

Abstract
Faster and more efficient approaches for radiolabeling biomolecules with short-lived 18F are in dire need. Herein we report a new 18F-labeled prosthetic group containing an acetylene function that permits the labeling of biomolecules via click chemistry. This template, propargyl 4-[18F]fluorobenzoate ([18F]PFB) was synthesized from a quaternary salt precursor in decay-corrected radiochemical yields of 58 ± 31%. Several model compounds containing an azide moiety—benzyl azide, two lysine derivatives and a transglutaminase-reactive peptide—were labeled using [18F]PFB via a click reaction in decay-corrected radiochemical yields of 88 ± 4%, 79 ± 33%, 75 ± 5%, and 37 ± 31%, respectively. Our results suggest that the novel agent [18F]PFB is a potentially useful template for the 18F-labeling of biomolecules via click chemistry.
Keywords: Fluorine-18, click reaction, positron emission tomography, defluorination
INTRODUCTION
Cutting edge molecular imaging techniques are increasingly becoming an integral part of the drug discovery process [1-3]. For example, microdosing is a new and promising approach that is used to obtain human pharmacokinetic data for investigational drugs that are in their early stage of development [4, 5]. Because of its potential for providing multi-organ, quantitative distribution and elimination data, molecular imaging should play a critical role in microdosing and some reports have already appeared in this regard [6-10].
Imaging techniques such as single photon emission computed tomography (SPECT) and positron emission tomography (PET) that utilize compounds labeled with radionuclides are particularly attractive because of their of high sensitivity and the fact that only miniscule amounts of labeled molecules need to be administered. PET is a tomographic imaging modality with excellent quantitation capabilities. Furthermore, it is a functional imaging method in that it can detect very early biochemical and physiological alterations even before the onset of anatomical changes [11]. Although there are a number of radionuclides that decay with the emission of positrons, and therefore potentially can be used for labeling biomolecules, 18F is by far the most widely available and versatile radionuclide utilized for PET applications [12].
The reaction conditions to introduce 18F onto molecules are often harsh, and this has necessitated the development of many prosthetic groups for the radiolabeling of sensitive molecules such as peptides and proteins with 18F [13, 14]. To-date, N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB), which was originally developed by us [15-17], is the most widely used agent for this purpose [18, 19]. The synthesis of [18F]SFB involves three steps, after which peptide conjugation must be performed, and in some cases, extra steps may be necessary to prevent the modification of pharmacologically sensitive functionalities on the peptide. Alternative strategies have been developed to overcome this problem [20-25]; however, none are ideal.
Recently, a two step strategy to label peptides with 18F has been developed by Marik and Sutcliffe utilizing click chemistry [26]. Click chemistry, so termed for its efficiency, involves the joining together of two molecules via heteroatom links utilizing the Huisgen 1,3-dipolar cycloaddition between an alkyne and an azide facilitated by copper catalysis [27, 28]. In their work, Marik and Sutcliffe generated T-[18F]fluoroalkynes in one step from tosylate precursors and conjugated them to peptides decorated with an azido function via click chemistry. A similar strategy but with the azide function on the 18F-labeled agent and the alkyne moiety on the peptides has been reported subsequently [29]. Sirion et al. have developed 18F-labeled alkynes containing a PEG spacer that are useful in the 18F-labeling of biomolecules via click chemistry [30]. Recently, a prosthetic group similar to this was employed to label a peptide containing the arginine-glycine-aspartic acid (RGD) motif and the labeled peptide was evaluated in vivo using microPET [31]. A common feature of all of these studies is that the 18F label was attached to an aliphatic carbon on the prosthetic group.
The goal of the current study was to develop a 18F-labeled prosthetic group for click chemistry conjugation with the 18F label attached to an aromatic carbon. Our rationale was to hopefully exploit the fact that the carbon-halogen bond strength is generally higher when the halogen is attached to a sp2 than to a sp3 hybridized carbon [32]. Thus, compounds in which fluorine is attached to a sp2 carbon are expected to be more inert to in vivo defluorination than those with the fluorine attached a sp3 carbon. Concordant with this, bone uptake of 18F, an indicator of defluorination, generally has been lower for compounds in which the fluorine label is on the aromatic ring rather than when it is attached to an aliphatic carbon [33-37]. Also, bone uptake of radioactivity was minimal for compounds containing a 4-[18F]fluorobenzoyl moiety [19, 38-42]. For this reason, we chose the propargyl ester of 4-fluorobenzoic acid as our target compound. The selection of this moiety was further motivated by the fact that prior studies have documented the rapid clearance from normal tissues of the potential catabolite 4-fluorobenzoic acid [43, 44] and its glycine conjugate (hippuric acid) [45, 46], which should help decrease normal tissue background after metabolism of labeled tracers. It should be pointed out that while our work was in progress, the development of a click labeling agent with 18F on an aromatic carbon has been reported [47]; however, the agent was derived from [18F]SFB in a process involving a total of four radiochemical steps.
Herein we report the synthesis of propargyl 4-fluorobenzoate (PFB, 1; Scheme 1) and the corresponding quaternary ammonium triflate precursor. Synthesis of 18F-labeled ester 1 was accomplished in a single step by the nucleophilic displacement of the quaternary function from the latter. The usefulness of the alkyne-containing prosthetic group was determined by evaluating by its click conjugation to model compounds including a peptide.
Scheme 1.
Synthesis of unlabeled and 18F-labeled propargyl 4-fluorobenzoate (1) and its quaternary salt precursor, 4-propargyloxycarbonyl N, N, N-trimethylanilinium trifluoromethanesulfonate (3).
EXPERIMENTAL PROCEDURES
General
All chemicals were purchased from Sigma-Aldrich unless otherwise noted and used without further purification. The reagent 6-azidohexanoic acid was prepared by modifying procedures reported for its synthesis [48, 49] and the product was characterized by NMR and mass spectrometry. S-H-Lys(Boc)-OtBu.HCl and silica-supported DCC were procured from Bachem (Torrrance, CA) and Silicycle (Quebec, Canada), respectively. Appropriately protected amino acids used in the peptide synthesis were obtained from Applied Biosystems (Foster City, CA) or from Bachem (Torrance, CA). Reagents for solid-phase peptide synthesis such as HBTU, DIEA and NMP were obtained from Applied Biosystems.
Aqueous [18F]fluoride was produced using the 18O(p, n)18F reaction by cyclotron irradiation of [18O]H2O in a silver target (1.5 mL). Except in a few cases, for which [18F]fluoride activity from dedicated runs was used, most of the labeling reactions reported herein were performed using the first or second wash of the target with regular deionized water (0.3 to 1.5 mL) following 18F runs for clinical level [18F]FDG production. In some cases, the activity was transferred directly from the cyclotron target bypassing the Teflon lines in place for delivering [18F]fluoride for clinical [18F]FDG synthesis. For one run, [18F]fluoride activity was obtained from PETNET Solutions (Raleigh-Durham Pharmacy). The [18F]fluoride activity (5-37 mCi) was mixed with a solution of Kryptofix® and K2CO3 (10 mg Kryptofix® in 1 mL acetonitrile and 1 mg K2CO3 in 5 :L water). The water from this mixture was removed by repeated (2-3 times) azeotropic evaporation with acetonitrile, and the radioactivity was resolubilized in 100 :L of acetonitrile.
Aluminum-backed sheets (Silica gel 60 F254) were used for analytical TLC, and normal-phase column chromatography was performed using silica gel 60, both obtained from EM Science (Gibbstown, NJ). Preparative thick layer chromatography was performed using 20 Η 20 cm, 1000 :m plates (Whatman, Clifton, NJ). Before applying the sample, the plates were run in ethyl acetate to remove any adsorbed impurities. Radio-TLC was analyzed using a System 200 Imaging Scanner (BioScan, Washington, DC) and in some cases, the sheets also were cut into strips and then counted using an automated gamma counter (LKB 1282, Wallac, Finland). Solid-phase peptide syntheses were carried out on a Perkin Elmer Applied Biosystems Model 433A automated peptide synthesizer employing Fast-Moc™ chemistry with HBTU activation of carboxyl groups. Two systems were used for performing high-pressure liquid chromatography. For radiochromatography, a Beckman System Gold HPLC equipped with a Model 126 programmable solvent module, a Model 166 NM variable wavelength detector, a Model 170 radioisotope detector, and a Model 406 analogue interface module was used. HPLC of unlabeled compounds and semi-preparative HPLC were performed using a Waters Model Delta 600 semi-preparative system with a Model 600 controller and a Model 2487 dual wavelength absorbance detector; data were acquired using Millenium software. Reversed-phase chromatography was performed using Waters XTerra C18 columns (analytical 4.6 H 250 mm, 5:; semi-preparative 19 H 150 mm). Unless indicated otherwise, the analytical column was eluted at a flow rate of 1 mL/min with a gradient consisting of 0.1% TFA in both water (solvent A) and acetonitrile (solvent B): the percent of solvent B was maintained at 10 for 5 min, then increased linearly to 100 in 30 min, and held at 100 for 5 min (standard gradient). The standard gradient conditions for most semi-preparative HPLC runs was the same as above except that a flow rate of 8 mL/min was used. A Zorbax Eclipse XDB-C18 column (4.6 H 250 mm, 5 :m; Agilent Technologies, Santa Clara, CA) was used for analyzing click reactions of peptide with the [18F]1. In this case, the column was eluted at a flow rate of 1 mL/min with a gradient consisting of 40 mM ammonium aceate, pH 4.73 (solvent A) and acetonitrile (solvent B); the percent of solvent B was kept at 5 for 5 min, then increased linearly to 100 over 30 min and held at 100 for 5 min. Infrared spectra were obtained on a Nicolet Avatar E.S.P. FT-IR spectrometer. Proton NMR spectra were obtained on a Varian Mercury 300 spectrometer. Chemical shifts are reported in δ units; solvent peaks were referenced appropriately. Mass spectra were obtained on a JEOL SX-102 high resolution mass spectrometer (FAB and EI), on an Applied Biosystems Voyager DE Pro (MALDI) or on a Shimadzu QP2010 GC/MS.
