

Abstract
Considerable preclinical and epidemiologic data suggest that vitamin D may play a role in the pathogenesis, progression and therapy of cancer. Numerous epidemiologic studies support the hypothesis that individuals with lower serum vitamin D levels have a higher risk of a number of cancers. Measures of vitamin D level in such studies include both surrogate estimates of vitamin D level (residence in more northern latitudes, history of activity and sun exposure) as well as measured serum 25(OH) cholecalciferol levels. Perhaps the most robust of these epidemiologic studies is that of Giovannucci and colleagues who developed and validated an estimate of serum 25(OH) cholecalciferol level and reported that among more than 40,000 individuals in the Health professionals Study an increase in 25(OH) cholecalciferol level of 62.5ng/mL was associated with a reduction in the risk of head/neck, esophagus, pancreas cancers and acute leukemia by >50%. Unfortunately very limited data are available to indicate whether or not giving vitamin D supplements reduces the risk of cancer. Many preclinical studies indicate that exposing cancer cells – as well as vascular endothelial cells derived from tumors - to high concentrations of active metabolites of vitamin D halts progression through cell cycle, induces apoptosis and will slow or stop the growth of tumors in vivo. There are no data that one type of cancer is more or less susceptible to the effects of vitamin D. Vitamin D also potentiates the antitumor activity of a number of types of cytotoxic anticancer agents in in vivo preclinical models. Vitamin D analogues initiate signaling through a number of important pathways, but the pathway(s) essential to the antitumor activities of vitamin D are unclear. Clinical studies of vitamin D as an antitumor agent have been hampered by the lack of a suitable pharmaceutical preparation for clinical study. All commercially available formulations are inadequate because of the necessity to administer large numbers of caplets and the poor “bioavailability” of calcitriol (the most carefully studied analogue) at these high doses. Preclinical data suggest that very high exposures to calcitriol are necessary for the antitumor effects. Clinical data do indicate that very high doses of calcitriol (>100mcg weekly, intravenously and 0.15mcg/kg weekly orally) can be given safely. The maximum tolerated dose (MTD) of calcitriol is unclear. While a 250 patient trial in men with castration resistant prostate cancer (CRPC) comparing docetaxel (36mg/sqm weekly) +/- calcitriol 0.15mcg/kg indicated that calcitriol was very safe, may have reduced to death rate, an adequately powered (1000 patients) randomized study of weekly docetaxel + calcitriol vs q3 week docetaxel was negative. The limitations of this trial were the unequal chemotherapy arms compared in this study and the failure to use an optimal biologic dose or MTD of calcitriol. In view of the substantial preclinical and epidemiologic data supporting the potential role of vitamin D in cancer, careful studies to evaluate the impact of vitamin D replacement on the frequency of cancer and the impact of an appropriate dose and schedule of calcitriol or other active vitamin D analogue on the treatment of established cancer are indicated.
A relationship between vitamin D and cancer has been suggested for many years, and is supported by two lines of research:
in vitro studies – numerous in vitro studies have demonstrated that exposure of tumor cells to high concentrations of vitamin D compounds inhibit their proliferation and in some settings induce differentiation (1-4).
Epidemiologic studies – many investigations have drawn the association between factors expected to lead to “low” vitamin D levels (e.g. geography, lifestyle/activity, history of sun exposure) in large populations and higher rates of several types of cancer (e.g. colon, breast, prostate) (5-7).
This paper will review briefly the biochemistry and physiology of vitamin D and describe the current status of studies seeking to define the role of vitamin D in cancer prevention and cancer therapy.
Vitamin D Synthesis and Metabolism
Vitamin D is actually a hormone. It is synthesized through a multistep process which begins in the skin (Fig. 1). Ultraviolet light of the appropriate wavelength (270-300nm) photocatalyzes the conversion of 7-dehydrocholesterol to cholecalciferol (vitamin D3). Vitamin D3 is further modified in the liver, predominantly by CYP2R1, to 25α hydroxyl cholecalciferol (25(OH)D3) and then 1α-hydroxylated in the kidney by CYP27B1 to 1,25 dihydroxycholcalciferol or calcitriol. Calcitriol is the most potent naturally occurring form of vitamin D. Vitamin D compounds are transported throughout the body by a specific binding protein, vitamin D binding protein (DBP). Vitamin D action is limited by catabolism, primarily by a 24-hydroxylase (CYP24A1) which results in 1,24,25(OH)3 D3, a compound with substantially lower affinity for the VDR; this compound is further metabolized to excreted products such as calcitroic acid. The metabolism of vitamin D is complex and tightly regulated. Rate limiting steps in the metabolism of vitamin D compounds are the activity of CYP2R1 which is induced by low 25(OH)D3 levels and the activity of CYP24A1 which is induced by 25(OH)D3 and 1,25(OH)D3. (8) While vitamin D metabolizing enzymes are located primarily in liver (CYP2R1) and kidney (CYP27B1 and CYP24), these enzymes are found in many tissues. Studies have demonstrated that potential vitamin D target tissues (e.g. colon, prostate, breast, lung, pancreas) can synthesize and degrade calcitriol. Local production and degradation of calcitriol have been suggested to be etiologic factors in several types of human cancer (9, 10).
Fig 1.
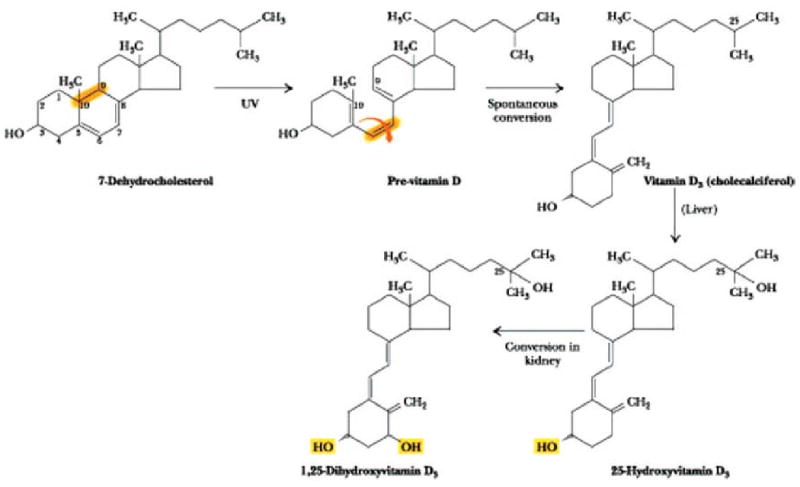
Synthesis of vitamin D
Vitamin D Effector Pathways
Calcitriol-Mediated Transcription of Target Genes
The dominant pathway through which calcitriol mediates its biologic effects is through binding to its specific receptor, the vitamin D receptor or VDR. Calcitriol bound to VDR forms heterodimers with the retinoid X receptor (RXR) and its ligand (9 cis-retinoic acid) and these dimers occupy specific binding sites on DNA (vitamin D response elements or VDREs). In conjunction with other transcription factors, this complex induces transcription of vitamin D responsive genes (11, 12). Cofactor proteins or transcription factors also modulate VDR-mediated gene expression; such proteins have intrinsic chromatin-modifying enzymatic activities and recruit basal transcription factors to the promoters. Calcitriol binding induces phosphorylation and conformational changes in VDR which causes the release of corepressors [e.g., Nuclear receptor Co-Repressors (NCoRs) and Silencing Mediator for Retinoid and the Thyroid hormone receptors (SMRT)/histone deacetylase (HDAC) complex]. Conformational changes also reposition the VDR activation function 2 (AF-2) domains to bind to stimulatory cofactors, such as Steroid Receptor Coactivators (SRCs), Nuclear Coactivator-62 kDa/SKI-interacting Protein (NCoA62/SKIP) and the chromatin modifiers, CREB binding protein (CBP)/p300 and polybromo- and SWI-2-related gene 1 associated factor (PBAF) (12). Once the chromatin is de-repressed, vitamin D receptor-interacting protein (DRIP) complexes bind to the AF-2 of VDR and interact with the transcription machinery, such as TFIIB and RNA polymerase II, and initiate transcription. Among the many genes that are transcriptionally activated by calcitriol are CYP24A1, BGLAP (osteocalcin), and CDKN1A (encoding p21Waf1/Cip1), the growth arrest and DNA-damage-inducible gene, GADD45 gene; the parathyroid hormone gene (PTH) is repressed by calcitriol (12). Calcitriol-mediated repression or activation of many proto-oncogenes or tumor suppressor genes has been described in normal and tumor tissues (13, 14); however, only a few such genes contain VDREs in the promoter regions and are under the direct transcriptional control of calcitriol (Cdk inhibitor p21Waf1/Cip143, CCNC (encoding cyclin C; a DR4-type VDRE) (15). This suggests that calcitriol exerts many of its effects indirectly by modulating signaling cascades or by as yet unknown non-genomic mechanisms.
Non-genomic Action of Calcitriol
Non-genomic actions of calcitriol are rapid and do not depend on transcription. Non-genomic signaling may indirectly affect transcription via cross-talk with other signaling pathway (16, 17). Data suggest that non-genomic effects begin at the plasma membrane and involve a nonclassical membrane receptor and a calcitriol-membrane-associated rapid-response steroid binding protein (1,25D3-MARRS).
