

Abstract
SIRT1 is a multifaceted, NAD+-dependent protein deacetylase that is involved in a wide variety of cellular processes from cancer to ageing. The function of SIRT1 in cancer is complex: SIRT1 has been shown to have oncogenic properties by downregulating p53 activity, but recent studies indicate that SIRT1 acts as a tumour suppressor in a mutated p53 background, raising intriguing questions regarding its mechanism of action. Here we discuss the current understanding of how SIRT1 functions in light of recent discoveries and propose that the net outcome of the seemingly opposite oncogenic and tumour-suppressive effects of SIRT1 depends on the status of p53.
SIRT1 is a class III histone deacetylase within the sirtuin family of related proteins that is uniquely dependent on NAD+ for catalysis. The enzyme has been conserved throughout evolution from yeast to human and is a crucial link between cell metabolism, longevity and stress response. An abundance of data link SIRT1 to cellular metabolic pathways (BOX 1), and it is clear that cellular metabolism is causally linked to health, longevity and diseases such as cancer. Recent data show that there are physiological benefits from the activation of SIRT1 in certain metabolic disorders1. However, whether or not modulating the activity of SIRT1 will improve the response of tumours to chemotherapy is unclear and is currently an intense area of research. SIRT1 levels are increased in a number of tumour types, and its functions in controlling cellular senescence and ageing are probably linked to tumour development and the dependence that tumour cells have on SIRT1 overexpression. Moreover, increasing evidence suggests that inhibiting SIRT1 has a direct effect on factors that are involved in the DNA damage response and the growth arrest of tumours in vivo2.
Box 1. SIRT1 and metabolism.
Restricting calorie intake, a reduction of calories by 20–40% known as caloric restriction, can increase lifespan in lower organisms56. Cellular ageing can often lead to the development of neoplastic lesions as the result of environmental insults and DNA stress to a cell, and caloric restriction has been shown to reduce tumour development in several cancer mouse models57-59. SIRT1 is induced by caloric restriction and is a crucial factor in the resistance to stress-induced apoptosis that occurs with ageing6,60. SIRT1 imparts deacetylase activity to a number of targets that are involved in metabolism and energy homeostasis. Deacetylation of the transcription co-activator PGC1α (peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1α) by SIRT1 activates gluconeogenic genes and increases hepatic glucose output during the caloric restriction response61. In addition, SIRT1 also deacetylates the liver X receptor and downregulates protein-tyrosine phosphatase 1B (PTP1B), leading to an increase in reverse cholesterol transport and decreased insulin resistance, respectively62,63. SIRT1 promotes insulin expression and secretion in pancreatic β-cells and can stimulate fat mobilization in white adipose tissue through deacetylation of PPARγ64-66.
The global cellular impact of SIRT1 on these metabolic pathways is multifaceted. Upregulation of SIRT1 during caloric restriction can lead to improved insulin use, decreased cholesterol absorption and reduced fat storage. In vivo evidence from Sirt1-null mice suggests that SIRT1 is required for the effects seen from caloric restriction. Calorie-restricted Sirt1-null mice are hypermetabolic, have altered liver mitochondrial function and use ingested calories inefficiently67. Therefore, SIRT1-dependent physiological changes under caloric restriction result in improved health and may contribute to the observed increase in longevity as well1. Systemic increases in SIRT1 expression may be more complex than originally thought, however, as caloric restriction induces a decrease in SIRT1 expression in the liver compared with white adipose tissue and muscle47. This may be beneficial for an organism, as reduced liver X receptor deacetylation in the liver will reduce fat synthesis.
In this Perspective we discuss the recent insights into the complex nature of SIRT1 regulation and function. Many questions remain about how SIRT1 affects tumour formation, and this might reflect the fact that the function of SIRT1 depends on the genetic context. In short, it might have differing functions in tumours depending on the genetic alterations that have occurred during tumorigenesis.
Regulators of SIRT1
The diverse roles of SIRT1 deacetylase activity in a number of cellular processes is underscored by its multilayered regulation (FIG. 1). The transcription, translation and post-translational modification of SIRT1 are regulated by a number of factors.
Figure 1. SIRT1 pathway overview.