Propargyl 4-fluorobenzoate (1)
Method A
Propargyl alcohol (61.3 μL; 1.05 mmol) was added to a mixture of DCC (206 mg; 1.00 mmol), 4-fluorobenzoic acid (148 mg; 1.05 mmol) and 4-N,N-dimethylaminopyridine (~1 mg) in 4 mL of anhydrous ethyl acetate in a flame-dried, argon-purged flask. The reaction mixture was stirred at room temperature overnight, and the precipitated DCU was filtered through a fritted funnel. The solvent from the filtrate was evaporated and the crude product was purified by preparative TLC using 10% ethyl acetate in hexanes to yield 32 mg (0.18 mmol, 18%) of a white solid (1).
Method B
A mixture of propargyl alcohol (2 mL), triethylamine (1.5 mL), and 4-fluorobenzoyl chloride (0.67 mL; 15.61 mmol) was stirred at room temperature for 1.5 h. The reaction mixture was partitioned between 50 mL each of water and ethyl acetate and the aqueous layer was extracted twice with ethyl acetate. The combined ethyl acetate layers were washed with brine, dried with sodium sulfate and concentrated to yield 907 mg (5.09 mmol; 90.7%) of 1: TLC 1:3 ethyl acetate:hexanes, Rf 0.28. HPLC tR = 23.6 min. 1H NMR (CDCl3) δ 2.52 (t, 1H, J = 2.4 Hz), 4.92 (d, 2H, J = 2.4 Hz), 7.0-7.2 (m, 2H), 8.0-8.2 (m, 2H). MS (EI+) m/z: 178.11 (M+), 149.11, 133.12, 123.10, 122.08, 95.07. HRMS (EI+) calcd for C10H7FO2 (M+): 178.0430. Found: 178.0428 ± 0.0003 (n = 2).
Propargyl 4-dimethylaminobenzoate (2)
Propargyl alcohol (2 mL) was added to 4-N,N-dimethylaminobenzoyl chloride (367 mg; 2.00 mmol) in 1 mL of dry methylene chloride in a flame-dried, argon-purged flask followed by 552 μL of triethylamine (4.0 mmol). The mixture was stirred at room temperature for 10-15 min and partitioned between ethyl acetate (50 mL) and water (25 mL). The aqueous layer was further extracted twice with ethyl acetate and the pooled ethyl acetate solutions were washed once with brine and then dried with sodium sulfate. Evaporation of ethyl acetate from the filtrate gave 315 mg (1.5 mmol, 77.5%) of an off-white solid (1.5 mmol; 77.5%), which was used in the next step without further purification: TLC 0.1% triethylamine in 1:3 ethyl acetate:hexanes Rf = 0.36. IR (ATR) ν (cm-1) 3232.63 (s), 2917.68 (br w), 2863.80 (w), 2821.97 (w), 2116.99 (m), 1690.84 (vs), 1607.49 (s), 1529.91 (s). 1H NMR (CDCl3) δ 2.48 (t, 1H, J = 2.3 Hz), 3.04 (s, 6H), 4.87 (d, 2H, J = 2.4), 6.64 (d, 2H, J = 9.0 Hz), 7.93 (d, 2H, J = .0 Hz). MS (EI+), m/z: 203.09 (M+), 174.09, 164.07, 148.07. HRMS (EI+) calcd for C12H13NO2 (M+): 203.0946. Found: 203.0941 ± 0.0005 (n = 4).
4-Propargyloxycarbonyl N, N, N-trimethylanilinium trifluoromethanesulfonate (3)
Methyl trifluoromethanesulfonate (295 μL; 2.6 mmol) was added to a stirred solution of 2 (265 mg; 1.3 mmol) in 2 mL anhydrous methylene chloride. The mixture was stirred at room temperature for 1.5 h and the methylene chloride was evaporated with a stream of argon. The residual solid was dissolved in 1 mL ethyl acetate and the product was precipitated with the addition of 25 mL diethyl ether, and then filtered, washed with ether, and dried to obtain 390 mg (1.06 mmol; 82%) of a solid: HPLC tR = 6.8 min. IR (ATR) ν (cm-1) 3239.92 (s), 3079.07 (w), 2124.95 (w), 1727.65 (s), 1608.05 (w). 1H NMR (CD3CN) δ 2.86 (t, 1H, J = 2.6 Hz), 3.56 (s, 9H), 4.95 (d, 2H, J = 2.6), 7.90 (d, 2H, J = 9.5 Hz), 8.21 (d, 2H, J = 9.5 Hz). MS (FAB+), m/z: 218.10 (M+), 203.07, 180.10. HRMS (FAB+) calcd for C13H16NO2 (M+): 218.1181. Found: 218.1190 ± 0.0001 (n = 4).
(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl 4-fluorobenzoate (4)
Copper sulfate (0.4 M, 50 :L) and sodium ascorbate (50:L 0.6 M) were added to a solution of benzyl azide (19.7 mg, 0.14 mmol, Alfa Aesar) and 1 (57 mg, 0.32 mmol) in 0.4 mL of tert-butanol and the mixture was stirred vigorously at room temperature for 1 h. The reaction mixture was partitioned between 5 mL each of ethyl acetate and water and the aqueous layer was extracted twice with ethyl acetate. The combined organic layer was dried over sodium sulfate, filtered and the filtrate evaporated. The crude product was purified by preparative thick layer chromatograpy using 1:3 ethyl acetate:hexanes to yield 27 mg (0.087 mmol, 58.7%) of a white powder: TLC 1:1 ethyl acetate:hexanes, Rf = 0.37. HPLC tR = 24.7 min. 1H NMR (CDCl3) δ 5.42 (s, 2H), 5.52 (s, 2H), 7.02 – 7.12 (m, 2H), 7.23 – 7.32 (m, 2H), 7.32 – 7.42 (m, 3H), 7.60 (s, 1H), 9.97 – 8.07 (m, 2H). MS (FAB+) m/z: 312.11 (MH+), 307.09, 289.08. HRMS (FAB+) calcd for C17H14FN3O2 (M+): 311.1070. Found: 311.1073 ± 0.0008 (n = 3).
Tert-butyl 5-(tert-butoxycarbonyl)-2-(4-azidobenzamido)pentylcarbamate (5)
A mixture of H-Lys(Boc)-OtBu·HCl (63 mg; 0.187 mmol)), 4-azidobenzoic acid (TCI, 97 mg; 0.373 mmol), and DCC on silica support (Silicycles, 500 mg; 0.560 mmol) in 2.5 mL anhydrous methylene chloride was stirred at room temperature under argon overnight. The reaction mixture was filtered and the solvent from the filtrate evaporated. The crude mixture was purified by preparative thin layer chromatography using 1:1 ethyl acetate:hexanes to yield 64.6 mg (0.14 mmol; 77.3%) of a foam: TLC 1:1 ethyl acetate:hexanes, Rf = 0.60. 1HNMR (CDCl3) δ 1.39 (s, 9H), 1.48 (s, 9H), 1.50 (m, 4H), 1.75 (m, 1H), 1.95 (m, 1H), 3.09 (d, 2H, J = 4.2 Hz), 4.65 (m, 2H), 6.79 (d, 1H, J = 7.5 Hz), 7.04 (d, 2H, J = 8.7 Hz), 7.81 (d, 2H, J = 8.7 Hz). MS (FAB+) m/z: 448.27 (M+), 422.27, 392.20, 348.21, 336.13, 318.13. HRMS (FAB+) calcd for C22H34N5O5 (MH+): 448.2562. Found: 448.2554 ± 0.0014 (n=4).
1-[4-(1-Tert-butoxycarbonyl-5-tert-butoxycarbonylamino-pentylcarbamoyl)-phenyl]-1H-[1,2,3]triazol-4-ylmethyl 4-fluorobenzoate (6)
Aqueous copper sulfate (0.4 M; 50 :L) and sodium ascorbate (0.6 M; 50 :L) were added to a solution of 5 (31.7 mg; 0.07 mmol) and 1 (25 mg; 0.14 mmol) in 400 :L tert-butanol in a small vial. The resultant heterogeneous mixture was stirred vigorously for 70 min and then partitioned between 10 mL ethyl acetate and 10 mL water. The aqueous layer was extracted once with 10 mL ethyl acetate and the combined organic layer was washed with brine and dried over sodium sulfate. The drying agent was filtered off and the ethyl acetate from the filtrate was evaporated. The residue was purified by preparative TLC using 1:1 ethyl acetate:hexanes to yield 38.9 mg (0.06 mmol; 88.4%) of an oil: TLC 1:1 ethyl acetate:hexane, Rf = 0.25. 1HNMR (CDCl3) δ 1.39 (s, 9H), 1.49 (s, 9H), 1.50 (m, 4H), 1.80 (m, 1H), 2.00 (m, 1H), 3.11 (d, 2H, J = 5.4 Hz), 4.65 (m, 2H), 5.54 (s, 2H), 6.92 (d, 1H, J = 7.2 Hz), 7.10 (t, 2H, J = 8.7 Hz), 7.83 (d, 2H, J = 8.7 Hz), 7.98 (d, 2H, J = 8.7 Hz), 8.02 – 8.12 (m, 2H), 8.19 (s, 1H). MS (FAB+) m/z: 626.3 (MH+), 570.2, 514.2, 470.2, 346.1, 302.2. HRMS (FAB+) calcd for C32H41FN5O7 (MH+): 626.2992. Found: 626.2987 ± 0.0012 (n=4).