The most well-described non-genomic effect of calcitriol is the rapid translocation of calcium across intestinal mucosal membranes. Binding of calcitriol to the proposed membrane receptor can result in the activation of numerous signaling cascades which can result in rapid opening of voltage-gated calcium channels and increase intracellular calcium (19). This may activate other growth regulatory pathways. (e.g. the Raf- MEK- MAPK- ERK described in skeletal muscle cells (18)). Such activation may result from increased calcium in normal colon and skeletal muscle cells. ERK can also enhance transcriptional activity of the VDR and non-genomic activation of PKC may stabilize VDR (via phosphorylation) (20). Non-genomic activation of these pathways may cooperate with the classical genomic pathway to transactivate VDR and elicit antiproliferative effects of calcitriol.
Antitumor Activity of Vitamin D
There are considerable data which indicate antitumor effects of vitamin D compounds in preclinical settings. Vitamin D compounds inhibit the growth and even kill cancer cells in vitro and in vivo and more recently the potentiating of the antitumor actions of a number of more “traditional” anticancer agents has been reported. In the Nkx3-1;Pten mutant mice, a putative model for prostate carcinogenesis calcitriol administration delays the development of prostate intraepithelial neoplasia (PIN); calcitriol has better antitumor activity when administered to mice with early-stage (PIN) rather than advanced stage prostate disease (21). In model systems of murine squamous cell carcinoma (SCC) (22), and human carcinomas arising in the prostate (23), lung (24), ovary (25), breast (26, 27), bladder (28) pancreas (29) and as well as neuroblastoma, calcitriol or calcitriol analogues have significant anticancer effects. Calcitriol and its derivatives act through the VDR to regulate differentiation, proliferation, apoptosis and angiogenesis.
Antiproliferative effects of calcitriol
Cell cycle perturbation appears to be central to calcitriol-mediated anti-proliferative activity in tumor cells. Progression through the cell cycle is regulated by cyclins, their association with cyclin dependent kinases (CDKs) and CDK inhibitors. Expression of the CKIs p21Waf1/Cip1 and p27Kip inhibits proliferation, in part by inducing G0/G1 cell cycle arrest. CDKN1A (encoding p21) and GADD45A contain a VDRE and are direct transcriptional targets of calcitriol-VDR. Vitamin D represses TYMS (encoding thymidylate synthetase) and TK1 (encoding thymidine kinase) (30) which are involved in DNA replication, and activates the INK4 family (31) of cyclin D-dependent kinase inhibitors, which mediate G1 cell cycle arrest; cyclin E-CDK2 and the Skp2 (S-phase kinase-associated protein 2) ubiquitin ligase, which targets CKIs to the proteasome, are downregulated (32, 33) by calcitriol. Calcitriol treatment also results in the repression of the proto-oncogene MYC (34).
Calcitriol has many indirect effects on cell cycle regulation through cross-talk with other pathways. Calcitriol treatment results in the upregulation of IGFBP3 (encoding insulin growth factor binding protein 3) and transforming growth factor-β (TGF-β)-SMAD3 signaling cascades and downregulates the epidermal growth factor receptor (EGFR) signaling pathway (35). While tumor cells treated with calcitriol are inhibited in progression through cell cycle, the molecular basis for this effect differs from one tumor cell type; no unifying hypothesis with regard to the exact mechanism of calcitriol-mediated cell cycle perturbation is possible.
Activation of the VDR by calcitriol can inhibit tumor cell proliferation by inducing differentiation in a variety of myeloid leukemia cell lines and freshly isolated leukemia cells (13). Such differentiation is dependent on the formation of activated VDR and PI3K complexes (36). In hematopoeitic progenitor cells, calcitriol inhibits differentiation through VDR-independent suppression of interleukin-12 (IL12) protein secretion and down-regulation of other co-stimulatory molecules (CD40, CD80, CD86). In head and neck, colon and prostate cell lines, administration of calcitriol or calcitriol analogues induces the expression of genes that are associated with the differentiated cell of origin (37, 38). Differentiation of colon cancer cells is associated with increased protein kinase C- (PKC) and JNK-dependent c-Jun activation (39) or differential regulation of the expression of inhibitor of DNA binding 1 and 2 (ID1 and ID2), which encode proteins that are transcriptional regulators of epithelial cell proliferation (ID2) and differentiation (ID1); the repression of ID2 may mediate the antiproliferative effects of calcitriol (40). Calcitriol promotes differentiation through the induction of CDH1 (encoding E-cadherin) in adenomatosis polyposis coli (APC)-mutated human colorectal cancer SW480 cells (41). CDH1 activation restrains cell growth by facilitating the translocation of β-catenin from the nucleus to the plasma membrane, hence inhibiting β-catenin-mediated transcription and allowing activated VDR to compete with β-catenin for transcription factor binding. There appears to be no cell-type specificity for calcitriol induction of differentiation in tumor cells.
Apoptosis
In addition to these antiproliferative effects, calcitriol modulates mediators of apoptosis. Calcitriol represses the expression of the anti-apoptotic, pro-survival proteins Bcl-2 and Bcl-XL and may induce the expression of pro-apoptotic proteins (Bax, Bak and Bad). Calcitriol may also directly activate caspase effector molecules. (41) Jiang et al. have recently reported that calcitriol destabilizes telomerase reverse transcriptase (TERT) mRNA, inducing apoptosis through telomere attrition and down-regulating telomerase activity (42). The diverse effects observed for calcitriol-mediated apoptosis complicate the development of a unifying mechanism(s) central to these pro-apoptotic activities.
Angiogenesis
Calcitriol inhibits the proliferation of endothelial cells in vitro and reduces angiogenesis in vivo (44-47). Vascular endothelial growth factor (VEGF)-induced endothelial cell tube formation and tumor growth are inhibited in vivo by calcitriol administration to mice with VEGF-overexpressing MCF-7 xenografts (48). Calcitriol can increase VEGF mRNA levels in vascular smooth muscle cells (49) and upregulate mRNA levels of the potent anti-angiogenic factor thrombospondin-1 (THSD1) in SW480-ADH human colon tumor cells (40). In SCC cells, calcitriol induces the angiogenic factor interleukin-8 (IL-8) (50) but in prostate cancer cells, calcitriol interrupts IL-8 signaling leading to inhibition of endothelial cell migration and tube formation (51). Significant inhibition of metastasis is observed in murine models of prostate and lung cancer treated with calcitriol; these effects may be based, at least in part, on anti-angiogenic effects (23, 24). Interestingly, in tumor-derived endothelial cells (TDECs), calcitriol induces apoptosis and cell cycle arrest; however, these effects are not seen in endothelial cells isolated from normal tissues or from Matrigel® (Matrigel®-derived endothelial cells) (44). Recently, Chung et al. (52) demonstrated that tumor-derived endothelial cells may be more sensitive to calcitriol due to novel epigenetic silencing of CYP24A1. Therefore, direct effects of calcitriol on endothelial cells may play a role in the calcitriol-mediated anti-tumor activity observed in animal models of cancer.
Preclinical Studies of Calcitriol in Combination Regimens
In vitro and in vivo analyses indicate that calcitriol acts synergistically with chemotherapeutic agents. Calcitriol potentiates the anticancer activity of platinum analogues (53, 54), taxanes (55) and DNA-intercalating agents (56). Optimal potentiation is seen when calcitriol is administered before or simultaneous with chemotherapy treatment; administration of calcitriol after the cytotoxic agent does not provide potentiation (55). The combination of calcitriol and cisplatin in SCC cells in vitro enhanced the apoptosis effects of calcitriol alone. The pro-apoptotic signaling molecule MEKK1 (mitogen-activated protein kinase kinase kinase 1), is up-regulated in both apoptotic and pre-apoptotic SCC cells treated with calcitriol (42), this up-regulation of MEKK1 was potentiated in combination with cisplatin treatment suggesting that calcitriol pretreatment commits cells to undergo apoptosis through specific molecular pathways and that this effect is enhanced when cells are treated with an additional genotoxic stimulus (53). Expression of the p53 homologue, p73, is enhanced by calcitriol treatment of several cell types; increased p73 levels enhance platinum analogues cytotoxicity in these cell types, providing yet another mechanism by which calcitriol may potentiate platinum analyogue cytotoxicity (56). Similar effects are seen in MCF-7 cells treated with the calcitriol analogue, ILX 23-7553 in combination with doxorubicin or ionizing radiation (57). In these studies, ILX 23-7553 enhanced doxorubicin cytotoxicity and blocked the induction of p53 expression. Enhanced anti-tumor activity with calcitriol and the taxane, paclitaxel is associated with a significant decrease in p21 expression, which sensitizes cells to both DNA damaging agents (e.g., cisplatin and doxorubicin) and microtubule disrupting agents (e.g., paclitaxel and docetaxel) (55). In SCC and PC-3 (prostate cancer) xenografts, pre-treatment with calcitriol results in an enhanced anti-tumor effect when treated in combination with paclitaxel. Similar results have also been observed in vivo with MCF-7 xenografts in which mice were treated with vitamin D3 analogues and paclitaxel. Calcitriol-mediated downregulation of COX-2 expression in prostate cancer cells leads to decreased prostaglandin activity, upregulation of 15-hydroxyprostaglandin dehydrogenase which degrade prostaglandins, and reduction of prostaglandin receptors (58). These findings support the rationale for clinical evaluation of a combination of calcitriol and non-steroidal anti-inflamatory drugs (NSAIDs) for prostate cancer therapy. Enhanced anti-tumor effects with calcitriol combination therapy offers the opportunity for clinical utilization of calcitriol across a number of tumor types where modest effects are observed with chemotherapy alone.