SIRT1 is an NAD+-dependent histone deacetylase that catalyses the removal of acetyl (Ac) groups from a number of non-histone targets. The downstream effects of target deacetylation include changes in cellular metabolism (lipid metabolism, insulin sensitivity, reverse cholesterol transport and gluconeogenesis) as well as cell survival and senescence effects (cell survival and DNA repair). Several protein regulators and small-molecule compounds that can activate or inhibit SIRT1 function have also been described. AROS, active regulator of SIRT1; DBC1, deleted in breast cancer 1; FOXO, forkhead box; HIC1, hypermethylated in cancer 1; LXR, liver X receptor; PGc1α, PPARγ coactivator 1α; PPARγ, peroxisome proliferator-activated receptor-γ; PTP1B, protein-tyrosine phosphatase 1B.
SIRT1 transcription
SIRT1 transcription is under the control of at least two negative feedback loops that keep its induction tightly regulated during cellular stress. The transcription factor E2F1 can induce SIRT1 expression. Indeed, etoposide-mediated DNA damage causes E2F1-dependent induction of SIRT1 expression3. E2F1 is known to induce the transcription of several apoptotic genes and can induce apoptosis after DNA damage events through both p53-dependent and p53-independent mechanisms4. Importantly, E2F1 is also a substrate of SIRT1 and deacetylation of E2F1 inhibits its activity as a transcriptional activator. Therefore, this SIRT1–E2F1 negative feedback loop might act as a regulatory switch that can determine the apoptotic fate of a cell. As E2F1 is a potent activator of apoptotic genes such as TP53, TP73 and APAF1, transient induction of SIRT1 by E2F1 may be one fail-safe mechanism for preventing apoptosis in response to DNA damage.
As well as being a direct effector of SIRT1 deacetylation, p53 can repress SIRT1 transcription through binding to two response elements within the SIRT1 promoter. Trp53-null mice have increased levels of SIRT1 in some tissue types and several p53-null tumour cell lines show SIRT1 overexpression5,6. SIRT1 and p53 also exist in a regulatory feedback loop: SIRT1-mediated deacetylation of p53 prevents p53-dependent transactivation of CDKN1A (which encodes p21) and BAX after DNA damage, and SIRT1 is capable of deacetylating all major p53 acetylation sites7 (W.G. and Y. Tam, unpublished data). These direct effects of SIRT1 on p53 transactivation are important for the function of p53 as a transcription factor: the acetylation status of p53 has been shown to be indispensable for its ability to repress cell growth and induce apoptosis8. Although p53 acetylation sites may be redundant for its activity as a transcriptional activator of CDKN1A8-12, a mutant lacking all major acetylation sites is transcriptionally inactive in vivo8-10. However, the regulation of p53 is complex, and unacetylated p53 has been shown to be active in the absence of MDM2 and MDM4 — the predominant negative regulators of p53 (REF. 8) — indicating that p53 acetylation is crucial for blocking the repression imposed by MDM2 and MDM4.
The transcriptional repressor hypermethylated in cancer 1 (HIC1) also negatively regulates SIRT1 transcription. HIC1, C terminal binding protein 1 (CTBP1) and SIRT1 form a co-repressor complex13 that binds enhancer elements upstream of the SIRT1 promoter and inhibits SIRT1 expression. HIC1 is a tumour suppressor gene: Hic1-heterozygous mice develop a variety of malignant tumours14. As cells age, HIC1 levels decrease owing to hypermethylation of its promoter15, and loss of HIC1 function promotes tumorigenesis by releasing the repression of SIRT1. In both mouse and human prostate cancer cells, as well as Hic−/− mouse embryonic fibroblasts, reduction or ablation of HIC1 is associated with an increase in SIRT1 expression levels16, indicating one possible explanation of the increased levels of SIRT1 during tumorigenesis. Increased levels of SIRT1 can deacetylate and inactivate p53, allowing the bypass of p53-mediated apoptosis and the promotion of cell survival after DNA damage events have occurred — a potentially tumorigenic scenario.