6-Amino-2-(4-azidobenzamido)hexanoic acid (7)
A solution of 5 (16.5 mg; 0.037 mmol) in 0.5 mL anhydrous methylene chloride was treated with 0.5 mL of anhydrous trifluoroacetic acid for 1 h at room temperature. The solvents were evaporated with a stream of argon and the mixture was triturated with diethyl ether and the residue dried for several hours under high vacuum to yield 7 as an oil in a nearly quantitative yield: HPLC tR = 12.5 min. 1HNMR (CD3OD) δ 1.56 (m, 2H), 1.72 (m, 2H), 1.88 (m, 1H), 2.04 (m, 1H), 2.93 (t, 2H, J = 7.5 Hz), 4.61 (q, 2H, J = 4.8 Hz), 7.15 (d, 2H, J = 9.0 Hz), 7.90 (d, 2H, J = 8.7 Hz). HRMS (FAB+) calcd for C13H18N5O3 (MH+): 292.1410. Found: 292.1416 ± 0.0003 (n=4).
1-[4-(5-Amino-1-carboxy-pentylcarbamoyl)-phenyl]-1H-[1,2,3]triazol-4-ylmethyl 4-fluorobenzoate (8)
Trifluoroacetic acid (0.5 mL) was added to a solution of 6 (14.1 mg; 0.023 mmol) in 0.5 mL of anhydrous methylene chloride and the solution was incubated at room temperature for 1 h. The solvents were evaporated with a stream of argon and the mixture was triturated with diethyl ether three times. The resultant residue was dried for several hours under high vacuum to yield 8 as an oil in a nearly quantitative yield: HPLC tR = 19.1 min. 1HNMR (CD3OD) δ 1.58 (m, 2H), 1.74 (m, 2H), 1.99 (m, 2H), 2.95 (t, 2H, J = 7.5 Hz), 4.64 (q, 2H, J = 4.8 Hz), 5.52 (s, 1H), 7.18 (t, 2H, J = 8.9 Hz), 7.99 (d, 2H, J = 8.4 Hz), 8.09 (m, 4H), 8.75 (s, 1H). MS (FAB+), m/z: 470.2 (M+), 413.3, 391.3, 389.3, 371.3. HRMS (FAB+) calcd for C23H25FN5O5 (MH+): 470.1841. Found: 470.1820 ± 0.0000 (n = 2).
Tert-butyl 5-(tert-butoxycarbonyl)-5-(6-azidohexanamido)pentylcarbamate (9)
A mixture of H-Lys(Boc)-OtBu·HCl (63 mg; 0.187 mmol), 6-azidohexanoic acid (59 mg; 0.373 mmol), and silica-supported DCC (500 mg; 0.560 mmol) and 2.5 mL of anhydrous methylene chloride was stirred vigorously at room temperature under argon overnight. The reaction mixture was filtered to remove the silica-bound carbodiimide and urea, and the filtrate was concentrated on a rotary evaporator. The crude mixture was purified by preparative TLC using 1:1 ethyl acetate:hexanes to yield 62.2 mg (71.5% after correcting for purity) of a foam. NMR indicated that this product was contaminated with 6-azidohexanoic acid (~5%). An analytical sample was obtained by further preparative TLC of the above sample. TLC 3:1 ethyl acetate:hexanes, Rf = 0.51 (visualized using 5% phosphomolybdic acid in ethanol). 1HNMR (CDCl3) 1.37 (m, 5H), 1.42 (s, 9H), 1.45 (s, 9H), 1.64 (m, 4H), 1.81 (m, 1H), 2.22 (t, 2H, J = 7.5 Hz), 3.07 (m, 2H), 3.26 (t, 2H, J = 6.9 Hz), 4.46 (m, 1H), 4.57 (br s, 1H), 6.07 (d, 2H, J = 6.9 Hz). HRMS (FAB+) calcd for C21H40N5O5 (MH+): 442.3031. Found: 442.3035 ± 0.0008 (n=4).
1-[5-(1-Tert-butoxycarbonyl-5-tert-butoxycarbonylamino-pentylcarbamoyl)-pentyl]-1H-[1,2,3]triazol-4-ylmethyl 4-fluorobenzoate (10)
Aqueous copper sulfate (0.4 M; 50 :L) and sodium ascorbate (0.4 M; 50 :L) were added to a solution of 9 (26.8 mg; 0.057 mmol based on 95% purity) and 1 (32 mg; 0.182 mmol) in 400 :L of tert-butanol and the resultant heterogeneous mixture was stirred vigorously for 90 min. The reaction mixture was partitioned between 10 mL each of ethyl acetate and water. The aqueous layer was extracted once with 10 mL ethyl acetate and the combined organic layers were washed once with 10 mL of brine, dried over sodium sulfate, filtered and concentrated. The crude mixture was purified by preparative TLC using 19:1 ethyl acetate:hexanes to yield 21.6 mg (0.03 mmol; 60.3%) of an oil: TLC 19:1 ethyl acetate:hexanes, Rf = 0.29. 1HNMR (CDCl3) δ 1.37 (m, 6H), 1.41 (s, 9H), 1.44 (s, 9H), 1.70 (m, 2H), 1.93 (qn, 2H, J = 7.5 Hz), 2.20 (t, 2H, J = 7.4 Hz), 3.08 (q, 2H, J = 6.5 Hz), 4.35 (t, 2H, J = 7.4 Hz), 4.45 (m, 1H), 4.64 (br s, 1H), 5.44 (s, 2H), 6.06 (d, 2H, J = 7.2 Hz), 7.08 (t, 2H, J = 8.7 Hz), 7.68 (s, 1H), 8.05 (m, 2H). HRMS (FAB+) calcd for C31H46FN5O7 (M +): 619.3381. Found: 619.3384 ± 0.0006 (n = 4).
6-Amino-2-(6-azidohexanamido)hexanoic acid (11)
A solution of 9 (16.0 mg; 0.036 mmol) in 0.5 mL of anhydrous methylene chloride was treated with 0.5 mL of trifluoroacetic acid at room temperature for 1 h. The solvents were evaporated with a stream of argon and the residue was triturated three times with 2 mL diethyl ether. The resultant oily residue was dried for several hours under high vacuum to give 11 in a nearly quantitative yield: 1HNMR (CD3CN) δ 1.35 (m, 4H), 1.68 (m, 8H), 2.20 (t, 2H, J = 7.4 Hz), 2.93 (m, 2H), 3.28 (t, 2H, J = 6.6 Hz), 4.30 (m, 1H), 6.58 (br s, 2H), 6.85 (d, 1H, J = 6.8 Hz), 10.39 (br s, 1H). HRMS (FAB+) calcd for C12H23N5O3 (M+): 285.1801. Found: 285.1802 ± 0.0010 (n = 4).
1-[5-(5-Amino-1-carboxy-pentylcarbamoyl)-pentyl]-1H-[1,2,3]triazol-4-ylmethyl 4-fluorobenzoate (12)
Trifluoroacetic acid (0.5 mL) was added to a solution of 10 (11.8 mg; 0.019 mmol) in 0.5 mL of anhydrous methylene chloride and the mixture was incubated at room temperature for 1 h. The solvent was evaporated with a stream of argon and the residue was triturated three times with 2 mL diethyl ether. The resultant oily residue was dried for several hours under high vacuum to give 12 in a nearly quantitative yield: HPLC tR = 17.7 min. 1HNMR (CD3CN) δ 1.59 (m, 12H), 2.22 (t, 2H, J = 7.1 Hz), 2.98 (m, 2H), 4.30 (m, 1H), 4.39 (t, 2H, J = 6.9 Hz), 5.43 (s, 2H), 6.69 (br s, 2H), 6.95 (d, 1H, J = 6.9 Hz), 7.95 (s, 1H), 8.07 (m, 2H). HRMS (FAB+) calcd for C22H30N5O5 (M+): 463.2231. Found 463.2233 ± 0.0011 (n = 4).
T-Azidohexanoic acid conjugated YQSIYVPDI (13)
Using the automated synthesizer, the following amino acids (N∀-Fmoc-protected) and T-azidohexanoic acid were sequentially attached to isoleucine anchored to HMP resin (0.25 mmol; Applied Biosystems): Asp(O-tBu), Pro, Val, Tyr(tBu), Ile, Ser(tBu), Gln(Trt), and Tyr(tBu). The linear peptide was cleaved from the resin (203 mg; 0.07 mmol) by treatment with a 95:2.5:2.5:100 (v/v/v/v) cocktail of TFA:water:triisopropylsilane:methylene chloride (2 mL). The mixture was gently stirred at room temperature for 1.5 h. The resin was filtered through a fritted funnel and washed with one volume of the above cocktail. The filtrate was evaporated with a stream of argon until a precipitate began to form, and then diluted with ether (10 mL). The precipitated peptide was isolated by centrifugation, washed with ether twice, and dried. The resultant residue was purified by semi-preparative HPLC using the general gradient described before. Evaporation of HPLC fractions gave 9.2 mg (0.007 mmol; 11.1%) of a solid: HPLC tR = 23.3 min. MS (MALDI), m/z: 1259.0 ([M+Na]+).
Peptide-click product (14)
Copper sulfate (25 :L; 0.4 M) and sodium ascorbate (25 :L; 0.6 M) were added to a mixture of 13 (11 mg; 8.9 :mol) and 1 (16 mg; 89 :mol) in 0.2 mL of DMSO. The resultant mixture was stirred vigorously at room temperature and the progress of the reaction was followed by HPLC. After 3 h, HPLC indicated the presence of both the azido peptide (tR = 23.3 min) and the alkyne (tR = 26.7 min); therefore, an additional 25 :L each of copper sulfate and sodium ascorbate were added. Two hours later, the reaction mixture was filtered through a glass wool plug, and ether (10 mL) was added to the filtrate. The precipitate that formed was isolated by centrifugation and washed several times with ether until an HPLC of the precipitate reconstituted in a solution of 3:2:1 (v/v/v) acetonitrile:water:acetic acid (0.6 mL) showed the complete removal of the alkyne 1 and a single peak corresponding to 14 (tR = 24.5 min). The dried precipitate weighed 7.6 mg (60%): MS (MALDI) m/z: 1435.6 ([M-H+Na]+), 1436.5 ([M+Na]+), 1451.5 ([M-H+K]+), 1452.5 ([M+K]+).
