

Abstract
Inhibitory and excitatory synapses play a fundamental role in information processing in the brain. Excitatory synapses usually are situated on dendritic spines, small membrane protrusions that harbor glutamate receptors and postsynaptic density components and help transmit electrical signals. In recent years, it has become evident that spine morphology is intimately linked to synapse function—smaller spines have smaller synapses and support reduced synaptic transmission. The relationship between synaptic signaling, spine shape, and brain function is never more apparent than when the brain becomes dysfunctional. Many psychiatric and neurologic disorders, ranging from mental retardation and autism to Alzheimer’s disease and addiction, are accompanied by alterations in spine morphology and synapse number. In this review, we highlight the structure and molecular organization of synapses and discuss functional effects of synapse pathology in brain disease.
Keywords: Synaptic plasticity, Spine morphology, Synapse pathology, Neurologic diseases, Psychiatric disorders
Introduction
The brain is the center of the nervous system and the most complex biological structure known. All thoughts, emotions, memories, behaviors, dreams, and other aspects of cognition arise within the brain. The brain coordinates the abilities to move, touch, smell, taste, hear, and see. It enables people to form words, understand mathematics, communicate with others, make decisions, compose and appreciate music, plan ahead, and even fantasize. It comes as no surprise that alterations in brain function account for many, if not most, neurologic and psychiatric disorders.
The human brain consists of more than 1011 (100 billion) neurons, which process and transmit information in the form of electrical signals. Communication between neurons occurs at specialized junctions called synapses. During the past century, basic neuroscience has taught us a great deal about the molecular and cellular mechanisms of synapse formation, stability, and function. Precise control of synaptic development and connectivity is critical for maintaining accurate neuronal network activity and normal brain function. Now it is widely believed that information in the brain can be stored in the form of altered structure and chemistry of synapses and/or by the formation of new synapses and the elimination of old ones [1]. This so-called plasticity of synapses is believed to be the basis of learning and memory in the brain.
It is not surprising that the inappropriate loss of synaptic stability may lead to the disruption of neuronal circuits and to brain diseases. Whether as the result of genetics, drug use, the aging process, viral infections, or other various causes, dysfunction in neuronal communication is almost certainly the underlying cause of many psychiatric and neurologic diseases, such as mental retardation [2], schizophrenia [3], Parkinson’s disease [4], autism [5], Alzheimer’s disease (AD) [6], compulsive behavior [7], and addiction [8]. Recent studies show that many neuropsychiatric diseases are characterized by synaptic pathology—including abnormal density and morphology of dendritic spines, synapse loss, and aberrant synaptic signaling and plasticity [9]. For AD, synaptic loss is the best current pathologic correlate of cognitive decline, and synaptic dysfunction is evident long before synapses and neurons are lost. The synapse thus constitutes an important target for treatments to slow progression and preserve cognitive and functional abilities in the disease. Understanding brain disorders is, at least in part, a matter of understanding the biochemical and cell biological basis of synaptic function and plasticity.
In this review, we discuss recent evidence that alteration in synapse structure and function underlies several psychiatric and neurologic disorders. We describe our current understanding of the molecular organization of excitatory and inhibitory synapses and propose that basic cell biological mechanisms link synapse function with neuropsychiatric health and disease.
Microanatomy of the Synapses
Chemical synapses consist of presynaptic axon terminals harboring synaptic vesicles and a postsynaptic region (usually on dendrites) containing neurotransmitter receptors (Fig. 1). The pre- and postsynaptic sites are separated by a gap of 20 to 25 nm, the synaptic cleft [10, 11]. A wide variety of cell adhesion molecules hold pre- and postsynaptic membranes together at the appropriate separation. Recently, several cell adhesion molecules, including N-cadherin, have been implicated in synapse formation and function [12, 13]. In humans, alterations in genes encoding the cell adhesion molecules neuroligin (NLGN) and neurexin (NRXN) recently were implicated in autism, directly linking synaptic proteins to cognition and its disorders [5].