SIRT1 translation
The tumour suppressor HUR (also known as ElAVL1) is an mRNA binding protein that binds the 3′ UTR of SIRT1 mRNA and helps to stabilize the transcript17. HUR also shows reduced expression as cells age and undergo cellular senescence, and this correlates with the reduced levels of SIRT1 expression in aged senescent cells (see below). An intriguing signalling link also exists between SIRT1 and HUR during DNA damage. After genotoxic stress occurs, the DNA damage-sensing kinase ataxia telangiectasia mutated (ATM) is activated to initiate a downstream signalling pathway that includes phosphorylation of CHK2. This protein, once activated, can phosphorylate HUR and cause disruption of the stabilization of SIRT1 mRNA by HUR17. In this regard, activation of the ATM pathway after a DNA damage event could decrease SIRT1 levels through HUR and promote a p53-mediated apoptotic outcome. Precise regulation of SIRT1 levels, as well as enzymatic activity, may therefore delicately balance the choice of cell cycle arrest or senescence over apoptosis.
Another downstream regulator of SIRT1, the microRNA miR-34a, also binds the 3′ UTR of SIRT1 mRNA18. In contrast to HUR, however, miR-34a prevents translation of SIRT1 and so inhibits SIRT1 deacetylase activity. This induces the accumulation of acetylated p53. Intriguingly, miR-34a can also regulate the expression of two p53 target genes — CDKN1A and PUMA (also known as BBC3) — and is itself regulated by p53 in a positive feedback loop, suggesting that miR-34a has a complex function in terms of regulating pathways upstream and downstream of p53.
Protein–protein interactions
Deleted in breast cancer 1 (DBC1, also known as KIAA1967) and active regulator of SIRT1 (AROS, also known as RPS19BP1) were recently described as direct negative and positive regulators of SIRT1 activity, respectively19-21. Assessment of the levels of p53 acetylation indicates that DBC1 directly inhibits SIRT1 function. Moreover, reduction of DBC1 inhibits p53-mediated apoptosis after induction of double-stranded DNA breaks owing to SIRT1-mediated p53 deacetylation21. Although little is known about the normal function of DBC1, its loss in several cancer cell lines and its inhibition of SIRT1 suggest it may have an important role in tumorigenesis.
AROS can activate SIRT1 and attenuate p53-dependent transcriptional activation. Reduction of AROS through an antisense expression vector increased cell susceptibility to apoptosis after DNA damage19. Both factors represent the first endogenous, direct regulators of SIRT1 function and as such might have important implications in the development of SIRT1-targeted therapeutics (see below).
Necdin was recently described as negatively regulating p53 by potentiating SIRT1-mediated deacetylation of p53 (REF. 22). located predominantly in post-mitotic neurons, necdin is a maternally imprinted gene that promotes cell differentiation and survival. Inhibition of p53 acetylation through a necdin–SIRT1–p53 complex prevents p53-mediated apoptosis in response to DNA damage.
Overall, expression of SIRT1 is subject to complex regulation, a reflection perhaps of its many functions in cell biology that, when deregulated, can influence the development of a number of pathologies. Its integration into the p53 pathway also indicates that it functions to regulate cell proliferation and growth arrest.
SIRT1 in growth arrest and senescence
SIRT1 has an important and direct role in the longevity and cellular senescence of most organisms. There is not yet a causal link between cellular senescence and ageing. However, a correlation exists, as the number of senescent cells increases in mammals as they age23. A large part of our understanding of cellular senescence mechanisms stems from both human cell cultures and mice (BOX 2). Cellular senescence can be divided into two types: replicative senescence, a permanent state of cell growth arrest that occurs after a limited number of cell divisions owing to telomere attrition, and premature senescence, a term used to describe a senescent-like state that can occur from aberrant oncogene expression.
Box 2. Replicative senescence in humans and mice.
In human cells, replicative senescence occurs owing to attrition of chromosomes at their telomere ends68. During S phase, this shortening is reversed by the enzyme telomerase. Proliferative cells extend their lifespan by combating telomeric shortening through this mechanism, though many human cell types exhibit progressive telomeric shortening with each cell division. It is thought that telomere shortening causes loss of specific DNA structures and binding proteins, a process called uncapping, that eventually triggers cell cycle checkpoint pathways leading to replicative senescence69. In mouse embryonic fibroblasts (MEFs), telomeric attrition is not the predominant mechanism for inducing replicative senescence. Instead, these cells undergo a process known as extrinsic senescence, whereby oxidative stress from cell culture conditions cause senescence to occur. MEFs tend to have long telomeres and high levels of telomerase, unlike human cells. Still, the mechanisms by which MEFs and human fibroblasts undergo replicative senescence have similarities. Human fibroblasts can undergo senescence in culture under various O2 conditions and telomeric shortening is accelerated under oxidative stress conditions23. In addition, Terc deficiency in mice enhances telomeric attrition and induces replicative senescence in later cell generations70.