Propargyl 4-[18F]fluorobenzoate ([18F]1)
A mixture of 3 (5-10 mg) and the 18F activity resolubilized in acetonitrile (1- 22 mCi in 100 :L) was heated at 130°C for 15 min. Standard analytical HPLC conditions were used for the determination of radiochemical yields. The reaction mixture was diluted with 3 mL anhydrous diethyl ether and passed through a silica Sep pak®, which was activated with 10 mL anhydrous diethyl ether. The reaction vessel was rinsed with an additional 3 mL anhydrous ether and the rinse passed through the same Sep pak®. The ether eluent was concentrated under a stream of argon to less than 200 :L. The activity was then transferred to a 1-mL Reacti® vial and the remaining ether was evaporated with a gentle stream of argon before use in click reactions. Caution! [18F]1 is rather volatile and significant amounts of radioactivity can be lost if the argon flow is strong or if the solution is allowed to dry completely.
(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl 4-[18F]fluorobenzoate ([18F]4)
A solution of benzyl azide in DMSO (1.9 mg in 0.1 mL) was added to [18F]1 (0.2 – 6.2 mCi) in a 1-mL Reacti® vial. Copper sulfate (12.5 :L; 0.4M), and sodium ascorbate (12.5 :L; 0.6 M) were added to the above solution and the mixture was stirred vigorously at room temperature for 30 min. Radiochemical yields were determined by HPLC using the standard analytical gradient conditions.
1-[4-(5-amino-1-carboxy-pentylcarbamoyl)-phenyl]-1H-[1,2,3]triazol-4-ylmethyl 4-[18F]fluorobenzoate ([18F]8)
A solution of 7 (3 – 8 mg) in tert-butanol (200 :L) was added to [18F]1 (0.6 – 1.7 mCi) in a 1-mL Reacti® vial. Copper sulfate (25 :L; 0.4M), and sodium ascorbate (25 :L; 0.6 M) were added to the above solution and the mixture was stirred vigorously at room temperature for 30 min. Radiochemical yields were determined by HPLC under the standard analytical gradient conditions.
1-[5-(5-amino-1-carboxy-pentylcarbamoyl)-pentyl]-1H-[1,2,3]triazol-4-ylmethyl 4-[18F]fluorobenzoate ([18F]12)
The synthesis of [18F]12 was performed essentially as described for [18F]8 starting with 7-10 mg of 11 and 2.5 – 3.0 mCi of [18F]1.
18F-labeled YQSIYVPDI derivative ([18F]14)
A solution of 13 (1 mg) in 100 :L of DMSO was added to [18F]1 (0.6 – 0.7 mCi) in a 1-mL Reacti® vial. Copper sulfate (12.5 :L; 0.4 M) and sodium ascorbate (12.5 :L; 0.6 M) were added to the above solution and the resultant mixture was stirred vigorously at room temperature for 30 min. To determine radiochemical yield, an aliquot of the reaction mixture was injected onto the Zorbax column; under these conditions, the tR of alkyne 1 and peptide 14 were 27.5 min and 19.3 min, respectively.
Identification of unlabeled byproducts generated in the synthesis of [18F]1
Mock reactions were conducted twice by replacing irradiated water in the synthesis of [18F]1 with deionized water. The ethereal eluents from the normal phase Sep-pak were pooled and evaporated. An aliquot of the residue was analyzed by HPLC to ensure the presence of two pseudo carriers and the remainder was subjected to preparative TLC using 1:9 ethyl acetate:hexanes. The product with an Rf value slightly higher than that of 2 was isolated. NMR and TLC indicated contamination of the product with a small amount of 2. NMR had the following peaks in addition to those of 2: 2.55 (m, 1H), 3.93 (s, 1H), 4. 76 (d, 2H), 4.90 (d, 2H), 7.02 (dd, 2H), 8.06 (dd, 2H). GCMS tR = 10.62 min: 214, 175, 159, 131, 141, 131, 121, 103, 92, 77, 63; tR = 11.23 min: 203, 148.
Additionally, the two pseudo carriers were isolated from an actual radiochemical synthesis of [18F]1 and after allowing the radioactivity to decay, GCMS was performed. The results were identical to that obtained above.
Feasibility of scavenging propargyl 4-dimethylaminobenzoate (2)
Polystyrene-bound p-toluenesulfonic acid (SigmaAldrich, cat # 532312; 100 mg, 0.2 – 0.3 mmol) was added to a solution of 2 (4 mg; 0.02 mmol) in 1 mL dry THF and the mixture was stirred gently at room temperature for 3 h. A 5 :L aliquot of the reaction mixture at 2 and 3 h each was analyzed by reversed-phase HPLC using the standard gradient elution conditions. The area of the peak corresponding to 2 from these runs was compared to the area from a HPLC run of a 5 :L solution of 2 before treating with the scavenger resin; 97% and 99% reduction in the area was seen after 2 h and 3 h, respectively.
Scavenging of 2 generated in the radiofluorination reaction
The dried activity of [18F]1 (~7 mCi) after removal of ether (see above) was reconstituted in 1 mL of THF and this solution was added to polystyrene-bound p-toluenesulfonic acid (116 mg), which was pre-swelled in THF for 30 min. The mixture was stirred gently at room temperature for 1 h and an aliquot of the supernatant (10 :L) was injected onto reversed-phase HPLC. As a control, the same volume of the solution of [18F]1 in THF before its treatment with the scavenging resin was also analyzed by HPLC.
Click reaction of peptide 13 with [18F]1 after its treatment with scavenging resin
The THF solution was carefully removed from the reaction mixture and the beads were rinsed with one volume of THF and the pooled solution of [18F]1 activity in THF was transferred to another test tube. THF was evaporated down to a small volume and after transferring the radioactivity to a Reacti® vial, THF was evaporated to dryness with a very gentle stream of argon. A solution of peptide 13 (0.65 mg) in 65 :L DMSO was added to the residual activity (2.96 mCi). Copper sulfate (0.4 M, 8.1 :L) and sodium ascorbate (0.6 M, 8.1 :L) were added to the above solution with stirring and the resultant mixture was stirred at room temperature for 30 min. The radiochemical yield was determined by analyzing a 10 :L aliquot of the reaction mixture by HPLC that was run under standard gradient conditions.
RESULTS AND DISCUSSION
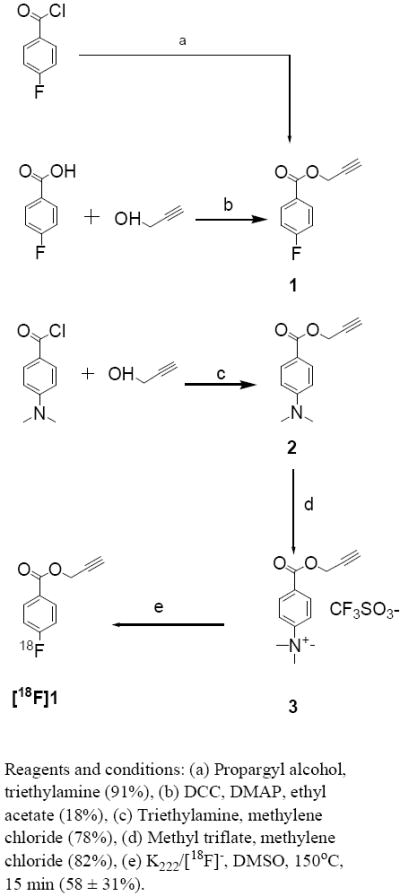
To exploit the potential enhanced in vivo stability of compounds with an aromatic carbon-fluorine bond and the favorable pharmacokinetic properties of the presumptive catabolite 4-fluorobenzoic acid, we set out to develop propargyl 4-fluorobenzoate as a template for the 18F labeling of biomolecules via click chemistry. Initially, propargyl 4-fluorobenzoate (1) was derived by the DCC-mediated esterification of 4-fluorobenzoic acid in 18% yield (Scheme 1). Treatment of the commercially available 4-fluorobenzoyl chloride with propargyl alcohol in the presence of a tertiary base provided 1 in 91% yield. The quaternary salt precursor 3 was synthesized from 4-dimethylaminobenzoyl chloride in two steps in 62% overall yield. Treatment of 3 with K222/[18F]- in acetonitrile at 130°C for 15 min yielded [18F]1 in 58 ± 31% (n = 15) decay-corrected radiochemical yields; if 4 outlier values were excluded, the yield was 75 ± 14%. In this study, we used a standard set of conditions and have not yet evaluated the effects of reaction variables such as time and temperature on radiochemical yield. In the HPLC of the reaction mixture there was essentially only one peak corresponding to 1 in the radioactivity trace (Figure 1). To investigate whether radioactive byproducts that are highly retained in the HPLC column may account for the less than ideal radiochemical yields, radio TLC of the reaction mixture was done. However, radio TLC also indicated that there was only one major peak corresponding to the product with an insignificant fraction at the origin (Figure 2). Thus, it is likely that some [18F]CH3F could have been generated (see below) and that it could have been lost by volatilization. The UV profile of HPLC (Figure 1) indicated that significant amounts of unlabeled materials were co-eluting with [18F]1. Initially we thought that this might be the unlabeled fluoro derivative that could be formed as a result of the presence of endogenous carrier fluoride in the [18F]fluoride activity [50-52] that we used or possibly due to the exchange of 19F from the relatively large amount of the triflate precursor used for the radiofluorination reaction although such exchange normally is not a problem [53]. It should be pointed out that the use of 10 mg or more of the triflate precursor in 18F-labeling reactions is not unusual [54, 55]. To investigate whether the carrier amounts in the radiolabeled preparations could be reduced by decreasing the amount of precursor used, radiofluorination reactions were performed using 1-2 mg of 3; however, very poor radiochemical yields were obtained.