In addition to studies demonstrating that 1,25(OH)2 D3 potentiates the antitumor effects of cytotoxic, biologic agents and receptor tyrosine kinase inhibitors, there are also data indicating that the antitumor effects of this compound can be potentiated by agents that inhibit the breakdown of 1,25(OH)2 D3. Azole antagonists of the primary catabolic enzyme (CYP24A1) responsible for vitamin D breakdown enhance the antitumor effects of 1,25(OH)2 D3 in vitro and in vivo (59). Ketoconazole is the most readily available of such agents and it is of interest that this drug has significant utility in the treatment of men with prostate cancer in whom disease progression has occurred despite androgen deprivation (so-called “androgen independent” or “castration resistant” prostate cancer). The activity of ketoconazole in tumor cells (prostate and non-prostate) which are apparently unresponsive to androgens supports the hypothesis that there are extra-androgenic mechanisms underlying ketoconazole activity. There are more specific inhibitors of CYP24, both azoles as well as secosteroid cholecalciferol analogues. These agents have antitumor activity in in vitro and in vivo models and potentiate that antitumor activity of 1,25(OH)2 D3 (60, 61).
Epidemiologic Studies and Studies of Vitamin D Replacement Regimens
There are many epidemiologic studies consistent with the concept that higher vitamin D levels are associated with suppression of tumor development and growth.
Geography and Cancer
Numerous investigators have drawn attention to the association between residence in northern latitudes - latitudes where incident sun exposure and hence, natural vitamin D synthesis is limited – and the mortality from numerous epithelial and hematopoietic cancers. Giovannucci and colleagues analyzed more than 40,000 individuals in the Health Professionals Study (HPS) (5). These workers developed, and prospectively validated, an algorithm, based on diet, physical activity and body mass index, that permitted estimation of serum 25(OH)D3 levels. They then applied this algorithm to the HPS participants and estimated that a 25ng/ml increase in 25(OH)D3 level reduced cancer risk 15-50%.
Cholecalciferol Replacement Studies
Many studies of vitamin D replacement have been analyzed and few indicate that the recommended daily allowance (RDA) cholecalciferol supplementation (400IU per day) has any impact on cancer risk or mortality. However, many have argued that this RDA is too low. (62) The recently analyzed Women's Health Initiative failed to show any impact of cholecalciferol (400IU per day) + calcium replacement (1500mg per day) on colorectal cancer risk and showed that this replacement dose of cholecalciferol resulted in no change in the serum level of 25(OH)D3. (63) A recent randomized study of vitamin D (cholecalciferol 1100IU per day) among postmenopausal women indicated that cancer diagnosis was substantially reduced in the vitamin D replacement group. (64)
Vitamin D Proteins as Prognostic Markers
There are a limited number of studies which have examined molecular markers of vitamin D pathway proteins or genes and cancer prognosis. Bises and colleagues have shown that high grade colorectal cancers have a lower expression of CYP27B than low grade tumors (65). Mimori and colleagues have shown that CYP24 gene expression is increased in esophageal cancers compared to normal mucosa and have suggested that CYP24 may be an “oncogene” (66). These studies suggest that reduced synthesis and/or enhanced breakdown of vitamin D in situ may be associated with tumor development. There are no prospective studies which exam D pathway genes or proteins and cancer prognosis. Diagnosis of cancer during months in which vitamin D levels would be expected to be the lowest (winter/spring) as well as low vitamin D levels at the time of cancer diagnosis have been reported to be an indicator of poor prognosis in lung, colorectal and breast cancer (67, 68). No clear mechanism for such an association has been developed. Taken together, these and many other studies suggest that vitamin D may play an important role in the genesis and course of cancer in humans and are consistent with molecular and cellular data demonstrating a role for vitamin D signaling in the growth of human and animal tumors.
Therapeutic Applications of these Preclinical Data
Single Agent 1,25(OH)2 D3 (Calcitriol)
Phase I Studies and Toxicity
The discussion of the therapeutic potential of vitamin D compounds will focus on the use of calcitriol since the great majority of clinical data have been developed with this compound – primarily because it is readily available as an injectable (Calcijex® - Abbott Pharmaceuticals) or oral (Rocaltrol® – Roche Laboratories Inc.) formulation. As discussed below neither formulation is ideally suited for use in cancer therapy, however. There are considerable in vivo data indicating that high doses of calcitriol may have therapeutic potential in patients with cancer. However, most discussions of the role of vitamin D in cancer therapy conclude with the aphorism that calcitriol is too toxic to be administered to patients with cancer. Careful examination of actual clinical trials experience demonstrates that this is not true. Administration of oral calcitriol on a daily schedule (1.5-2.5mcg/day, weekly “dose intensity” ∼10.5-17.5mcg/week) is associated with a 20-30% frequency of hypercalcemia in men with prostate cancer as well as in individuals in whom calcitriol is administered to prevent osteoporosis (69, 70). However, continuous oral administration is not the schedule that has been used in preclinical studies in which clear antitumor effects have been seen; intermittent dosing regimens have been studied. Careful clinical trials have clearly shown that high dose, intermittent calcitriol is safe and feasible in patients with advanced cancer.
Our own studies have evaluated calcitriol administered by mouth daily for three days every week. In a phase I study 28mcg daily X3 days + dexamethasone 4mg daily X4 weekly were administered without toxicity in men with advanced prostate cancer, before this study was stopped because of unacceptable pharmacokinetic characteristics of the oral agent (see below). In this study a weekly “dose intensity” of calcitriol of 84mcg/week was achieved (71). We also conducted a phase one study of escalating doses of calcitriol (QDX3 weekly) + paclitaxel (80mg/kg weekly X4 weeks). In this study we were able to administer 38mcg daily X3 weekly for 4 weeks without limiting toxicity (72). Pharmacokinetic studies in these phase I trials demonstrated that calcitriol as Rocaltrol® was unsuitable for such high dose administration both because of inconvenience (38mcg requires the administration of 76 caplets) and pharmacokinetic “failure” (73, 74). Administration of higher and higher doses of this formulation does not lead to a proportional increase in serum levels or systemic exposure. While the mechanism of this “saturable absorption” is unclear, identical findings were noted by Beer and colleagues who have studied a once weekly oral regimen (74). It appears that the proportional increase in serum calcitriol with increasing dose is lost at doses of 16-20mcg. Because of the lack of a suitable preparation of oral calcitriol, a small biotechnology company, Novocea Pharmaceuticals, formed with the express purpose to develop a more suitable formulation. Their formulation, called DN-101, does have a linear relationship between dose and exposure up to doses 165mcg (75, 76). This agent has been tested in phase I, II and III trials (see below).
Fakih and colleagues have studied intravenous calcitriol (Calcijex, Roche Pharmaceutical Corporation). A phase I trial of weekly intravenous calcitriol + gefitinib was conducted based on preclinical data that calcitriol potentiates the antitumor effects of this EGF receptor tyrosine kinase (RTK) inhibitor (77). While this study failed to demonstrate potentiation of the RTK inhibitor, as assessed by changes in phospho-EGF-R and phosph-AKT in serially obtained skin biopsies, this trial did demonstrate that very high doses of calcitriol can be administered safely. The dose limiting toxicity of weekly intravenous calcitriol + gefitinib was grade 3 hypercalcemia at a dose of 98mcg/week. The phase II dose of this regimen is 77mcg weekly. While the criteria for dose limiting toxicity were met in this trial, it is noteworthy that the calcitriol AUC was higher at 77mcg than 98mcg and those patients with dose limiting toxicity at 98mcg all had diseases in which parathormone (PTH) or parathormone-related peptide (PTH-RP) was increased before calcitriol was given. These data indicate that further dose escalation of calcitriol in patients without a predisposition to develop hypercalcemia may be possible. Once the MTD of weekly intravenous calcitriol + gefitinib was determined, this trial continued to determine the MTD of calcitriol + gefitinib + dexamethasone. The component of the trial was based on our preclinical data that glucocorticoids potentiate the antitumor effects of calcitriol and block hypercalcemia. The MTD of weekly calcitriol + gefitinib + dexamethasone was 120mcg weekly. It is important to note that the systemic exposure of calcitriol following 98mcg is approximately 30ng.hr/24hr which is in the range of exposure we have reported in murine models in which calcitriol has clearcut antitumor activity (78).
Beer and colleagues have also studied high dose oral calcitriol. Using a weekly schedule they treated patients up to 2.6mcg/kg weekly (74). No dose limiting toxicity was noted, but pharmacokinetic studies showed a loss of proportional increase in exposure at doses higher than 0.5mcg/kg. These investigators concluded that a reasonable dose of oral calcitriol using this formulation was 0.5mcg/kg, weekly and continued the exploration of this regimen in prostate cancer (see below). Studies with DN-101 demonstrated that 45mcg weekly was safe and well tolerated, that 165mcg on week one, followed by 45mcg weekly produced no toxicity and that a linear relationship between dose administered and AUC was maintained up to 165mcg (75, 76).