SIRT1 in acute and chronic DNA stress
SIRT1 levels have been shown to decrease in normal human fibroblasts during senescence17. In addition, an increase in the acetylation levels of p53 correlates with replicative or oncogene-induced senescence24. Aberrant expression of oncogenes such as HRAS induces expression of the tumour suppressors ARF and INK4A and halts cell cycle progression through the p53 and RB signalling pathways, respectively23. Although overexpression of SIRT1 can inhibit oncogene-induced senescence as a result of direct deacetylation of p53, removing SIRT1 does not have the opposite effect. Despite Sirt1-null mouse embryonic fibroblasts having hyperacetylated p53, they show increased proliferative capacity at chronic sublethal doses of oxidative stress. Though the mechanisms are unclear, these cells seem to be unable to mount an adequate DNA damage response by upregulating either ARF or p53 under these conditions25-27. Sirt1-null embryos exhibit an impaired DNA damage response and have a deficiency in DNA repair, suggesting that SIRT1 serves an important role in helping a cell recover from DNA damage events28. Recent evidence also shows that SIRT1 relocalizes from repetitive DNA sequences to sites of DNA breaks in response to DNA damage to help promote repair29. SIRT1 also deacetylates NBS1, a component of the MRN (MRE11–RAD50–NBS1) DNA repair complex, further supporting its function in the DNA damage repair pathway30. In mammalian cells, SIRT1 may promote cellular senescence in cells exposed to constant, low-level DNA damage events, such as low-level oxidative stress, as a mechanism for promoting longevity. This is a well-established characteristic attributed to SIRT1 in organisms ranging from yeast to mammals1. Therefore, removal of SIRT1 under these conditions would allow for the continuous expression of acetylated, activated p53 and potentially induce premature senescence and early ageing phenotypes. However, there is a balance with regard to cell growth control: prolonging the presence of senescent cells in a neoplastic microenvironment may promote malignant progression of adjacent epithelial cells, as senescent cells are known to secrete factors that stimulate cell growth and transformation31.
Keeping in mind that the functions of SIRT1 in senescence are complex and still largely undefined, a key unresolved question is whether activation of SIRT1 is beneficial for an organism with regard to ageing and cancer. In the context of senescence and ageing, mammalian cells exhibit antagonistic pleiotropy; that is, cellular senescence can be viewed as beneficial for a young organism to suppress aberrant mutations or combat exposure to oxidative stress, but may be detrimental for an older organism as it promotes phenotypes associated with old age and potentially contributes to tumorigenesis31. Not only is senescence a potent mechanism for tumour suppression, but also it can limit cell growth during times of physical or mechanical stress: hepatic stellate cells undergo senescence upon injury as a way of reducing fibrogenic response to acute tissue damage32. SIRT1-mediated induction of cellular senescence may therefore provide a fail-safe mechanism to prevent continued growth of cells exposed to non-lethal levels of oxidative stress. The ability of SIRT1 to induce senescence in response to other forms of genotoxic stress remains to be elucidated.
SIRT1 and cancer
The roles and functions of SIRT1 in cancer development have become increasingly complex and are still not well understood. SIRT1 clearly has a repressive effect on the tumour suppressor p53 and other genes involved with the stress response, including KU70 and members of the forkhead box (FOXO) transcription factor family33. Deacetylation of FOXO3 in response to DNA double-stranded breaks increases the ability of this transcription factor to induce cell cycle arrest and simultaneously reduce FOXO-mediated apoptosis34,35. Deacetylation of FOXO3 by SIRT1 also provides resistance to oxidative stress. SIRT1 therefore seems to alter the transcriptional profile of FOXO-responsive genes by promoting the transcription of cell cycle arrest genes over apoptosis-inducing genes. In theory, this would promote cell cycle arrest, allow DNA repair and aid the longevity of the organism35. This could also be true for the E2F1–SIRT1 pathway, as discussed earlier. Taken together, these data indicate that SIRT1 has a specific bias towards inducing cell growth arrest after DNA damage events. Its promotion of a DNA repair programme over an apoptotic fate suggests that the physiological role of SIRT1 in mammalian cells is to prevent tumorigenesis and ensure cellular longevity. However, the continuous prevention of apoptosis may aid in the development of neoplastic transformation in cells that are not repairable and that would have otherwise entered an apoptotic fate.