Fig. (1).
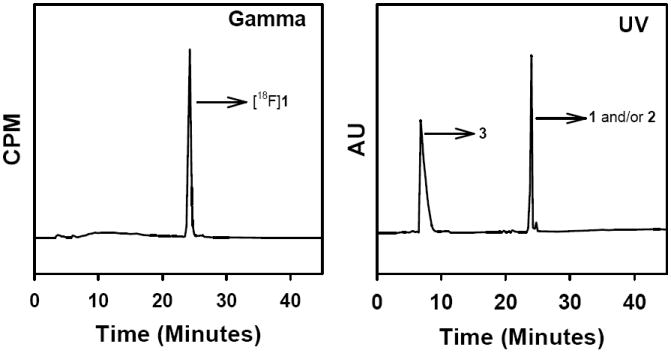
A typical HPLC profile of the reaction mixture from the synthesis of propargyl 4-[18F]fluorobenzoate ([18F]1).
Fig. (2).
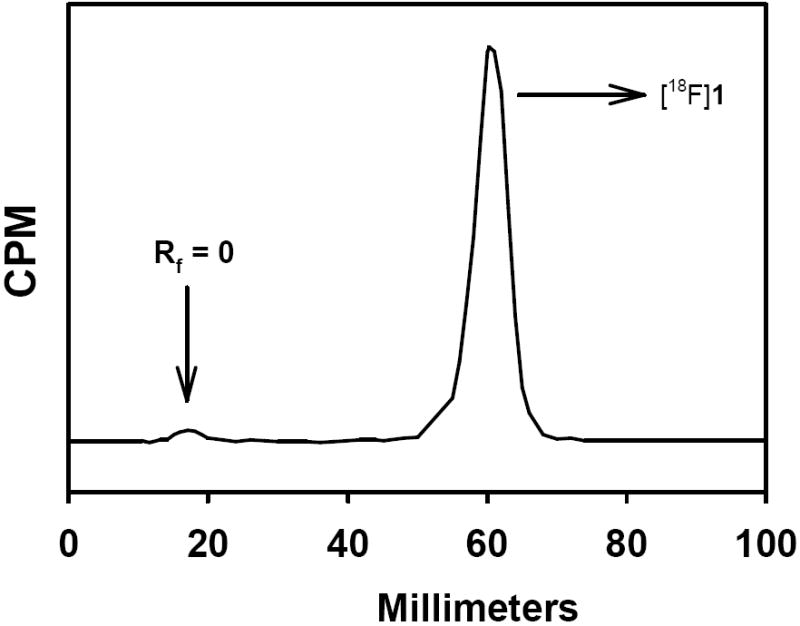
Radio thin layer chromatography profile of the reaction mixture from the synthesis of propargyl 4-[18F]fluorobenzoate ([18F]1).
To probe whether carrier fluoride present in the irradiated water contributed to the problem, we conducted radiofluorination reactions using [18F]fluoride that was either transferred directly from the cyclotron target to the reaction vessel, bypassing the Teflon delivery lines (a potential source of carrier fluoride) used in routine [18F]FDG production, or obtained from a commercial vendor. In both cases, the spurious UV peaks in the HPLC profile reduced in intensity only to small degree. Fortuitously, the retention time of compound 2 under these HPLC conditions was almost same as that of 1. Formation of methyl [18F]fluoride, formed via the nucleophilic attack of [18F]fluoride on one of the methyl groups of the trimethylanilinium triflate moiety of quaternary salt precursors, has been implicated in the low radiochemical yields of intended 18F-labeled compounds [56]. Radiochemical yield of 1 was generally good and even if this side reaction occurred in our case, the amount of 2 formed as a result should be quite low. The co-eluting unlabeled product was identified as 2 from the NMR and mass spectral data of this compound isolated from a mock reaction without radioactivity as well as from the GCMS of the compounds isolated from an actual reaction (mass spectrum was obtained after the decay of 18F). Based on these results, we conclude that some nucleophile other than [18F]fluoride itself must be the cause for the formation of significant amounts of 2.
The feasibility of scavenging 2 from the reaction mixture by using a solid-phase-bound sulfonic acid derivative was investigated. Polystyrene-bound sulfonic acid has been employed to scavenge amines [57, 58] and we envisaged that it may be possible to remove 2 from the mixture containing [18F]1. HPLC data indicated that treatment of a THF solution of 2 with the scavenging resin removed 97% of the dimethylamine compound in 2 h. It is likely that almost complete removal of 2 occurred much earlier; however, 2 h is the first time point we studied. Figure 3 shows the HPLC profile of the reaction mixture before and after treatment with the scavenging resin demonstrating that it is possible to remove the byproduct 2. Further studies are needed to optimize conditions including time for removal of this byproduct.
Fig. (3).
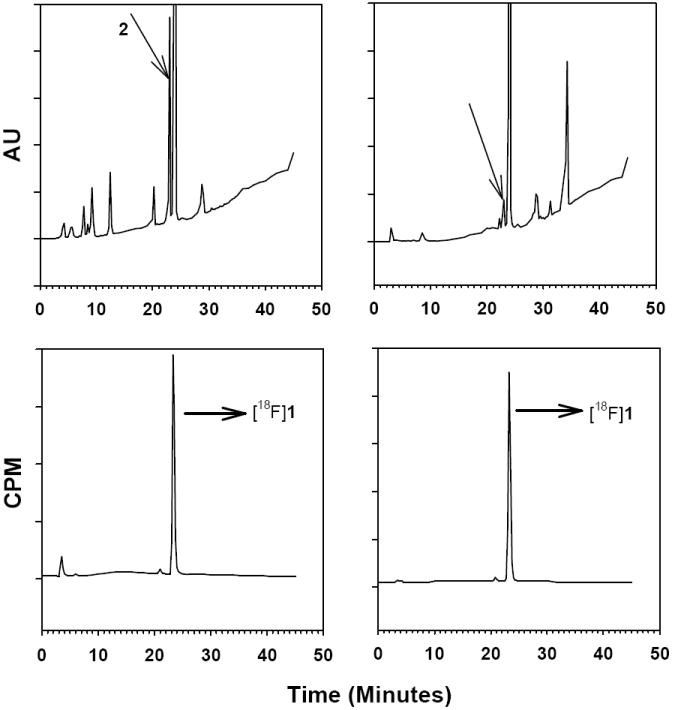
HPLC of reaction mixture from the preparation of [18F]1 before and after treatment with polystyrene-bound sulfonic acid. Reaction mixture was reconstituted in THF as described in the text and a 10 :L aliquot was injected onto HPLC (left panel). Then the THF solution was treated with the sulfonic acid derivative for 1 h and 10 :L aliquot was injected again (right panel). Note the disappearance of the peak corresponding to compound 2 pointed out by arrows.
We next investigated the usefulness of the labeled alkyne [18F]1 in click reactions using four model compounds including a peptide. As model compounds we selected the commercially available benzyl azide and two lysine derivatives decorated with azide functions. We chose the former to facilitate comparison with a similar compound (4-methoxybenzyl azide) that was used by Sirion et al. [30] and the lysine derivatives served as simple peptide mimics. The standard of click product of the reaction of benzyl azide and propargyl ester 1 was obtained in about 60% yield under typical click conditions (Scheme 2). The decay-corrected radiochemical yield for the synthesis of [18F]4 from benzyl azide and [18F]1 under conditions adapted from Sirion et al. [30] was 88 ± 4% (n = 3). In one of the lysine derivatives used, the ∀-amino group was modified with a 4-azidobenzoyl group (Scheme 3). The substrate 7 was derived from the bis-protected lysine by acylation with 4-azidobenzoic acid using silica-supported DCC followed by removal of the protecting groups in 70% overall yield. A standard of the clicked product 8 was obtained by subjecting the intermediate 5 to click reaction and subsequent removal of the protecting groups from the resultant 6 in 66% overall yields for two steps. For the synthesis of [18F]8 by reacting 7 with [18F]1, the average decay-corrected radiochemical yield was 79 ± 33% (n = 3).
Scheme 2.
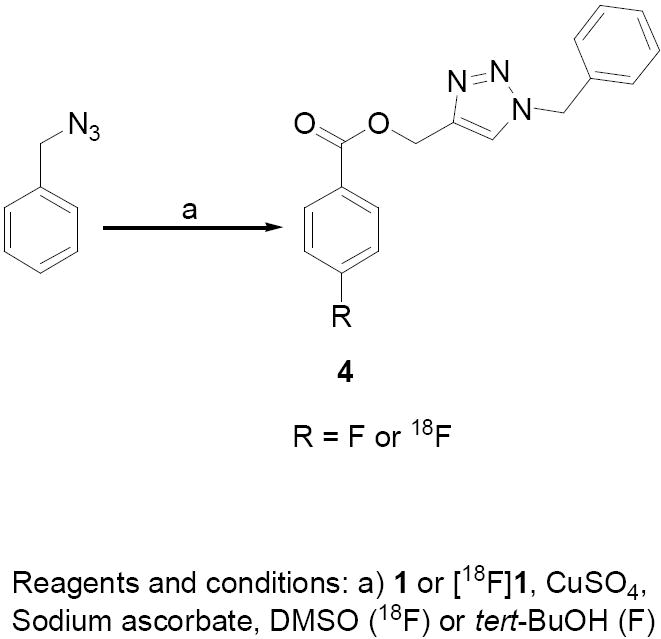
Synthesis of unlabeled and 18F-labeled (1-benzyl-1H-1,2,3-triazol-4-yl)methyl 4-fluorobenzoate.
Scheme 3.