Taken together these data indicate that:
high dose calcitriol therapy is possible and safe up to a MTD of at least 100mcg weekly intravenously and perhaps higher using the oral formulation (DN-101) developed by Novocea
with intravenous and oral administration (DN-101) systemic exposure of calcitriol in humans is similar to the exposure achieved in preclinical models in which distinct antitumor activity is seen
there is no commercially available oral preparation that is suitable for high dose oral administration
there is not a well defined MTD for oral calcitriol
there is no information regarding a biologically optimal dose of calcitriol
EB1089 (seocalcitol)
Few data exist exploring the administration of other vitamin D analogues as cancer therapy. Phase I and phase II studies of seocalcitol (EB-1089) an analogue of calcitriol have been conducted (79-81). EB-1089 was administered by mouth everyday and dose limiting toxicity was determined by hypercalciuria. Using this regimen and with a phase II dose determined in this manner no clinical activity of EB-1089 was seen in pancreatic cancer or hepatocellular carcinoma (80, 81). Hypercalciuria likely is an unnecessarily conservative dose-limiting toxicity in developing an agent for the treatment of patients with advanced cancer.
1 alpha vitamin D2
Wilding's group has carefully studied 1α hydroxyvitamin D2 (1αD2). They have completed phase I trials using a daily regimen as well as phase II trials in prostate cancer as a single agent and in combination with docetaxel (82, 83, 84). Only limited evidence of antitumor activity was seen, but, again, the daily regimen is likely to be a less active than an intermittent high dose schedule.
Inecalcitol
Inecalcitol is a novel vitamin D analogue (19 nor-, 14 epi-, 23-ync, 1,25 dihydroxy vitamin D3) which is being developed by Hybrigenics Corporation. The analogue appears to express less propensity to induce hypercalcemia while maintaining antitumor activity. A pilot trial presented at the meeting of the American Society of Clinical Oncology in May 2009 indicates that oral doses up to 1000 mcg daily + docetaxel are safe (85).
Paricalcitol
Paricalcitol is 19 nor, 1 alpha, 25 dihdroxy vitamin D2 and also has less potential for hypercalcemia than calcitriol. Paricalcitol, marketed as Zemplar® by Abbott Laboratories, appears to be superior to calcitriol in the therapy of secondary hyperparathyroidism and chronic renal failure. Patients receiving paricalcitol survive longer than those receiving calcitriol (86). Schwartz and colleagues have defined a 3× weekly intravenous regimen for men with prostate cancer. No clear responses were seen in this trial though in many men PSA reductions were seen (87).
Consideration of the spectrum of side effects of high dose, intermittent vitamin D analogues in cancer patients is in order. Dose-limiting hypercalcemia has been encountered only at doses ∼100mcg following intravenous administration; transient increase in serum calcium (11-13mg/dL) does occur 1-3 days after completion of a single or daily X3 schedule. However, only at doses achieving an AUC∼30ng.hr/ml has dose-limiting hypercalcemia been encountered (77). Hypercalciuria is universal following administration of high dose calcitriol. Studies both by our group and those of Beer's group fail to clearly demonstrate that dietary calcium restriction reduces hypercalciuria. Substantial dietary calcium restriction is difficult for cancer patients and then there are few consequences of hypecalciuria in cancer patients. Careful studies have revealed no deterioration of renal function in patients receiving high dose intermittent calcitriol for more than 12 months. Radiographic monitoring for urinary tract stones (ultrasound or CT) in our studies suggests that newly discovered urinary tract stones occur in 1-3% of patients (71, 72, 77).
While in a number of phase I and II trials of single agent calcitriol and other vitamin D analogues, partial response and PSA response (in prostate cancer) has been seen, effective clinical antitumor benefit is small. Is this worth emphasizing that “modest” single agent antitumor response is the rule for many new agents (e.g. bevacizumab, sorafinib, sunitinib)? In view of the many unresolved questions regarding the MTD, optimal biologic dose, optimal schedule and pharmaceutical concerns about the available vitamin D formulations, it is not surprising that limited antitumor activity has been seen in phase I and II trials. Another important concern is the uncertainty regarding the mechanism of calcitriol antitumor effects. As reviewed above, antitumor effects may be mediated by a number of direct antiproliferative and proapoptotic effects as well as effects directed against tumor-associated endothelial cells.
1,25(OH)2 D3 (Calcitriol) Combination Studies
Phase I Studies and Toxicity
Our preclinical studies and those of others clearly demonstrate that calcitriol in high doses potentiates the antitumor activity of a wide variety of cytotoxic agents. In fact, in our in vivo studies based on clonogenic assays we examined many classes of agents (Table 1) and found that calcitriol synergistically enhanced clonogenic cell kill when used with every class of agent except nitrosoureas. Further examination of the reasons for the lack of synergy with the nitrosurea, BCNU (bis chloroethylnitrosourea, Carmustine®) may shed additional light on the chemopotentiation activity of calcitriol. Based on data such as these, a number of phase I clinical trials of calcitriol in combination with cytotoxic agents have been completed. Interpretation of the results of phase I and II clinical trials are hampered by the same challenges which limit our knowledge with regard to the interpretation of studies of calcitriol used as a single agent: lack of clear delineation of an optimal biologic dose and limited data on the maximum tolerated dose of calcitriol. The following reviews these combination studies and points out the findings and limitations of these trials:
Table 1. Potentiation* of Cytotoxic Agents by Calcitriol.
| Synergistic Combinations | ||
| Platinums | Anthracyclines | |
| carboplatin | doxorubicin | |
| cisplatin | mitoxantrone | |
| Taxanes | Antimetabolites | |
| docetaxel | cytosine arabinoside | |
| paclitaxel | gemcitabine | |
| 5-fluorouracil | ||
| Topoisomerase inhibitors | ||
| irinotecan | ||
| etoposide | ||
| Antagonistic Combinations | ||
| carmustine (BCNU) |
potentiation was determined by in vivo clonogenic assay with the determination of synergy and antagonism according to the method of Chou and Talalay (Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55.
Taxanes
Docetaxel
Beer and colleagues have studied the combination of calcitriol + docetaxel with the intent of applying these studies to the treatment of men with advanced prostate cancer progressing despite castration, so-called androgen independent prostate cancer (AIPC) or castration resistant prostate cancer (CRPC). These investigators used the commercially available oral formulation of calcitriol (Rocaltrol®) and based on the finding that calcitriol + docetaxel were at least additively cytotoxic in the human prostate cell line, PC-3, they conducted a phase II trial of weekly docetaxel (36mg/sqm, weekly X6) on day 2 + their pragmatically defined phase II dose of calcitriol (0.5mcg/kg orally weekly) on day 1 (88, 89). No unusual toxicity was seen in this trial and PSA response (≥50% reduction, on 2 successive measurements maintained for >28 days) was seen in 30 of 37 patients (81%; 95% confidence interval [CI], 68% to 94%). Twenty-two patients (59%; 95% CI, 43% to 75%) had a confirmed > 75% reduction in PSA. Eight of the 15 patients with measurable disease (53%; 95% CI, 27% to 79%) had a confirmed partial response. Median time to progression was 11.4 months (95% CI, 8.7 to 14 months), and median survival was 19.5 months (95% CI, 15.3 months to incalculable). Overall survival at 1 year was 89% (95% CI, 74% to 95%). Treatment-related toxicity was generally similar to that expected with single-agent docetaxel. Pharmacokinetics of neither calcitriol nor docetaxel were affected by the presence of its companion drug in an exploratory substudy. These results were encouraging and provided important momentum which lead Novocea to develop a new formulation of calcitriol (DN-101) and to undertake two studies: first, a large randomized, double-blinded, “phase II” trial of docetaxel +/- DN-101(ASCENT I = AIPC Study of Calcitriol Enhancing Taxotere); the endpoint of this trial was PSA response. This trial was followed by a large randomized, double-blind, placebo-controlled phase III trial in which survival was the primary endpoint (ASCENT II). ASCENT I enrolled 250 patients. In ASCENT I, PSA response rates were 63% (DN-101) and 52% (placebo), p = .07. Patients in the DN-101 group had a hazard ratio for death of 0.67 (P = .04) in a secondary multivariate analysis that included baseline hemoglobin and performance status. Median survival was not reached for the DN-101 arm and was estimated to be 24.5 months, compared with 16.4 months for placebo. Grade 3/4 adverse events occurred in 58% of DN-101 patients and in 70% of placebo-treated patients (p = .07). The most common grade 3/4 toxicities were: [DN-101 versus placebo] neutropenia (10% v 8%), fatigue (8% v 16%), infection (8% v 13%), and hyperglycemia (6% v 12%). This study suggests that DN-101 treatment was associated with improved survival – but no improvement in PSA response. The addition of weekly DN-101 did not increase the toxicity of weekly docetaxel. These preliminary results were very encouraging and led to ASCENT II, a 900 patient randomized, double-blind, placebo-controlled phase III trial, in which survival was the endpoint. The goal of ASCENT II was to define the survival advantage of calcitriol treatment in combination with docetaxel with the goal to achieve FDA approval of this combination, if the results of ASCENT I were confirmed. Unfortunately, in designing ASCENT II, 2 issues were unaddressed which ultimately prove to be problematic in the interpretation of ASCENT II.