Despite the fact that increased expression of SIRT1 has a clear, negative effect on the tumour suppressors p53 and FOXO by inhibiting their transcriptional functions7,13,34,35, several recent publications have indicated that SIRT1 has tumour-suppressive functions in vivo. In the ApcMin mouse model of colon cancer, increased SIRT1 levels as a result of ectopic expression or calorie restriction resulted in reduced cell proliferation and tumour formation36. Activation of SIRT1 by resveratrol was also shown to limit cell growth and reduce tumour formation in BRCA1-deficient tumour cells as well as Trp53+/−;Sirt1+/− mice28,37. Further, Sirt1-null embryos exhibit an altered DNA damage response and increased genomic instability in a Trp53-null background28. These reports also showed reduced levels of SIRT1 in several human tumour types. This contrasts with previous data that have shown increased levels of SIRT1 in some tumour types16,28,38-40. It is intriguing that SIRT1 does not fit the classical model of a tumour suppressor: deleterious point mutations and gene deletions have not been described in human tumours. In addition, SIRT1 expression in cell culture does not induce cell growth arrest as would be expected of a bone fide tumour suppressor7,41. Moreover, resveratrol also has several cellular targets in addition to SIRT1, and the cell growth effects seen after resveratrol treatment could be attributed to other downstream effects in conjunction with SIRT1 (REF. 42). However, recent evidence does support a role for SIRT1 in enabling DNA repair after DNA damage as opposed to inducing apoptosis.
The data described to date suggest that the precise effect of SIRT1 is highly dependent on the genetics of the cell or tumour in question and the presence or absence of p53. In mammalian cells retaining wild-type p53, SIRT1 promotes cellular senescence and limits cell proliferation, particularly in cells exposed to chronic, non-lethal forms of genotoxic stress, as discussed above. However, in cells that have lost p53 or other tumour suppressors that can directly inhibit SIRT1 expression, such as HIC1, repression of SIRT1 is lost. Several tumour cell lines deficient in p53 overexpress SIRT1 and, more importantly, undergo apoptosis after small interfering RNA knockdown of SIRT1 (REFS 5,17). Dependence on SIRT1 overexpression in these tumour cell lines suggests p53-independent functions that are necessary for continued proliferation. Constitutive FOXO deacetylation could help tumour cells to avoid apoptotic pathways and promote their continued longevity. SIRT1 also deacetylates lys16 on histone H4, and loss of acetylation at this site is an important marker for cancer development43,44. overexpression of SIRT1 would maintain histones in a deacetylated state and provide access for transcription factor complexes, enabling continued gene transcription.
SIRT1 also has significant regulatory effects on metabolic pathways within the cell (BOX 1) and, as metabolic homeostasis is an important element of tumorigenesis, it remains possible that SIRT1 is indirectly affecting cell growth and tumour formation through metabolic pathways.
Together, these data suggest that SIRT1 is involved in a complex regulatory network and may have both tissue- and context-specific functions. The regulatory role of SIRT1 in both tumour suppressor pathways and metabolic pathways suggests that the net effect seen may represent both direct and indirect downstream regulation and is likely to depend on the presence or absence of functional p53.
SIRT1: a chemotherapeutic target?
Intense focus on understanding SIRT1 regulation at the molecular level has led to interest in isolating and developing small-molecule inhibitors and activators of SIRT1 activity to treat a variety of diseases.