Fig. 1.
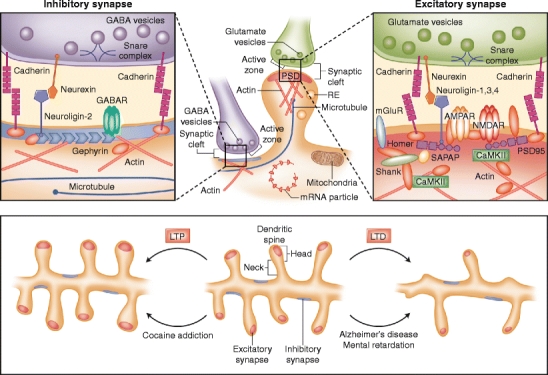
Molecular architecture of inhibitory and excitatory synapses. The top panels show excitatory and inhibitory synapses. Excitatory synapses target on mature mushroom-shaped spines containing a prominent postsynaptic density (PSD), and inhibitory synapses are present along the dendritic shaft lacking postsynaptic thickening. Various organelles support the synapse; mitochondria provide energy, polyribosomes and RNA particles allow local protein synthesis, recycling endosomes (REs) transport internalized synaptic receptors back to the plasma membrane, and the cytoskeleton regulates spine dynamics. The abundant actin cytoskeleton is connected to the PSD and is the primary determinant of spine shape and motility. Transient invasion of dynamic microtubule into dendritic spines can regulate formation of spine head protrusions and rapid spine growth. Excitatory and inhibitory synapses contain a unique set of channels, scaffolding proteins, and other postsynaptic molecules. The microanatomy of the inhibitory and excitatory synapses and their organization of proteins and protein–protein interactions are depicted in the left and right panels, respectively. Major families of postsynaptic proteins are shown, including scaffolding proteins, adhesion molecules, and receptors. The lower panel shows major morphologic events occurring in dendritic spines upon long-term potentiation (LTP; left) or long-term depression (LTD; right). In Alzheimer’s disease and mental retardation, signaling cascades are triggered similar to LTD, leading to thinner, immature spines. In contrast, cocaine addiction shows similarities to LTP, resulting in bigger, mushroom-shaped, mature spines. The molecular and morphologic changes in the synapse are hallmarks of the disease pathology and are responsible for the cognitive alterations in neuropsychiatric diseases. CamKII—Ca2+/calmodulin-dependent kinase II; AMPAR—amino-3-hydroxy-5-methyl-4-isoazolepropionate receptor; GABA—γ-aminobutyric acid; GABAR—GABA receptor; NMDAR—N-methyl-d-aspartate receptor; mGluR—metabotropic glutamate receptor; SAPAP—synapse-associated protein 90/PSD-95-associated protein
Presynaptic Structure and Function
The nerve impulse, or action potential, traveling along the axonal membrane of the presynaptic neuron cannot cross the synaptic cleft to communicate with postsynaptic neurons. Therefore, the electric signal is carried at the synapse by neurotransmitters, such as glutamate or γ-aminobutyric acid (GABA). These neurotransmitters are made by the presynaptic neuron and stored in synaptic vesicles at presynaptic terminals (Fig. 1). Synaptic vesicles make contact with a thickening of the presynaptic plasma membrane, named the active zone, where vesicle fusion and exocytosis of neurotransmitters occur [14]. Genetic and biochemical studies from mice, Caenorhabditis elegans, and Drosophila have identified numerous proteins involved in controlling synaptic vesicle fusion and neurotransmitter release [15]. The docking and fusion of vesicles at the presynaptic membrane is at least controlled by the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex [14]. Many other presynaptic proteins bind to SNAREs and regulate the formation or disassembly of this complex, whereas others control Ca2+-dependent neurotransmitter release [15]. Other studies show a critical role for presynaptic molecules in the pathology of neurodegenerative disease. For example, the presynaptic protein α-synuclein is involved in the pathogenesis of Parkinson’s disease [16].