In the interval between the completion of the first, single institution, phase II study by Beer et al, in which weekly docetaxel + calcitriol indicated substantial activity as measured by PSA response rate (89), and the initiation of ASCENT II, two large randomized trials provided evidence that weekly docetaxel was inferior to every 3 week (q 3 week) docetaxel in men with AIPC and that q 3 week docetaxel therapy improved the survival in men with AIPC (90, 91). This led to FDA to approve docetaxel, 75mg/sqm q 3 weeks+ prednisone 10mg daily in AIPC on May 19, 2004. Since q 3 week docetaxel became the standard therapy for AIPC it was the requisite control arm in ASCENT II. ASCENT II which entered its first patient in January 2006 was designed as a randomized study comparing docetaxel (q3weeks, 75mg/sqm) + prednisone (QD, 10mg) + placebo vs docetaxel (weekly, 36mg/sqm) + prednisone (QD, 10mg) + calcitriol (DN-101, 0.5mcg/kg 1 day before docetaxel). This design violates one of the primary tenets of randomized trial design – that is seek to eliminate all variables between standard and experimental arms, except one.
The dose of calcitriol (DN-101) selected for ASCENT 1 and 2 was based on the results of the small phase II trial of Beer and colleagues. This dose was chosen based on the study of calcitriol in a different formulation, a formulation shown by both Beer et al and Muindi et al to be pharmaceutically inadequate. In addition, even at the present time, there are no data which define either the optimal or maximal dose of oral calcitriol. The 0.5mcg/kg weekly oral dose was a dose of convenience. A dose ∼ 77mcg (>1mcg/kg in a 70kg patient) calcitriol is required intravenously to achieve the AUC which is associated with antitumor effect in mice. The dose of calcitriol utilized in ASCENT 1 and 2 was 0.5mcg/kg by mouth. The bioavailability of calcitriol has never been studied in humans, but is approximately 60-75% in dogs (see below).
With these concerns in mind, perhaps it is not surprising that ASCENT 2 was halted in November 2007 when the data safety monitoring committee noted that the death rate in the investigational arm (weekly docetaxel + calcitriol + prednisone) was greater than in the standard therapy arm (q3week docetaxel + placebo + prednisone). Subsequent analysis of this trial through June 2008 indicates that all deaths in this study were due to progressive prostate cancer and there was not an excess of toxicity related to administration of calcitriol (personal communication, Novacea Pharmaceuticals). The result of ASCENT 2 is a discouraging finding in the quest to define a role for high dose calcitriol in cancer therapy. However, there are several unaddressed questions in the development of calcitriol as a cancer therapy. The negative findings in ASCENT 2 may be related to inappropriate trial design rather than failure of the overall concept.
Paclitaxel
We have shown that calcitriol + paclitaxel result in synergistic cell killing in vitro and in vivo in several model systems. We completed a phase I trial of weekly paclitaxel + calcitriol (oral, 38mcg QD X3). No limiting toxicity was seen in this study which was terminated when the unfavorable pharmacokinetics of oral calcitriol using this formulation was delineated (72).
Platinum Analogues
In vitro and in vivo, calcitriol produces synergistic antitumor activity with both cisplatin and carboplatin. Flaig et al and Beer et al have conducted clinical trials of oral calcitriol (0.5mcg/kg) weekly X4 and q 4weeks with carboplatin AUC=2 and AUC=6 respectively (92, 93). No unusual toxicity was seen and antitumor responses were documented. However, the antitumor effects were not clearly different than might have been expected with carboplatin alone. The dose of calcitriol employed was chosen based on prior clinical trials of Beer et al. In our laboratory, the extent of potentiation of cisplatin by calcitriol exceeds that seen with carboplatin. Cisplatin has not been studied in clinical trials in humans. Rassnick et al completed a phase I study in dogs with spontaneous tumors (primarily sarcomas and skin tumors). Cisplatin (60mg/sqm) and escalating doses of intravenous calcitriol were given every 4 weeks. The MTD of intravenous calcitriol was 3.75 mcg/kg calcitriol in combination with cisplatin, and hypercalcemia was the dose-limiting toxicity. The relationship between calcitriol dosage and both Cmax and AUC was linear. Calcitriol dosages >1.5 mcg/kg achieved Cmax ≥9.8 ng/mL and dosages >1.0 mcg/kg achieved AUC ≥45 nghr/mL. AUC at the MTD was greater than the AUC which in mice was associated with antitumor effects in a squamous cell model. Complete responses were seen in 5 of 12 dogs treated; this was higher than expected with cisplatin alone in such patients. However, this was a small trial and conclusions regarding any findings other than the safety of this regimen are hazardous (94). A phase I clinical trial in humans of calcitriol (intravenous) + docetaxel + cisplatin is underway (Ramnath, personal communication).
While there are preclinical data that would support the study of combinations of calcitriol and a number of other antitumor agents including antimetabolites (methotrexate, cytosine arabinoside, gemcitabine), anthracyclines and anthracenediones and topoisomerase inhibitors, no clinical trials of such combinations have been conducted.
Conclusion
Considerable data suggest that there is a role for vitamin D in cancer therapy and prevention. While the preclinical data are persuasive and the epidemiologic data intriguing, no well designed trial of optimal administration vitamin D as a cancer therapy has ever been conducted. Had there been the opportunity – and insight- to develop calcitriol as any other cancer drug, the following studies would have been completed:
definition of the MTD
definition of a phase II dose – as a single agent and in combination with cytotoxic agents
studies to define an optimal dose
conduct of phase II (probably randomized phase II) studies of calcitriol alone and chemotherapy +/- calcitriol
Then, randomized phase III trials would be conducted – and designed such that the only variable was the administration of calcitriol
Prerequisites 1-5 have not been completed for calcitriol. Preclinical data provide considerable rationale for continued development of vitamin D analogue based cancer therapies. Design of future studies should be informed by good clinical trials design principles – and the mistakes of the past – to delineate whether or not there is a role for vitamin D analogues in cancer therapy.
Acknowledgments
Financial Support: NCI grants CA067267, CA085142, CA095045 and P30 CA016056-32, as well as DOD grant PC040238.
References
- 1.Rubin D, Levij IS. Suppression by vitamins D2 and D3 of hamster cheek pouch carcinoma induced with 9,10-dimethyl-1,2-benzanthracene with a discussion of the role of intracellular calcium in the development of tumors. Pathol Microbiol (Basel) 1973;39(6):446–60. [PubMed] [Google Scholar]
- 2.Eisman JA, Martin TJ, MacIntyre I, Frampton RJ, Moseley JM, Whitehead R. 1,25-Dihydroxyvitamin D3 receptor in a cultured human breast cancer cell line (MCF 7 cells) Biochem Biophys Res Commun. 1980 Mar 13;93(1):9–15. doi: 10.1016/s0006-291x(80)80238-5. [DOI] [PubMed] [Google Scholar]
- 3.Eisman JA, Macintyre I, Martin TJ, Frampton RJ, King RJ. Normal and malignant breast tissue is a target organ for 1,25-(0H)2 vitamin D3. Clin Endocrinol (Oxf) 1980 Sep;13(3):267–72. doi: 10.1111/j.1365-2265.1980.tb01053.x. [DOI] [PubMed] [Google Scholar]
- 4.Abe E, Miyaura C, Sakagami H, Takeda M, Konno K, Yamazaki T, Yoshiki S, Suda T. Differentiation of mouse myeloid leukemia cells induced by 1 alpha,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1981 Aug;78(8):4990–4. doi: 10.1073/pnas.78.8.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannucci E, et al. Prospective study of predictors of vitamin D status and cancer incidence and mortality in men. J Natl Cancer Inst. 2006;98:451–459. doi: 10.1093/jnci/djj101. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz GG, Skinner HG. Vitamin D status and cancer: new insights. Curr Opin Clin Nutr Metab Care. 2007;10:6–11. doi: 10.1097/MCO.0b013e328011aa60. [DOI] [PubMed] [Google Scholar]