The requirement for NAD+ not only links SIRT1 activity with cellular metabolism and energy levels, but also adds a layer of regulatory control to its vast array of functions. Nicotinamide phosphoribosyltransferase (NAMPT) catalyses an increase in cellular NAD+ levels in response to stress and was shown to enhance SIRT1 activity and extend replicative lifespan45. The cell-specific redox state under various physiological conditions (such as high-fat diet or calorie restriction) also affects the NAD+:NADH ratio, and SIRT1 levels correlate with the redox state of cells46. It has also been suggested that measuring the NAD+:NADH ratio is a better indicator of SIRT1 activity in vivo when comparing different cell types47.
Given the recent data discussed above regarding the potential of SIRT1 to suppress tumour development, activators of SIRT1, such as resveratrol and sirtuin-activating compounds, would be advantageous for treating human tumours with mutated p53 as well as metabolic conditions.
In the treatment of specific tumours that seem to be addicted to SIRT1 overexpression, potent inhibitors would be warranted to block SIRT1 activity and potentially induce apoptosis (FIG. 2). Nicotinamide is a precursor to the NAD+ synthesis pathway and blocks SIRT1 function by sequestering a key intermediate48. As this compound is present in most mammalian cells, it acts as a physiological inhibitor of SIRT1 and has been shown to block replicative senescence of primary cells49. Small-molecule inhibitors of SIRT1, such as sirtinol, splitomycin, suramin and dihydrocoumarin, either act non-specifically or have molecular characteristics that are unfavourable for drug development. Other SIRT1-inhibiting compounds, including EX-527 and cambinol, show potent increases of p53 activity but require the combination of DNA-damaging agents for their full effect50-52.
Figure 2. Chemotherapeutic targeting of the p53–SIRT1 pathway.
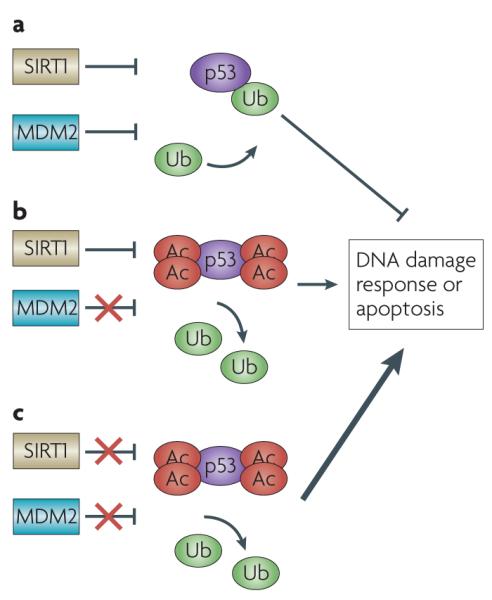
a | MDM2 and SIRT1 negatively regulate p53 and prevent upregulation of p53 under normal conditions. b | Small-molecule inhibitors of MDM2 have been shown to inhibit cell growth and induce apoptosis in some tumour cell lines. c | Use of small-molecule inhibitors of SIRT1 in tumour cells that overexpress the protein may sensitize the cells to a combination of therapeutics, inducing a stronger p53 response and promoting apoptosis of tumour cells. Ac, acetyl; Ub, ubiquitin.
One family of small-molecular inhibitors recently described, called tenovins, inhibit SIRT1 function at single-digit micromolar concentrations and prevent tumour growth in vivo in a p53-dependent manner as single agents53. Tenovin 6 is an attractive drug development candidate for tumour cells overexpressing SIRT1, as it potently inhibits SIRT1 at low concentrations in vivo and has favourable chemical properties for small-molecule drug design.
Chemotherapeutic inhibition of SIRT1 in tumours overexpressing the protein may also be advantageous when used in combination with other compounds that target the p53 pathway. Small-molecule inhibitors of MDM2 cause activation of p53 and exhibit significant growth suppression in several tumour cell lines54. As acetylation of p53 also inhibits the p53–MDM2 interaction, an inhibitor of SIRT1 such as tenovin 6 would promote p53 activation and further sensitize tumour cells to the MDM2 inhibitor. A double hit on this pathway would push the equilibrium toward acetylated, activated p53 and may induce a DNA damage response sufficient for apoptosis in tumour cells overexpressing SIRT1.
SIRT1 has been proposed as a biomarker for tumorigenesis in humans55, so development of potent inhibitors may change the current oncology landscape for treating a wide variety of tumours. However, the recent data implicating SIRT1 as a tumour suppressor indicates that these drugs will need to be pursued with caution. Good biomarkers for tumours that are reliant on SIRT1 for survival need to be established, such that SIRT1 inhibitory drugs are only used in patients who will derive benefit and not harm from these agents.