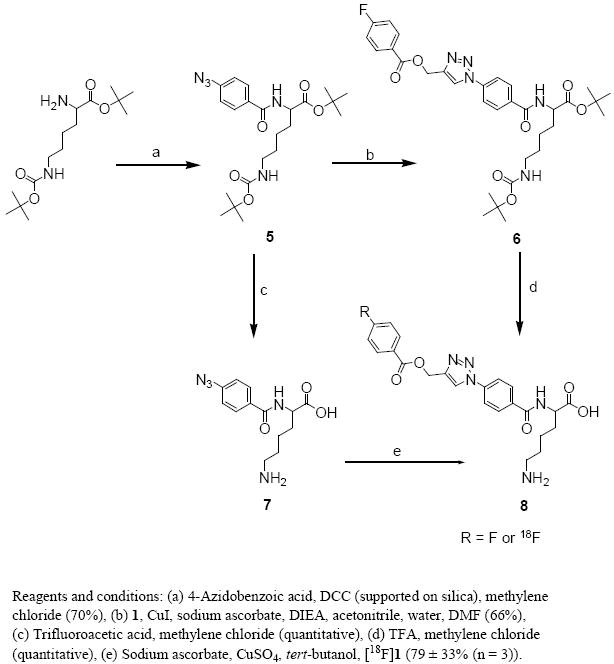
Synthesis of 6-amino-2-(4-azidobenzamido)hexanoic acid (7) and, labeled and unlabeled 1-[4-(5-amino-1-carboxy-pentylcarbamoyl)-phenyl]-1H-[1,2,3]triazol-4-ylmethyl 4-fluorobenzoate (8).
As another model, lysine modified with an azidohexanoyl function was synthesized (Scheme 4). The final target 12 was generated from bis-protected lysine in 3 steps in an overall yield of 48%. The substrate for the radiochemical click reaction was synthesized from the bis-protected lysine in two steps in 80% overall yield. Radiolabeled 12 was synthesized in 75 ± 5% (n = 3) decay-corrected radiochemical yields under conditions similar to those used for the synthesis of [18F]8.
Scheme 4.
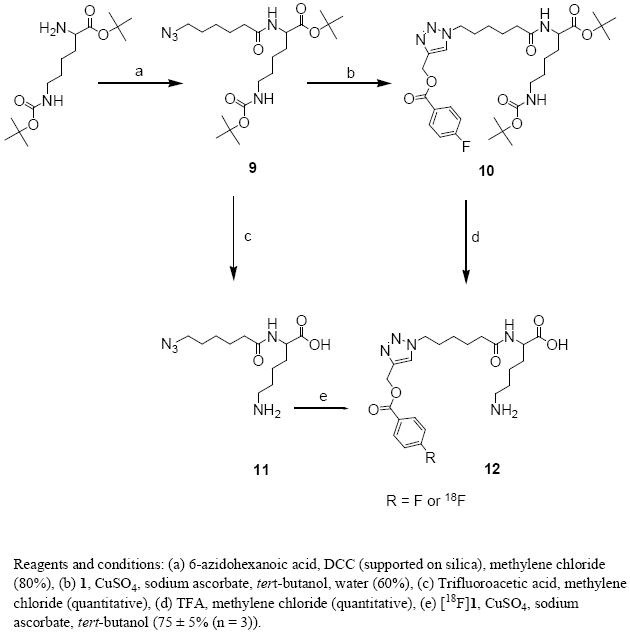
Synthesis of 6-amino-2-(6-azidohexamido)hexanoic acid (11) and, labeled and unlabeled 1-[5-(5-amino-1-carboxy-pentylcarbamoyl)-pentyl]-1H-[1,2,3]triazol-4-ylmethyl 4-fluorobenzoate (12).
Finally, the suitability of [18F]1 for labeling a peptide was investigated. We are working on a project that involves in vivo imaging of the enzyme transglutaminase (TG). TG is an important tissue stabilizing enzyme that catalyses the formation of covalent cross-links between proteins of the extracellular matrix. While it is expressed in a number of normal cells, it has been shown that TG is extensively expressed in certain tumors, such as human mammary carcinoma [59]. Also, its expression is up-regulated in drug-resistant and metastatic breast cancer cells and it can serve as a valuable prognostic marker for these phenotypes [60]. Recently, optimal substrate peptides for TG were derived by phage display [61] and, based on this work, we chose the sequence YQSIYVPDI as a possible peptide for targeting TG. To facilitate click labeling, we initially chose to derivatize this sequence with the simple azidohexanoic acid; however, in future studies, we plan to use an azido alkanoic acid containing a suitable PEG chain. The azidohexanoic acid-modified sequence was synthesized by automated solid-phase peptide synthesis, and from this, a standard of the click product 14 (Scheme 5) was derived. MALDI mass spectrometric data of 13 and 14 are consistent with their structures.
Scheme 5.
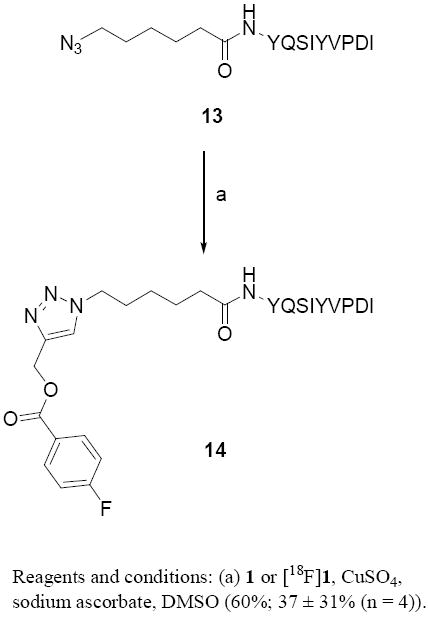
Synthesis of labeled and unlabeled 14.
Fluorine-18 click labeling of this peptide (13) was performed using DMSO as the solvent simply because the peptide was not very soluble in tert-butanol. In any case, Sirion et al. [30] have shown that, in tandem with sodium ascorbate and copper sulfate, DMSO was the best among the three solvents evaluated for click reactions, including tert-butanol. The average decay-corrected radiochemical yield for this coupling was quite variable, 37 ± 31% (n = 4). A typical HPLC profile of this reaction is shown in Figure 4. The two major peaks corresponded to unreacted 1 and the product peptide; however, there were two other minor byproducts, which have not been identified.
Fig. (4).
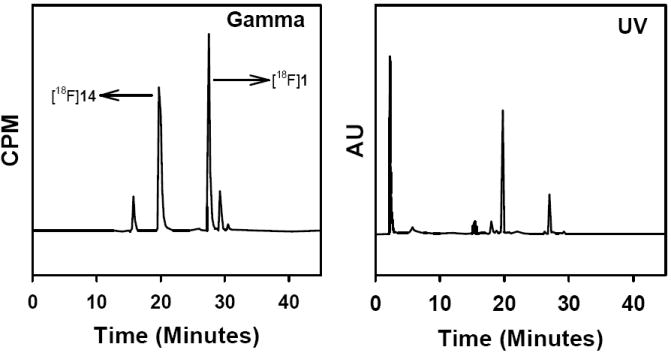
HPLC profile of the click reaction between azidohexanoyl-YQSIYVPDI and [18F]1.
The radiochemical yields for the click labeling of the simple model compounds were high as reported for similar 18F labeling reactions [26, 29, 30]. The radiochemical yield for the peptide labeling was rather low except in one run when an 80% yield was obtained. This is contrary to the consistently high yields reported for peptide labeling by other investigators [26, 29]. The lack of sufficient amount of precursor peptide is not likely to have been the reason for the relatively low and variable yields because Marik and Sutcliffe [26] used only 0.3 pmol of their peptides for their labeling reactions and obtained more favorable results. On the other hand, Glaser et al. [29] used 2 mg (3-4 :mol) of the alkyne peptide and Li et al. [31] used 1 mg (0.7 umole) of a RGD peptide with an azide function. It is likely that the low radiochemical yield is as a result of the presence of substantially large amounts of pseudo carriers in the [18F]1 preparations. In addition to the formation of 2 that was discussed earlier, another unlabeled compound with a tR slightly higher than that of 1 also was present in radiofluorination reaction mixtures. Although NMR and mass spectral data were obtained for this species, it was not possible to discern the exact identity of this compound from these data. NMR data indicates that it may be a 1,4-disubstituted benzene derivative with a propargyl function. The amount of this compound was substantially smaller and insignificant in some earlier labeling runs (Figure 1) but was observed consistently in quantities seemingly higher than that of 2 (based on HPLC peak area; see Figure 3-UV profiles) in later (and most) runs. While the polystyrene-bound sulfonic acid efficiently removed 2, the amount of the compound with the higher tR peak in the reaction mixture was unaffected. A click reaction of the peptide was conducted after scavenging 2 from the reaction mixture without any improvement in the radiochemical yields (~30%). It is tempting to speculate that the unidentified product may contain the propargyl function intact (NMR indicates this may be the case) and therefore it can compete for [18F]1 in the click reactions. One possibility is that this is formed by the nucleophilic displacement of the trimethylamine, as in the intended 18F reaction, by the same unknown nucleophile which generates 2 by its attack on the methyl group. We are currently attempting to identify this nucleophile and then determine how its presence can be avoided so that [18F]1 can be obtained in high specific activities. We have developed a method for the synthesis of the radioiodinated analogue of 1, propargyl 4-[125/131I]iodobenzoate at a no-carrier-added level by the iododestannylation of a tin precursor and have shown that much higher radiochemical yields (82%) can be obtained for its click reaction with peptide 13 (will be published elsewhere) corroborating the fact that the low radiochemical yields seen for the click reaction of [18F]1 with 13 indeed is due to the presence of a large amount of pseudo carriers. Although it should be possible to isolate [18F]1 from these pseudo carriers by HPLC and/or scavenging resins, these additional steps will reduce the effective radiochemical yields. Further work is needed to address these problems which currently detract from the practical merit of this approach for labeling biomolecules with 18F.