- 7.Garland CF, Comstock GW, Garland FC, Helsing KJ, Shaw EK, Gorham ED. Serum 25-hydroxyvitamin D and colon cancer: eight-year prospective study. Lancet. 1989 Nov 18;2(8673):1176–8. doi: 10.1016/s0140-6736(89)91789-3. [DOI] [PubMed] [Google Scholar]
- 8.Haussler MR, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz GG, Eads D, Rao A, Cramer SD, Willingham MC, Chen TC, Jamieson DP, Wang L, Burnstein KL, Holick MF, Koumenis C. Pancreatic cancer cells express 25-hydroxyvitamin D-1 alpha-hydroxylase and their proliferation is inhibited by the prohormone 25-hydroxyvitamin D3. Carcinogenesis. 2004 Jun;25(6):1015–26. doi: 10.1093/carcin/bgh086. Epub 2004 Jan 23. [DOI] [PubMed] [Google Scholar]
- 10.Cross HS. Extrarenal vitamin D hydroxylase expression and activity in normal and malignant cells: modification of expression by epigenetic mechanisms and dietary substances. Nutr Rev. 2007 Aug;65(8 Pt 2):S108–12. doi: 10.1111/j.1753-4887.2007.tb00334.x. [DOI] [PubMed] [Google Scholar]
- 11.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlberg C, Dunlop TW, Frank C, Vaisanen S. In: Vitamin D. 2. Feldman D, Pike JW, Glorieux FH, editors. Vol. 1. Elsevier Inc.; San Diego: 2005. [Google Scholar]
- 13.Muto A, et al. 1,25-Dihydroxyvitamin D3 induces differentiation of a retinoic acid-resistant acute promyelocytic leukemia cell line (UF-1) associated with expression of p21(WAF1/CIP1) and p27(KIP1) Blood. 1999;93:2225–2233. [PubMed] [Google Scholar]
- 14.Yanagisawa J, et al. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 1999;283:1317–1321. doi: 10.1126/science.283.5406.1317. [DOI] [PubMed] [Google Scholar]
- 15.Sinkkonen L, Malinen M, Saavalainen K, Vaisanen S, Carlberg C. Regulation of the human cyclin C gene via multiple vitamin D3-responsive regions in its promoter. Nucleic acids research. 2005;33:2440–2451. doi: 10.1093/nar/gki502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norman AW. Vitamin D receptor: new assignments for an already busy receptor. Endocrinology. 2006;147:5542–5548. doi: 10.1210/en.2006-0946. [DOI] [PubMed] [Google Scholar]
- 17.Nemere I, et al. Ribozyme knockdown functionally links a 1,25(OH)2D3 membrane binding protein (1,25D3-MARRS) and phosphate uptake in intestinal cells. Proc Natl Acad Sci U S A. 2004;101:7392–7397. doi: 10.1073/pnas.0402207101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morelli S, Buitrago C, Boland R, de Boland AR. The stimulation of MAP kinase by 1,25(OH)(2)-vitamin D(3) in skeletal muscle cells is mediated by protein kinase C and calcium. Molecular and cellular endocrinology. 2001;173:41–52. doi: 10.1016/s0303-7207(00)00435-4. [DOI] [PubMed] [Google Scholar]
- 19.Wali RK, Baum CL, Sitrin MD, Brasitus TA. 1,25(OH)2 vitamin D3 stimulates membrane phosphoinositide turnover, activates protein kinase C, and increases cytosolic calcium in rat colonic epithelium. The Journal of Clinical Investigation. 1990;85:1296–1303. doi: 10.1172/JCI114567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh JC, et al. Human vitamin D receptor is selectively phosphorylated by protein kinase C on serine 51, a residue crucial to its trans-activation function. Proc Natl Acad Sci U S A. 1991;88:9315–9319. doi: 10.1073/pnas.88.20.9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banach-Petrosky W, et al. Vitamin D inhibits the formation of prostatic intraepithelial neoplasia in Nkx3.1;Pten mutant mice. Clin Cancer Res. 2006;12:5895–5901. doi: 10.1158/1078-0432.CCR-06-1039. [DOI] [PubMed] [Google Scholar]
- 22.McElwain MC, et al. Antiproliferative effects in vitro and in vivo of 1,25-dihydroxyvitamin D3 and a vitamin D3 analog in a squamous cell carcinoma model system. Mol Cell Differ. 1995;3:31–50. [Google Scholar]
- 23.Getzenberg RH, et al. Vitamin D inhibition of prostate adenocarcinoma growth and metastasis in the Dunning rat prostate model system. Urology. 1997;50:999–1006. doi: 10.1016/S0090-4295(97)00408-1. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa K, Kawaura A, Kato S, Takeda E, Okano T. 1 alpha,25-Dihydroxyvitamin D(3) is a preventive factor in the metastasis of lung cancer. Carcinogenesis. 2005;26:429–440. doi: 10.1093/carcin/bgh332. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, et al. Growth suppression of ovarian cancer xenografts in nude mice by vitamin D analogue EB1089. Clin Cancer Res. 2005;11:323–328. [PubMed] [Google Scholar]
- 26.Colston KW, Chander SK, Mackay AG, Coombes RC. Effects of synthetic vitamin D analogues on breast cancer cell proliferation in vivo and in vitro. Biochemical pharmacology. 1992;44:693–702. doi: 10.1016/0006-2952(92)90405-8. [DOI] [PubMed] [Google Scholar]
- 27.Welsh J, et al. Vitamin D-3 receptor as a target for breast cancer prevention. The Journal of nutrition. 2003;133:2425S–2433S. doi: 10.1093/jn/133.7.2425S. [DOI] [PubMed] [Google Scholar]
- 28.Ma Yingyu, Yu Wei-Dong, Trump Donald L, Johnson Candace S. 1,25D3 enhances antitumor activity of gemcitabine and cisplatin in human bladder cancer models. doi: 10.1002/cncr.25059. submitted 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colston KW, James SY, Ofori-Kuragu EA, Binderup L, Grant AG. Vitamin D receptors and anti-proliferative effects of vitamin D derivatives in human pancreatic carcinoma cells in vivo and in vitro. British journal of cancer. 1997;76:1017–1020. doi: 10.1038/bjc.1997.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer HG, et al. Genetic signatures of differentiation induced by 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003;63:7799–7806. [PubMed] [Google Scholar]
- 31.Wang QM, Jones JB, Studzinski GP. Cyclin-dependent kinase inhibitor p27 as a mediator of the G1-S phase block induced by 1,25-dihydroxyvitamin D3 in HL60 cells. Cancer Res. 1996;56:264–267. [PubMed] [Google Scholar]
- 32.Jiang F, Li P, Fornace AJ, Jr, Nicosia SV, Bai W. G2/M arrest by 1,25-dihydroxyvitamin D3 in ovarian cancer cells mediated through the induction of GADD45 via an exonic enhancer. J Biol Chem. 2003;278:48030–48040. doi: 10.1074/jbc.M308430200. [DOI] [PubMed] [Google Scholar]
- 33.Li P, et al. p27(Kip1) stabilization and G(1) arrest by 1,25-dihydroxyvitamin D(3) in ovarian cancer cells mediated through down-regulation of cyclin E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2 ubiquitin ligase. J Biol Chem. 2004;279:25260–25267. doi: 10.1074/jbc.M311052200. [DOI] [PubMed] [Google Scholar]
- 34.Caligo MA, Cipollini G, Petrini M, Valentini P, Bevilacqua G. Down regulation of NM23.H1, NM23.H2 and c-myc genes during differentiation induced by 1,25 dihydroxyvitamin D3. Leukemia research. 1996;20:161–167. doi: 10.1016/0145-2126(95)00122-0. [DOI] [PubMed] [Google Scholar]
- 35.Yanagisawa J, et al. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 1999;283:1317–1321. doi: 10.1126/science.283.5406.1317. [DOI] [PubMed] [Google Scholar]
- 36.Hmama Z, et al. 1alpha,25-dihydroxyvitamin D(3)-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J Exp Med. 1999;190:1583–1594. doi: 10.1084/jem.190.11.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Zhang J, Studzinski GP. AKT pathway is activated by 1, 25-dihydroxyvitamin D3 and participates in its anti-apoptotic effect and cell cycle control in differentiating HL60 cells. Cell Cycle. 2006 Feb;5(4):447–51. doi: 10.4161/cc.5.4.2467. [DOI] [PubMed] [Google Scholar]
- 38.Aguilera O, Peña C, García JM, Larriba MJ, Ordóñez-Morán P, Navarro D, Barbáchano A, López de Silanes I, Ballestar E, Fraga MF, Esteller M, Gamallo C, Bonilla F, González-Sancho JM, Muñoz A. The Wnt antagonist DICKKOPF-1 gene is induced by 1alpha,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis. 2007 Sep;28(9):1877–84. doi: 10.1093/carcin/bgm094. [DOI] [PubMed] [Google Scholar]