Future progress
Significant strides have been made in understanding SIRT1 regulation and function in the context of cell metabolism, cellular senescence and cancer over the past 20 years. Nevertheless, the function of SIRT1 in cancer remains complex, with many questions still to be answered. Perhaps most pressing in the light of recent data is understanding how SIRT1 function is influenced during tumour evolution. Why can SIRT1 overexpression be a useful therapeutic target in some tumours and yet have a tumour-suppressive effect in others? It seems likely that SIRT1 overexpression is oncogenic in cells expressing wildtype p53 but has the opposite effect in cells with mutated p53. The correlation between SIRT1 expression and p53 mutation in human tumours needs to be elucidated, especially as p53 is mutated in more than 50% of human tumours, and it remains feasible that activating SIRT1 alone might be sufficient to induce tumour suppression in human cancers with mutated p53. What is also not clear is how important SIRT1 is for induction of growth arrest as a tumour-suppressive mechanism and how easily this can be subverted in tumour cells that have engaged other mechanisms to avoid senescence. Future work will probably refine our understanding of how SIRT1 is regulated during cellular stress and of the mechanism by which cells choose cellular senescence or apoptotic outcomes. SIRT1 is a crucial molecular component of a variety of pathways that are important in human health and longevity and it will certainly remain a focus of therapeutic research for years to come.
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=geneAPAF1|BAX|CDKN1A|miR-34a|PUMA|Terc|TP73
National cancer institute Drug Dictionary: http://www.cancer.gov/drugdictionary/etoposide|resveratrol
Pathway interaction Database: http://pid.nci.nih.gov/p53
FURTHER INFORMATION
W. Gu’s homepage: http://icg.cpmc.columbia.edu/faculty_Gu.htm
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Acknowledgements
This work was supported in part by grants from NIH/NCI to W.G. W.G. is an Ellison Medical Foundation Senior Scholar in Aging.
References
- 1.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nature Rev. Mol. Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 2.Campisi J. Suppressing cancer: the importance of being senescent. Science. 2005;309:886–887. doi: 10.1126/science.1116801. [DOI] [PubMed] [Google Scholar]
- 3.Wang C, et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nature Cell Biol. 2006;8:1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- 4.Nahle Z, et al. Direct coupling of the cell cycle and cell death machinery by E2F. Nature Cell Biol. 2002;4:859–864. doi: 10.1038/ncb868. [DOI] [PubMed] [Google Scholar]
- 5.Ford J, Jiang M, Milner J. Cancer-specific functions of SIRT1 enable human epithelial cancer cell growth and survival. Cancer Res. 2005;65:10457–10463. doi: 10.1158/0008-5472.CAN-05-1923. [DOI] [PubMed] [Google Scholar]
- 6.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 7.Luo J, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 8.Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krummel KA, Lee CJ, Toledo F, Wahl GM. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc. Natl Acad. Sci. USA. 2005;102:10188–10193. doi: 10.1073/pnas.0503068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 13.Chen WY, et al. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 14.Chen WY, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nature Genet. 2003;33:197–202. doi: 10.1038/ng1077. [DOI] [PubMed] [Google Scholar]
- 15.Chen W, et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell. 2004;6:387–398. doi: 10.1016/j.ccr.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 16.Huffman DM, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 17.Abdelmohsen K, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell. 2007;25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl Acad. Sci. USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 20.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 21.Zhao W, et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J. Neurosci. 2008;28:8772–8784. doi: 10.1523/JNEUROSCI.3052-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lombard DB, et al. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 24.Pearson M, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 25.Cheng HL, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl Acad. Sci. USA. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chua KF, et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell. Metab. 2005;2:67–76. doi: 10.1016/j.cmet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Langley E, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang RH, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oberdoerffer P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan Z, Zhang X, Sengupta N, Lane WS, Seto E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell. 2007;27:149–162. doi: 10.1016/j.molcel.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krtolica A, Campisi J. Cancer and aging: a model for the cancer promoting effects of the aging stroma. Int. J. Biochem. Cell Biol. 2002;34:1401–1414. doi: 10.1016/s1357-2725(02)00053-5. [DOI] [PubMed] [Google Scholar]