CONCLUSIONS
We have developed the synthesis of 18F-labeled propargyl 4-fluorobenzoate and demonstrated its usefulness in click labeling. This could ultimately be another tool in the armamentarium for the labeling of biomolecules with the short lived 18F radionuclide. Because 18F is attached to an aryl sp2 carbon, it is expected that compounds labeled with this prosthetic group will be more inert to in vivo defluorination than other agents reported for 18F labeling via click chemistry. However, approaches to prevent the formation of or to remove the formed pseudo carriers from [18F]1 preparations are needed.
Acknowledgments
The authors want to thank Michael Dailey and Sean Murphy for providing 18F. This work is supported by Grants from CA42324 and NS20023 from the National Cancer Institute.
References
- 1.Weissleder R. Molecular imaging in cancer. Science. 2006;312:1168–71. doi: 10.1126/science.1125949. [DOI] [PubMed] [Google Scholar]
- 2.Kelloff GJ, Krohn KA, Larson SM, Weissleder R, Mankoff DA, Hoffman JM, Link JM, Guyton KZ, Eckelman WC, Scher HI, O’Shaughnessy J, Cheson BD, Sigman CC, Tatum JL, Mills GQ, Sullivan DC, Woodcock J. The progress and promise of molecular imaging probes in oncologic drug development. Clin Cancer Res. 2005;11:7967–85. doi: 10.1158/1078-0432.CCR-05-1302. [DOI] [PubMed] [Google Scholar]
- 3.Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nat Rev Drug Discov. 2003;2:123–31. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- 4.Sparreboom A. Unexplored pharmacokinetic opportunities with microdosing in oncology. Clin Cancer Res. 2007;13:4033–4. doi: 10.1158/1078-0432.CCR-07-0540. [DOI] [PubMed] [Google Scholar]
- 5.Singh SS. Preclinical pharmacokinetics: an approach towards safer and efficacious drugs. Curr Drug Metab. 2006;7:165–82. doi: 10.2174/138920006775541552. [DOI] [PubMed] [Google Scholar]
- 6.Lever JR. PET and SPECT imaging of the opiod system: receptors, radioligands and avenues for drug discovery and development. Curr Pharm Des. 2007;13:33–49. doi: 10.2174/138161207779313821. [DOI] [PubMed] [Google Scholar]
- 7.Bauer M, Langer O, Dal-Bianco P, Karch R, Brunner M, Abrahim A, Lanzenberger R, Hofmann A, Joukhadar C, Carminati P, Ghirardi O, Piovesan P, Forloni G, Corrado ME, Lods N, Dudczak R, Auff E, Kletter K, Muller M. A positron emission tomography microdosing study with a potential antiamyloid drug in healthy volunteers and patients with Alzheimer’s disease. Clin Pharmacol Ther. 2006;80:216–27. doi: 10.1016/j.clpt.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Lundqvist H, Antoni G, Langstrom B. Genotoxic hazard of radiopharmaceuticals in humans: chemical and radiation aspects coupled to microdosing. Eur J Clin Pharmacol. 2007;63:641–5. doi: 10.1007/s00228-007-0304-6. [DOI] [PubMed] [Google Scholar]
- 9.Saleem A, Aboagye EO, Matthews JC, Price PM. Plasma pharmacokinetic evaluation of cytotoxic agents radiolabeled with positron emitting radioisotopes. Cancer Chemother Pharmacol. 2008;61:865–73. doi: 10.1007/s00280-007-0552-2. [DOI] [PubMed] [Google Scholar]
- 10.Bauer M, Wagner CC, Langer O. Microdosing studies in humans: the role of positron emission tomography. Drugs R D. 2008;9:73–81. doi: 10.2165/00126839-200809020-00002. [DOI] [PubMed] [Google Scholar]
- 11.Rohren EM, Turkington TG, Coleman RE. Clinical applications of PET in oncology. Radiology. 2004;231:305–32. doi: 10.1148/radiol.2312021185. [DOI] [PubMed] [Google Scholar]
- 12.Couturier O, Luxen A, Chatal JF, Vuillez JP, Rigo P, Hustinx R. Fluorinated tracers for imaging cancer with positron emission tomography. Eur J Nucl Med Mol Imaging. 2004;31:1182–206. doi: 10.1007/s00259-004-1607-9. [DOI] [PubMed] [Google Scholar]
- 13.Okarvi SM. Recent progress in fluorine-18 labelled peptide radiopharmaceuticals. Eur J Nucl Med. 2001;28:929–38. doi: 10.1007/s002590100508. [DOI] [PubMed] [Google Scholar]
- 14.Wilbur DS. Radiohalogenation of proteins: an overview of radionuclides, labeling methods, and reagents for conjugate labeling. Bioconjugate Chem. 1992;3:433–70. doi: 10.1021/bc00018a001. [DOI] [PubMed] [Google Scholar]
- 15.Vaidyanathan G, Bigner DD, Zalutsky MR. Fluorine-18 –labeled monoclonal antibody fragments: a potential approach for combining radioimmunoscintigraphy and positron emission tomography. J Nucl Med. 1992;33:1535–41. [PubMed] [Google Scholar]
- 16.Page RL, Garg PK, Vaidyanathan G, Zalutsky MR. Preclinical evaluation and PET imaging of 18F-labeled Mel-14 F(ab’)2 fragment in normal dogs. Nucl Med Biol. 1994;21:911–9. doi: 10.1016/0969-8051(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 17.Vaidyanathan G, Zalutsky MR. Synthesis of N-succinimidyl 4-[18F]fluorobenzoate, an agent for labeling proteins and peptides with 18F. Nature Protocols. 2006;1:1655–61. doi: 10.1038/nprot.2006.264. [DOI] [PubMed] [Google Scholar]
- 18.Shively JE. 18F labeling for immuno-PET: where speed and contrast meet. J Nucl Med. 2007;48:170–2. [PubMed] [Google Scholar]
- 19.Wester HJ, Hamacher K, Stocklin G. A comparative study of N.C.A. fluorine-18 labeling of proteins via acylation and photochemical conjugation. Nucl Med Biol. 1996;23:365–72. doi: 10.1016/0969-8051(96)00017-0. [DOI] [PubMed] [Google Scholar]
- 20.Berndt M, Pietzsch J, Wuest F. Labeling of low-density lipoproteins using 18F-labeled thiol-reactive reagent N-[6-(4-[18F]fluorobenzylidene)aminoxyhexyl]maleimide. Nucl Med Biol. 2007;34:5–15. doi: 10.1016/j.nucmedbio.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Cai W, Zhang X, Wu Y, Chen X. A thiol-reactive 18F-labeling agent, N-[2-(4-18F-fluorobenzamido)ethyl]maleimide, and synthesis of RGD peptide–based tracer for PET imaging of alpha v beta 3 integrin expression. J Nucl Med. 2006;47:1172–80. [PMC free article] [PubMed] [Google Scholar]
- 22.de Bruin B, Kuhnast B, Hinnen F, Yaouancq L, Amessou M, Johannes L, Samson A, Boisgard R, Tavitian B, Dolle F. 1-[3-2-[18F]fluoropyridin-3-yloxy)propyl]pyrrole-2,5-dione: design, synthesis, and radiosynthesis of a new [18F]fluoropyridine-based maleimide reagent for the labeling of peptides and proteins. Bioconjugate Chem. 2005;16:406–20. doi: 10.1021/bc0497463. [DOI] [PubMed] [Google Scholar]
- 23.Lee YS, Jeong JM, Kim HW, Chang YS, Kim YJ, Hong MK, Rai GB, Chi DY, Kang WJ, Kang JH, Lee DS, Chung JK, Lee MC, Suh YG. An improved method of 18F peptide labeling: hydrazone formation with HYNIC-conjugated c(RGDyK) Nucl Med Biol. 2006;33:67783. doi: 10.1016/j.nucmedbio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Poethko T, Schottelius M, Thumshirn G, Hersel U, Herz M, Henriksen G, Kessler H, Schwaiger M, Wester HJ. Two-step methodology for high-yield routine radiohalogenation of peptides: 18F-labeled RGD and octreotide analogs. J Nucl Med. 2004;45:892–902. [PubMed] [Google Scholar]
- 25.Schirrmacher R, Bradtmoller G, Schirrmacher E, Thews O, Tillmanns J, Siessmeier T, Buchholz HG, Bartenstein P, Waengler B, Niemeyer CM, Jurkschat K. F-18-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew Chem Int Ed. 2006;45:6047–50. doi: 10.1002/anie.200600795. [DOI] [PubMed] [Google Scholar]
- 26.Marik J, Sutcliffe JL. Click for PET: rapid preparation of [F-18]fluoropeptides using Cu-I catalyzed 1,3,-dipolar cycloaddition. Tetrahedron Lett. 2006;47:6681–84. [Google Scholar]
- 27.Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array fo building blocks. Angew Chem Int Ed. 2002;41:1053–7. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 28.Kolb HC, Finn MG, Sharpless KB. Click chemistry: Diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40:2004–21. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 29.Glaser M, Arstad E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjugate Chem. 2007;18:989–93. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]
- 30.Sirion U, Kim HJ, Lee JH, Seo JW, Lee BS, Lee SJ, Oh SJ, Chi DY. An efficient F-18 labeling method for PET study: Huisgen 1,3-dipolar cycloaddition of bioactive substances and F-18-labeled compounds. Tetrahedron Lett. 2007;48:3953–7. [Google Scholar]