Postsynaptic Structure and Function
Neurotransmitters released from the presynaptic terminal act on neurotransmitter receptors on the membrane of the postsynaptic neuron. Whether a synapse is excitatory or inhibitory determines the postsynaptic current displayed, which in turn is a function of the type of receptors and neurotransmitters operating at the synapse (Fig. 1). There are two types of postsynaptic receptors that recognize neurotransmitters: ligand-gated ion channels (ionotropic receptors) and G protein-coupled (metabotropic) receptors. The binding of glutamate to amino-3-hydroxy-5-methyl-4-isoazolepropionate (AMPA)-type and N-methyl-d-aspartate (NMDA)-type ionotropic glutamate receptors leads to excitatory synaptic transmission [17], whereas the interaction of GABA and ionotropic GABAA receptors allows an influx of negatively charged chloride ions and provides the major form of inhibitory synaptic transmission [18]. Recently, the architecture and atomic structure of the ionotropic glutamate receptor were resolved [19].
Neuronal signal processing is mediated by integration of excitatory and inhibitory synaptic inputs. A single neuron usually has hundreds or thousands of excitatory and inhibitory synapses at its dendrites or cell body, and whether this neuron fires an action potential depends on the total input of all these synapses. If the postsynaptic neuron receives many strong inhibitory synaptic inputs, the likelihood of the cell firing an action potential is very low. Therefore, precise regulatory mechanisms must exist to maintain the balance of excitatory and inhibitory synaptic transmission, the so-called E/I balance. Alteration in the E/I synapse balance has been proposed for many brain disorders, including autism and schizophrenia [5, 18].
Immediately behind the postsynaptic membrane is an elaborate complex of interlinked proteins called the postsynaptic density (PSD), in which adhesion molecules, receptors, and their associated signaling proteins are highly concentrated (Fig. 1). The presence of a prominent PSD is characteristic of excitatory synapses; in contrast, inhibitory synapses lack postsynaptic thickening [10, 11]. The PSD is identified by electron microscopy as electron-dense material approximately 20 to 30 nm thick and around 300 nm wide. Recent electron micrographic studies are beginning to reveal the precise three-dimensional organization of the PSD and its constituent protein complexes [20]. PSDs are composed primarily of glutamate receptors, ion channels, cell adhesion molecules, and signaling enzymes, as well as membrane trafficking, cytoskeletal, and scaffolding proteins [11]. Key among these are NMDA and AMPA receptors, PSD-95, Ca2+/calmodulin-dependent kinase II (CamKII), NLGN, Shank family proteins, synapse-associated protein-associated protein (SAPAP), and actin. The PSD functions primarily as a postsynaptic organizing structure—it clusters receptors, adhesion molecules, and channels and assembles a variety of signaling molecules at the postsynaptic membrane [21, 22].
Glutamate receptors and PSD proteins play a central role in excitatory synaptic plasticity. Current models show that intense NMDA receptor activation triggers a signaling cascade in the PSD that induces recruitment of AMPA receptors into the postsynaptic membrane, leading to long-term potentiation (LTP) of synaptic strength, whereas weaker prolonged activation of NMDA receptors leads to removal of postsynaptic AMPA receptors and hence long-term depression (LTD) [17]. Thus, it is of key importance that the trafficking of synaptic AMPA receptors is controlled carefully to modify synaptic strength during plasticity. Misregulation of synaptic trafficking may contribute to various brain disorders by preventing appropriate synaptic signaling and plasticity [9].