- 32.Krizhanovsky V, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saunders LR, Verdin E. Sirtuins: critical regulators at the crossroads between cancer and aging. Oncogene. 2007;26:5489–5504. doi: 10.1038/sj.onc.1210616. [DOI] [PubMed] [Google Scholar]
- 34.Motta MC, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 35.Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 36.Firestein R, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS ONE. 2008;3:e2020. doi: 10.1371/journal.pone.0002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang RH, et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol. Cell. 2008;32:11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bradbury CA, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- 39.Hida Y, Kubo Y, Murao K, Arase S. Strong expression of a longevity-related protein, SIRT1, in Bowen’s disease. Arch. Dermatol. Res. 2007;299:103–106. doi: 10.1007/s00403-006-0725-6. [DOI] [PubMed] [Google Scholar]
- 40.Stunkel W, et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007;2:1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 41.Huang J, et al. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE. 2008;3:e1710. doi: 10.1371/journal.pone.0001710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fulda S, Debatin KM. Resveratrol modulation of signal transduction in apoptosis and cell survival: a mini-review. Cancer Detect. Prev. 2006;30:217–223. doi: 10.1016/j.cdp.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 43.Vaquero A, et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 44.Fraga MF, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 45.van der Veer E, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2007;282:10841–10845. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Q, et al. Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc. Natl Acad. Sci. USA. 2007;104:829–833. doi: 10.1073/pnas.0610590104. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Chen D, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 49.Lim CS, Potts M, Helm RF. Nicotinamide extends the replicative life span of primary human cells. Mech. Ageing Dev. 2006;127:511–514. doi: 10.1016/j.mad.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Nayagam VM, et al. SIRT1 modulating compounds from high-throughput screening as anti-inflammatory and insulin-sensitizing agents. J. Biomol. Screen. 2006;11:959–967. doi: 10.1177/1087057106294710. [DOI] [PubMed] [Google Scholar]
- 51.Heltweg B, et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006;66:4368–4377. doi: 10.1158/0008-5472.CAN-05-3617. [DOI] [PubMed] [Google Scholar]
- 52.Solomon JM, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 2006;26:28–38. doi: 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lain S, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell. 2008;13:454–463. doi: 10.1016/j.ccr.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim CS. Human SIRT1: a potential biomarker for tumorigenesis? Cell Biol. Int. 2007;31:636–637. doi: 10.1016/j.cellbi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 56.Westphal CH, Dipp MA, Guarente L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem. Sci. 2007;32:555–560. doi: 10.1016/j.tibs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 57.Jones JM, et al. Absence of p53 in a mouse mammary tumor model promotes tumor cell proliferation without affecting apoptosis. Cell Growth Differ. 1997;8:829–838. [PubMed] [Google Scholar]
- 58.Mai V, et al. Calorie restriction and diet composition modulate spontaneous intestinal tumorigenesis in ApcMin mice through different mechanisms. Cancer Res. 2003;63:1752–1755. [PubMed] [Google Scholar]
- 59.Berrigan D, Perkins SN, Haines DC, Hursting SD. Adult-onset calorie restriction and fasting delay spontaneous tumorigenesis in p53-deficient mice. Carcinogenesis. 2002;23:817–822. doi: 10.1093/carcin/23.5.817. [DOI] [PubMed] [Google Scholar]
- 60.Cohen HY, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 61.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 62.Sun C, et al. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell. Metab. 2007;6:307–319. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 63.Li X, et al. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 64.Picard F, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moynihan KA, et al. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell. Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 66.Banks A, et al. SIRT1 gain of function increases energy efficient and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boily G, et al. SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS ONE. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21:532–540. doi: 10.1038/sj.onc.1205080. [DOI] [PubMed] [Google Scholar]
- 69.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J. Clin. Invest. 2004;113:8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Espejel S, Blasco MA. Identification of telomere-dependent “senescence-like” arrest in mouse embryonic fibroblasts. Exp. Cell Res. 2002;276:242–248. doi: 10.1006/excr.2002.5533. [DOI] [PubMed] [Google Scholar]