- 39.Chen A, Davis BH, Bissonnette M, Scaglione-Sewell B, Brasitus TA. 1,25-Dihydroxyvitamin D(3) stimulates activator protein-1-dependent Caco-2 cell differentiation. J Biol Chem. 1999;274:35505–35513. doi: 10.1074/jbc.274.50.35505. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Garcia NI, et al. 1alpha,25-Dihydroxyvitamin D3 regulates the expression of Id1 and Id2 genes and the angiogenic phenotype of human colon carcinoma cells. Oncogene. 2005;24:6533–6544. doi: 10.1038/sj.onc.1208801. [DOI] [PubMed] [Google Scholar]
- 41.Palmer HG, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. The Journal of cell biology. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGuire TF, Trump DL, Johnson CS. Vitamin D(3)-induced apoptosis of murine squamous cell carcinoma cells. Selective induction of caspase-dependent MEK cleavage and up-regulation of MEKK-1. J Biol Chem. 2001;276:26365–26373. doi: 10.1074/jbc.M010101200. [DOI] [PubMed] [Google Scholar]
- 43.Jiang F, Bao J, Li P, Nicosia SV, Bai W. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down-regulation of telomerase. J Biol Chem. 2004;279:53213–53221. doi: 10.1074/jbc.M410395200. [DOI] [PubMed] [Google Scholar]
- 44.Chung I, Han G, Seshadri M, Gillard BM, Yu WD, Foster BA, Trump DL, Johnson CS. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009 Feb 1;69(3):967–75. doi: 10.1158/0008-5472.CAN-08-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernardi RJ, Johnson CS, Modzelewski RA, Trump DL. Antiproliferative effects of 1alpha,25-dihydroxyvitamin D(3) and vitamin D analogs on tumor-derived endothelial cells. Endocrinology. 2002;143:2508–2514. doi: 10.1210/endo.143.7.8887. [DOI] [PubMed] [Google Scholar]
- 46.Merke J, et al. Identification and regulation of 1,25-dihydroxyvitamin D3 receptor activity and biosynthesis of 1,25-dihydroxyvitamin D3. Studies in cultured bovine aortic endothelial cells and human dermal capillaries. The Journal of clinical investigation. 1989;83:1903–1915. doi: 10.1172/JCI114097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pendás-Franco N, García JM, Peña C, Valle N, Pálmer HG, Heinäniemi M, Carlberg C, Jiménez B, Bonilla F, Muñoz A, González-Sancho JM. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene. 2008 Jul 24;27(32):4467–77. doi: 10.1038/onc.2008.88. [DOI] [PubMed] [Google Scholar]
- 48.Mantell DJ, Owens PE, Bundred NJ, Mawer EB, Canfield AE. 1 alpha,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circulation research. 2000;87:214–220. doi: 10.1161/01.res.87.3.214. [DOI] [PubMed] [Google Scholar]
- 49.Cardus A, et al. 1,25-Dihydroxyvitamin D3 stimulates vascular smooth muscle cell proliferation through a VEGF-mediated pathway. Kidney Int. 2006;69:1377–1384. doi: 10.1038/sj.ki.5000304. [DOI] [PubMed] [Google Scholar]
- 50.Lin R, et al. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Mol Endocrinol. 2002;16:1243–1256. doi: 10.1210/mend.16.6.0874. [DOI] [PubMed] [Google Scholar]
- 51.Bao BY, Yao J, Lee YF. 1{alpha}, 25-dihydroxyvitamin D3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis. 2006 doi: 10.1093/carcin/bgl041. [DOI] [PubMed] [Google Scholar]
- 52.Chung I, et al. Epigenetic silencing of CYP24 in tumor-derived endothelial cells contributes to selective growth inhibition by calcitriol. J Biol Chem. 2007 doi: 10.1074/jbc.M608894200. [DOI] [PubMed] [Google Scholar]
- 53.Hershberger PA, et al. Cisplatin potentiates 1,25-dihydroxyvitamin D3-induced apoptosis in association with increased mitogen-activated protein kinase kinase kinase 1 (MEKK-1) expression. Mol Cancer Ther. 2002;1:821–829. [PubMed] [Google Scholar]
- 54.Moffatt KA, Johannes WU, Miller GJ. 1Alpha,25dihydroxyvitamin D3 and platinum drugs act synergistically to inhibit the growth of prostate cancer cell lines. Clin Cancer Res. 1999;5:695–703. [PubMed] [Google Scholar]
- 55.Hershberger PA, et al. Calcitriol (1,25-dihydroxycholecalciferol) enhances paclitaxel antitumor activity in vitro and in vivo and accelerates paclitaxel-induced apoptosis. Clin Cancer Res. 2001;7:1043–1051. [PubMed] [Google Scholar]
- 56.Ma Y, Yu WD, Hershberger PA, Flynn G, Kong RX, Trump DL, Johnson CS. 1alpha,25-Dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol Cancer Ther. 2008 Sep;7(9):3047–55. doi: 10.1158/1535-7163.MCT-08-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaudhry M, Sundaram S, Gennings C, Carter H, Gewirtz DA. The vitamin D3 analog, ILX-23-7553, enhances the response to adriamycin and irradiation in MCF-7 breast tumor cells. Cancer Chemother Pharmacol. 2001;47:429–436. doi: 10.1007/s002800000251. [DOI] [PubMed] [Google Scholar]
- 58.Krishnan AV, et al. Novel pathways that contribute to the anti-proliferative and chemopreventive activities of calcitriol in prostate cancer. J Steroid Biochem Mol Biol. 2007 doi: 10.1016/j.jsbmb.2006.12.051. [DOI] [PubMed] [Google Scholar]
- 59.Muindi JR, Yu WD, Ma Y, Engler KL, Kong RX, Trump DL, Johnson CS. CYP24A1 inhibition enhances the antitumor activity of calcitriol. doi: 10.1210/en.2009-1156. submitted, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lechner D, Manhardt T, Bajna E, Posner GH, Cross HS. A 24-phenylsulfone analog of vitamin D inhibits 1alpha,25-dihydroxyvitamin D(3) degradation in vitamin D metabolism-competent cells. J Pharmacol Exp Ther. 2007 Mar;320(3):1119–26. doi: 10.1124/jpet.106.115451. [DOI] [PubMed] [Google Scholar]
- 61.Kahraman M, Sinishtaj S, Dolan PM, Kensler TW, Peleg S, Saha U, Chuang SS, Bernstein G, Korczak B, Posner GH. Potent, selective and low-calcemic inhibitors of CYP24 hydroxylase: 24-sulfoximine analogues of the hormone 1alpha,25-dihydroxyvitamin D(3) J Med Chem. 2004 Dec 30;47(27):6854–63. doi: 10.1021/jm040129+. 1. [DOI] [PubMed] [Google Scholar]
- 62.Vieth R, Bischoff-Ferrari H, Boucher BJ, Dawson-Hughes B, Garland CF, Heaney RP, Holick MF, Hollis BW, Lamberg-Allardt C, McGrath JJ, Norman AW, Scragg R, Whiting SJ, Willett WC, Zittermann A. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr. 2007 Mar;85(3):649–50. doi: 10.1093/ajcn/85.3.649. [DOI] [PubMed] [Google Scholar]
- 63.Wactawski-Wende J, Kotchen JM, Anderson GL, Assaf AR, Brunner RL, O'Sullivan MJ, Margolis KL, Ockene JK, Phillips L, Pottern L, Prentice RL, Robbins J, Rohan TE, Sarto GE, Sharma S, Stefanick ML, Van Horn L, Wallace RB, Whitlock E, Bassford T, Beresford SA, Black HR, Bonds DE, Brzyski RG, Caan B, Chlebowski RT, Cochrane B, Garland C, Gass M, Hays J, Heiss G, Hendrix SL, Howard BV, Hsia J, Hubbell FA, Jackson RD, Johnson KC, Judd H, Kooperberg CL, Kuller LH, LaCroix AZ, Lane DS, Langer RD, Lasser NL, Lewis CE, Limacher MC, Manson JE, Women's Health Initiative Investigators Calcium plus vitamin D supplementation and the risk of colorectal cancer. N Engl J Med. 2006 Feb 16;354(7):684–96. doi: 10.1056/NEJMoa055222. Erratum in. [DOI] [PubMed] [Google Scholar]; N Engl J Med. 2006 Mar 9;354(10):1102. [Google Scholar]
- 64.Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk:results of a randomized trial. Am J Clin Nutr. 2007 Jun;85(6):1586–91. doi: 10.1093/ajcn/85.6.1586. [DOI] [PubMed] [Google Scholar]
- 65.Bises G, Kállay E, Weiland T, Wrba F, Wenzl E, Bonner E, Kriwanek S, Obrist P, Cross HS. 25-hydroxyvitamin D3-1alpha-hydroxylase expression in normal and malignant human colon. J Histochem Cytochem. 2004 Jul;52(7):985–9. doi: 10.1369/jhc.4B6271.2004. [DOI] [PubMed] [Google Scholar]
- 66.Mimori K, Tanaka Y, Yoshinaga K, Masuda T, Yamashita K, Okamoto M, Inoue H, Mori M. Clinical significance of the overexpression of the candidate oncogene CYP24 in esophageal cancer. Ann Oncol. 2004 Feb;15(2):236–41. doi: 10.1093/annonc/mdh056. [DOI] [PubMed] [Google Scholar]
- 67.Moan J, Porojnicu A, Lagunova Z, Berg JP, Dahlback A. Colon cancer: prognosis for different latitudes, age groups seasons in Norway. J Photochem Photobiol B. 2007 Dec 14;89(2-3):148–55. doi: 10.1016/j.jphotobiol.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Zhou W, Heist RS, Liu G, Asomaning K, Neuberg DS, Hollis BW, Wain JC, Lynch TJ, Giovannucci E, Su L, Christiani DC. Circulating 25-hydroxyvitamin D levels predict survival in early-stage non-small-cell lung cancer patients. J Clin Oncol. 2007 Feb 10;25(5):479–85. doi: 10.1200/JCO.2006.07.5358. [DOI] [PubMed] [Google Scholar]
