Some effects of monoamine oxidase inhibitors on the metabolism of gamma-aminobutyric acid in rat brain. Journal of Neurochemistry. 1969;16:899–907. doi: 10.1111/j.1471-4159.1969.tb08978.x. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Goldman-Rakic PS. Cytoarchitectonic definition of prefrontal areas in the normal human cortex: I. quantitative criteria for distinguishing areas 9 and 46. Cerebral Cortex. 1995;4:307–322. doi: 10.1093/cercor/5.4.307. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–482. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochet T, Kopp N, Vedrinne J, Deluermoz S, Debilly G, Miachon S. Benzodiazepine binding sites and their modulators in hippocampus of violent suicide victims. Biological Psychiatry. 1992;32:922–931. doi: 10.1016/0006-3223(92)90181-x. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Fenton LR, Fasula MK, Rothman DL, Levin Y, Krystal JH, Mason GF. Cortical gamma-aminobutyric acid concentrations in depressed patients receiving cognitive behavioral therapy. Biological Psychiatry. 2006;59:284–286. doi: 10.1016/j.biopsych.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, Krystal JH, Mason GF. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Archives of General Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Behar KL, Hyder F, Petroff OA, Berman RM, Charney DS, Krystal JH. Reduced cortical gamma-aminobutyric acid levels in depressed patients determined by proton magnetic resonance spectroscopy. Archives of General Psychiatry. 1999;56:1043–1047. doi: 10.1001/archpsyc.56.11.1043. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Hyder F, Ciarcia JJ, Ostroff RB, Berman RM, Krystal JH. Increased cortical GABA concentrations in depressed patients receiving ECT. American Journal of Psychiatry. 2003;160:577–579. doi: 10.1176/appi.ajp.160.3.577. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Rothman DL, Krystal JH. Increased occipital cortex GABA concentrations in depressed patients after therapy with selective serotonin reuptake inhibitors. American Journal of Psychiatry. 2002;159:663–665. doi: 10.1176/appi.ajp.159.4.663. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Saricicek A. GABAergic contributions to the pathophysiology of depression and the mechanism of antidepressant action. CNS & Neurological Disorders Drug Targets. 2007;6:127–140. doi: 10.2174/187152707780363294. [DOI] [PubMed] [Google Scholar]
- Smith WT, Londborg PD, Glaudin V, Painter JR. Short-term augmentation of fluoxetine with clonazepam in the treatment of depression: a double-blind study. American Journal of Psychiatry. 1998;155:1339–1345. doi: 10.1176/ajp.155.10.1339. [DOI] [PubMed] [Google Scholar]
- Zhu H, Karolewicz B, Nail E, Stockmeier CA, Szebeni K, Ordway GA. Normal [3H]flunitrazepam binding to GABAA receptors in the locus coeruleus in major depression and suicide. Brain Research. 2006;1125:138–146. doi: 10.1016/j.brainres.2006.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]