- 31.Li ZB, Wu Z, Chen K, Chin FT, Chen X. Click chemistry of 18F-labeling of RGD peptides and microPET imaging of tumor integrin alpha v beta 3 expression. Bioconjugate Chem. 2007;18:1987–94. doi: 10.1021/bc700226v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coenen HH, Moerlein SM, Stocklin G. No-carrier-added radiohalogenation methods with heavy halogens. Radiochim Acta. 1983;34:47–68. [Google Scholar]
- 33.Cheng DF, Yin DZ, Zhang L, Wang MW, Li GC, Wang YX. Preparation of the novel fluorine-18-labeled VIP analog for PET imaging studies using two different synthesis methods. J Fluorine Chem. 2007;128:196–201. [Google Scholar]
- 34.Carson RE, Wu YJ, Lang LX, Ma Y, Der MG, Herscovitch P, Eckelman WC. Brain uptake of the acid metabolite of F-18-labeled WAY 100635 analogs. J Cereb Blood Flow Metab. 2003;23:249–60. doi: 10.1097/01.WCB.0000046145.31247.7A. [DOI] [PubMed] [Google Scholar]
- 35.Magata Y, Lang LX, Kiesewetter DO, Jagoda EM, Channing MA, Eckelman WC. Biologically stable [F-18]-labeled benzyl fluoride derivatives. Nucl Med Biol. 2000;27:163–8. doi: 10.1016/s0969-8051(99)00108-0. [DOI] [PubMed] [Google Scholar]
- 36.Lee SY, Choe YS, Kim DH, Park BN, Kim SE, Choi Y, Lee KH, Lee JW, Kim BT. A simple and efficient in vitro method for metabolism studies of radiotracers. Nucl Med Bio. 2001;28:391–5. doi: 10.1016/s0969-8051(01)00203-7. [DOI] [PubMed] [Google Scholar]
- 37.Ryu EK, Choe YS, Kim DH, Ko BH, Choi Y, Lee KH, Kim BT. In vitro metabolism studies of F-18 labeled 1-phenylpiperazine using mouse liver S9 fraction. Nucl Med Biol. 2006;33:165–72. doi: 10.1016/j.nucmedbio.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Chen X, Park R, Hou Y, Khankaldyyan V, Gonzales-Gomez I, Tohme M, Bading JR, Laug WE, Conti PS. MicroPET imaging of brain tumor angiogenesis with 18F-labeled PEGylated RGD peptide. Eur J Nucl Med Mol Imaging. 2004;31:1081–9. doi: 10.1007/s00259-003-1452-2. [DOI] [PubMed] [Google Scholar]
- 39.Bergmann R, Helling R, Heichert C, Scheunemann M, Mäding P, Wittrisch H, Johannsen B, Henle Th. Radiofluorination and positron emission tomography (PET) as a new approach to study the in vivo distribution and elimination of the advanced glycation endproducts N’-carboxymethyllysine (CML) and N’-carboxyethyllysine (CEL) Nahrung/Food. 2001;45:182–8. doi: 10.1002/1521-3803(20010601)45:3<182::AID-FOOD182>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 40.Fredriksson A, Ekberg K, Ingvar M, Johansson BL, Wahren J, Stone-Elander S. In vivo biodistribution and pharmacokinetics of 18F-labeled human C-peptide: evaluation in monkeys using positron emission tomography. Life Sci. 2002;71:1361–70. doi: 10.1016/s0024-3205(02)01859-3. [DOI] [PubMed] [Google Scholar]
- 41.Hausner SH, DiCara D, Marik J, Marshall JF, Sutcliffe JL. Use of a peptide derived from foot-and-mouth disease virus for the noninvasive imaging of human cancer. Generation and evaluation of 4-[F-18]fluorobenzoyl A20FMDV2 for in vivo imaging of integrin alpha(v) beta(6) expression with positron emission tomography. Cancer Res. 2007;67:7833–40. doi: 10.1158/0008-5472.CAN-07-1026. [DOI] [PubMed] [Google Scholar]
- 42.Zhang X, Cai W, Cao F, Schreibmann E, Wu Y, Wu JC, Xing L, Chen X. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J Nucl Med. 2006;47:492–501. [PubMed] [Google Scholar]
- 43.Yagle KJ, Eary JF, Tait JF, Grierson JR, Link JM, Lewellen B, Gibson DF, Krohn KA. Evaluation of F-18-annexin V as a PET imaging agent in an animal model of apoptosis. J Nucl Med. 2005;46:658–66. [PubMed] [Google Scholar]
- 44.Sutcliffe-Goulden JL, O’Doherty MJ, Marsden PK, Hart IR, Marshall JF, Bansal SS. Rapid solid phase synthesis and biodistribution of F-18-labelled linear peptides. Eur J Nucl Med Mol Imaging. 2002;29:754–9. doi: 10.1007/s00259-001-0756-3. [DOI] [PubMed] [Google Scholar]
- 45.Lang L, Jagoda E, Schmall B, Vuong BK, Adams HR, Nelson DL, Carson RE, Eckelman WC. Development of fluorine-18-labeled %-HT1A anatagonists. J Med Chem. 1999;42:1576–86. doi: 10.1021/jm980456f. [DOI] [PubMed] [Google Scholar]
- 46.Pietzsch J, Bergmann R, Wuest F, Pawelke B, Hultsch C, van den Hoff J. Catabolism of native and oxidized low density lipoproteins: in vivo insights from small animal positron emission tomography studies. Amino Acids. 2005;29:389–404. doi: 10.1007/s00726-005-0203-z. [DOI] [PubMed] [Google Scholar]
- 47.Ramenda T, Bergmann R, Wuest F. Synthesis of 18F-labeled neurotensin (8-13) via copper-mediated 1,3-dipolar (3+2) cycloaddition reaction. Lett Drug Design Disc. 2007;4:279–85. [Google Scholar]
- 48.Grandjean C, Boutonnier A, Guerreiro C, Fournier JM, Mulard LA. On the preparation of carbohydrate-protein conjugates using the traceless Staudinger ligation. J Org Chem. 2005;70:7123–32. doi: 10.1021/jo0505472. [DOI] [PubMed] [Google Scholar]
- 49.Parrish B, Emrick T. Soluble camptothecin derivatives prepared by click cycloaddition chemistry on functional aliphatic polyesters. Bioconjugate Chem. 2007;18:263–7. doi: 10.1021/bc060201d. [DOI] [PubMed] [Google Scholar]
- 50.Kiesewetter DO, Kilbourn MR, Landvatter SW, Heiman DF, Katzenellenbogen JA, Welch MJ. Preparation of 4 fluorine-18-labeled estrogens and their selective uptakes in target tissues of immature rats. J Nucl Med. 1984;25:1212–21. [PubMed] [Google Scholar]
- 51.Nishijima K, Kuge Y, Tsukamoto E, Seki K, Ohkura K, Magata Y, Tanaka A, Nagatsu K, Tamaki N. Increased [F-18]2-fluoro-2-deoxy-D-glucose ([F-18]FDG) yied with recycled target [O-18]water: factors affecting [F-18]FDG yield. Appl Radiat Isot. 2002;57:43–9. doi: 10.1016/s0969-8043(02)00070-2. [DOI] [PubMed] [Google Scholar]
- 52.Schlyer DJ, Firouzbakht ML, Wolf AP. Impurities in the [O-18]water target and their effect on the yield of an aromatic displacement reaction with [F-18] fluoride. Appl Radiat Isot. 1993;44:1459–65. doi: 10.1016/0969-8043(93)90099-v. [DOI] [PubMed] [Google Scholar]
- 53.Jalilian AR, Sheikhha M, Mirzaei M, Aslani GR, Shafiee A. Radiosynthesis of [F-18]-5-[2-(2-chlorophenoxy)phenyl]-1,3,4-oxadiazole-2-4-fluorobenzoate: A labeled ligand for benzodiazepine receptors. J Radioanal Nucl Chem. 2004;260:373–7. [Google Scholar]
- 54.Mading P, Fuchtner F, Johannsen B, Steinbach J, Hilger CS, Friebe M, Halks-Miller M, Horuk R, Mohan R. F-18-labelling of a potent nonpeptide CCR1 antagonist: synthesis of 1-(5-chloro-2-{2-[(2R)-4-(4-F-18]fluorobenzyl)-2-methylpiperazin-1-yl]-2-oxoethoxy}phenyl)urea in an automated module. J Label Compd Radiopharm. 2006;49:253–62. [Google Scholar]
- 55.Wadsak W, Wirl-Sagadin B, Mitterhauser M, Mien LK, Ettlinger DE, Keppler BK, Dudczak R, Kletter K. NCA nucleophilic radiofluorination on substituted benzaldehydes for the preparation of [18F]fluorinated aromatic amino acids. Appl Radia Isot. 2006;64:355–9. doi: 10.1016/j.apradiso.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 56.Sun HR, DiMagno SG. Competitive demethylation and substitution in N,N,N-trimethylanilinium fluorides. J Fluorine Chem. 2007;128:806–12. [Google Scholar]
- 57.Chen J, Dixon BR, Dumas J, Brittelli D. Protocols for amide high-speed analoging. Preparation of novel, small molecule cathepsin D inhibitors. Tetrahedron Lett. 1999;40:9195–99. [Google Scholar]
- 58.Weidner JJ, Parlow JJ, Flynn DL. Polymer-assisted solution phase synthesis: a general method for sequestration of byproducts formed from activated acyl-transfer reactants. Tetrahedron Lett. 1999;40:239–42. [Google Scholar]
- 59.Hettasch JM, Bandarenko N, Burchette JL, Lai TS, Marks JR, Haroon ZA, Peters K, Dewhirst MW, Iglehart JD, Greenberg CS. Tissue transglutaminase expression in human breast cancer. Lab Invest. 1996;75:637–45. [PubMed] [Google Scholar]
- 60.Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin Cancer Res. 2004;10:8068–76. doi: 10.1158/1078-0432.CCR-04-1107. [DOI] [PubMed] [Google Scholar]
- 61.Sugimura Y, Hosono M, Wada F, Yoshimura T, Maki M, Hitomi K. Screening for the preferred substrate sequence of transglutaminase using a phage-displayed peptide library-identification of peptide substrates for TGase 2 and Factor XIIIA. J Biol Chem. 2006;281:17699–706. doi: 10.1074/jbc.M513538200. [DOI] [PubMed] [Google Scholar]