- 69.Osborn JL, Schwartz GG, Smith DC, Bahnson R, Day R, Trump DL. Phase II trial of oral 1,25-dihydroxyvitamin D (calcitriol) in hormone refractory prostate cancer. Urologic Oncology: Seminars and Original Investigations. 1995 September-October;1(5):195–198. doi: 10.1016/1078-1439(95)00061-5. [DOI] [PubMed] [Google Scholar]
- 70.Gallagher JC, Goldgar D. Treatment of postmenopausal osteoporosis with high doses of synthetic calcitriol. A randomized controlled study. Ann Intern Med. 1990 Nov 1;113(9):649–55. doi: 10.7326/0003-4819-113-9-649. [DOI] [PubMed] [Google Scholar]
- 71.Trump DL, Hershberger PA, Bernardi RJ, Ahmed S, Muindi J, Fakih M, Yu WD, Johnson CS. Anti-tumor activity of calcitriol: pre-clinical and clinical studies. J Steroid Biochem Mol Biol. 2004 May;89-90(1-5):519–26. doi: 10.1016/j.jsbmb.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 72.Muindi JR, Peng Y, Potter DM, Hershberger PA, Tauch JS, Capozzoli MJ, Egorin MJ, Johnson CS, Trump DL. Pharmacokinetics of high-dose oral calcitriol: results from a phase 1 trial of calcitriol and paclitaxel. Clin Pharmacol Ther. 2002 Dec;72(6):648–59. doi: 10.1067/mcp.2002.129305. [DOI] [PubMed] [Google Scholar]
- 73.Muindi JR, Potter DM, Peng Y, Johnson CS, Trump DL. Pharmacokinetics of liquid calcitriol formulation in advanced solid tumor patients: comparison with caplet formulation. Cancer Chemother Pharmacol. 2005 Nov;56(5):492–6. doi: 10.1007/s00280-005-1015-2. Epub 2005 May 26. [DOI] [PubMed] [Google Scholar]
- 74.Beer TM, Munar M, Henner WD. A Phase I trial of pulse calcitriol in patients with refractory malignancies: pulse dosing permits substantial dose escalation. Cancer. 2001 Jun 15;91(12):2431–9. [PubMed] [Google Scholar]
- 75.Beer TM, Javle M, Lam GN, Henner WD, Wong A, Trump DL. Pharmacokinetics and tolerability of a single dose of DN-101, a new formulation of calcitriol, in patients with cancer. Clin Cancer Res. 2005 Nov 1;11(21):7794–9. doi: 10.1158/1078-0432.CCR-05-0552. [DOI] [PubMed] [Google Scholar]
- 76.Beer TM, Javle MM, Ryan CW, Garzotto M, Lam GN, Wong A, Henner WD, Johnson CS, Trump DL. Phase I study of weekly DN-101, a new formulation of calcitriol, in patients with cancer. Cancer Chemother Pharmacol. 2007 Apr;59(5):581–7. doi: 10.1007/s00280-006-0299-1. [DOI] [PubMed] [Google Scholar]
- 77.Fakih MG, Trump DL, Muindi JR, Black JD, Bernardi RJ, Creaven PJ, Schwartz J, Brattain MG, Hutson A, French R, Johnson CS. A phase I pharmacokinetic and pharmacodynamic study of intravenous calcitriol in combination with oral gefitinib in patients with advanced solid tumors. Clin Cancer Res. 2007 Feb 15;13(4):1216–23. doi: 10.1158/1078-0432.CCR-06-1165. [DOI] [PubMed] [Google Scholar]
- 78.Muindi JR, Modzelewski RA, Peng Y, Trump DL, Johnson CS. Pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 in normal mice after systemic exposure to effective and safe antitumor doses. Oncology. 2004;66(1):62–6. doi: 10.1159/000076336. [DOI] [PubMed] [Google Scholar]
- 79.Gulliford T, English J, Colston KW, Menday P, Moller S, Coombes RC. A phase I study of the vitamin D analogue EB 1089 in patients with advanced breast and colorectal cancer. Br J Cancer. 1998 Jul;78(1):6–13. doi: 10.1038/bjc.1998.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Evans TR, Colston KW, Lofts FJ, Cunningham D, Anthoney DA, Gogas H, de Bono JS, Hamberg KJ, Skov T, Mansi JL. A phase II trial of the vitamin D analogue Seocalcitol (EB1089) in patients with inoperable pancreatic cancer. Br J Cancer. 2002 Mar 4;86(5):680–5. doi: 10.1038/sj.bjc.6600162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dalhoff K, Dancey J, Astrup L, Skovsgaard T, Hamberg KJ, Lofts FJ, Rosmorduc O, Erlinger S, Bach Hansen J, Steward WP, Skov T, Burcharth F, Evans TR. A phase II study of the vitamin D analogue Seocalcitol in patients with inoperable hepatocellular carcinoma. Br J Cancer. 2003 Jul 21;89(2):252–7. doi: 10.1038/sj.bjc.6601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu G, Oettel K, Ripple G, Staab MJ, Horvath D, Alberti D, Arzoomanian R, Marnocha R, Bruskewitz R, Mazess R, Bishop C, Bhattacharya A, Bailey H, Wilding G. Phase I trial of 1alpha-hydroxyvitamin d(2) in patients with hormone refractory prostate cancer. Clin Cancer Res. 2002 Sep;8(9):2820–7. [PubMed] [Google Scholar]
- 83.Liu G, Wilding G, Staab MJ, Horvath D, Miller K, Dresen A, Alberti D, Arzoomanian R, Chappell R, Bailey HH. Phase II study of 1alpha-hydroxyvitamin D(2) in the treatment of advanced androgen-independent prostate cancer. Clin Cancer Res. 2003 Sep 15;9(11):4077–83. [PubMed] [Google Scholar]
- 84.Attia S, Eickhoff J, Wilding G, McNeel D, Blank J, Ahuja H, Jumonville A, Eastman M, Shevrin D, Glode M, Alberti D, Staab MJ, Horvath D, Straus J, Marnocha R, Liu G. Randomized, double-blinded phase II evaluation of docetaxel with or without doxercalciferol in patients with metastatic, androgen-independent prostate cancer. Clin Cancer Res. 2008 Apr 15;14(8):2437–43. doi: 10.1158/1078-0432.CCR-07-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Medioni J, Deplanque G, Maurina T, Ferrero JM, Rodier JM, Raymond E, Allyon J, Kalla S, Dufour-Lamartinie JF, Oudar S. Dose finding and safety analysis of inecalcitol in combination with docetaxel-prednisone regimen in hormone-refractory prostate cancer (HRPC) patients. Proceedings ASCO. 2009 [Google Scholar]
- 86.Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003 Jul 31;349(5):446–56. doi: 10.1056/NEJMoa022536. [DOI] [PubMed] [Google Scholar]
- 87.Schwartz GG, Hall MC, Stindt D, Patton S, Lovato J, Torti FM. Phase I/II study of 19-nor-1alpha-25-dihydroxyvitamin D2 (paricalcitol) in advanced, androgen-insensitive prostate cancer. Clin Cancer Res. 2005 Dec 15;11(24 Pt 1):8680–5. doi: 10.1158/1078-0432.CCR-05-1237. [DOI] [PubMed] [Google Scholar]
- 88.Beer TM, Eilers KM, Garzotto M, Egorin MJ, Lowe BA, Henner WD. Weekly high-dose calcitriol and docetaxel in metastatic androgen-independent prostate cancer. J Clin Oncol. 2003 Jan 1;21(1):123–8. doi: 10.1200/jco.2003.05.117. [DOI] [PubMed] [Google Scholar]
- 89.Beer TM, Ryan CW, Venner PM, Petrylak DP, Chatta GS, Ruether JD, Redfern CH, Fehrenbacher L, Saleh MN, Waterhouse DM, Carducci MA, Vicario D, Dreicer R, Higano CS, Ahmann FR, Chi KN, Henner WD, Arroyo A, Clow FW, ASCENT Investigators Double-blinded randomized study of high-dose calcitriol plus docetaxel compared with placebo plus docetaxel in androgen-independent prostate cancer: a report from the ASCENT Investigators. J Clin Oncol. 2007 Feb 20;25(6):669–74. doi: 10.1200/JCO.2006.06.8197. [DOI] [PubMed] [Google Scholar]
- 90.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004 Oct 7;351(15):1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 91.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA. TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004 Oct 7;351(15):1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 92.Flaig TW, Barqawi A, Miller G, Kane M, Zeng C, Crawford ED, Glodé LM. A phase II trial of dexamethasone, vitamin D, and carboplatin in patients with hormone-refractory prostate cancer. Cancer. 2006 Jul 15;107(2):266–74. doi: 10.1002/cncr.21982. [DOI] [PubMed] [Google Scholar]
- 93.Beer TM, Garzotto M, Katovic NM. High-dose calcitriol and carboplatin in metastatic androgen-independent prostate cancer. Am J Clin Oncol. 2004 Oct;27(5):535–41. doi: 10.1097/01.coc.0000136020.27904.9c. [DOI] [PubMed] [Google Scholar]
- 94.Rassnick KM, Muindi JR, Johnson CS, Balkman CE, Ramnath N, Yu WD, Engler KL, Page RL, Trump DL. In vitro and in vivo evaluation of combined calcitriol and cisplatin in dogs with spontaneously occurring tumors. Cancer Chemother Pharmacol. 2008 Feb 2; doi: 10.1007/s00280-008-0678-x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]