Spine Morphology Controls Synaptic Function
Dendritic spines are small membranous protrusions that contain the postsynaptic machinery, including glutamate receptors, the PSD, the actin cytoskeleton, and a wide variety of membrane-bound organelles, such as smooth endoplasmic reticulum, mitochondria, and endosomes (Fig. 1) [11]. Typical spines have a bulbous head connected to the dendritic shaft through a thin spine neck. Electron microscopy studies identified several categories of spines based on their shape and size: thin, stubby, cup, and mushroom shaped [10]. Live imaging studies showed that spines are remarkably dynamic, changing size and shape over timescales of seconds to minutes and of hours to days [23]. Dynamic changes in spine morphology are closely linked to changes in strength of synaptic connections [1]. Mushroom spines have larger, more complex PSDs with a higher density of glutamate receptors and are more sensitive to glutamate [24]. The size of the spine head is correlated with the dimensions of the PSD and the size of the presynaptic active zone [10].
Spine morphology is subject to rapid alteration depending on neuronal activity and glutamate receptor activation. Induction of LTP causes enlargement of spine heads, whereas activity patterns that induce LTD cause spine head shrinkage (Fig. 1) [1, 24]. Long-term in vivo two-photon fluorescence imaging showed that dendritic spines undergo structural changes in size and shape after novel sensory experience [23]. Interestingly, abnormal spine structures often are associated with various neurologic disorders, such as fragile X syndrome (FXS), Rett’s syndrome, and Down syndrome [9].
Recent studies have identified several cell biological pathways that regulate dendritic spine morphology [25, 26]. Trafficking of recycling endosomes (REs) by motor protein myosin Vb (MyoVb) leads to spine enlargement [27••, 28]. Transient invasion of dynamic microtubule into dendritic spines is associated with the formation of spine head protrusions and rapid spine growth [29••, 30]. Most signaling pathways controlling spine shape seem to directly or indirectly regulate the actin cytoskeleton [25, 26]. Therefore, it is not surprising that several genes that encode factors involved in spine structure and organization have been found mutated in human brain disease. GTPase-activating proteins and guanosine exchange factors are mutated in individuals with mental retardation and autism [31]. LIM kinase 1, a serine/threonine kinase controlling actin dynamics, and CYLN2, encoding for the microtubule plus-end binding protein CLIP-115, are hemizygously deleted in Williams-Beuren syndrome (WBS), leading to abnormal thin spines, mental retardation, and visuospatial cognitive deficits [32]. Consequently, there now is considerable interest in understanding the underlying molecular mechanisms of spine pathology and the relationship between spine alterations and cognitive deficits.
Neuropsychiatric Disorders and Synapse Alteration
Neurodegenerative diseases, such as AD, Parkinson’s disease, and Huntington’s disease, are caused by gradual neuronal death, leading to decline in movement control, memory, and cognition. The role of synaptic pathology in AD is particularly interesting because β-amyloid (Aβ) oligomers, which are formed after proteolytic cleavage of the amyloid precursor protein (APP) may interfere with basic synaptic mechanisms at an early disease stage [6, 33]. Moreover, alterations in synaptic receptor trafficking, abnormal spine morphology, and defects in synaptic function have been reported in animal models of neuropsychiatric disorders including addiction and schizophrenia [8, 34], as well as in models of mental retardation, such as FXS [35]. Recently, autism was associated with mutations in synaptic adhesion and scaffolding molecules, which most likely have important consequences for E/I balance [5]. Here, we discuss in more detail the relationship between these disorders and synapse pathology.
Autism Spectrum Disorders
Autism spectrum disorders (ASDs), including autism, Asperger syndrome, and pervasive developmental disorder-not otherwise specified, are diagnosed on the basis of three behavioral features: deficits in social communication, absence or delay in language, and stereotypy. ASDs are brain disorders typically diagnosed before the second or third year of life, during the time of human synapse formation and maturation [36]. ASDs have a strong genetic basis and are the most heritable (∼80%), suggesting that the disease is largely determined by genes and not by the environment. Despite the high heritability, the identification of genetic factors in ASDs has proved difficult, at least partly because ASDs are characterized by a high degree of genetic heterogeneity.
It has been widely speculated that alterations at the synaptic level make individuals more vulnerable to developing ASDs. This theory was confirmed by the identification of mutations affecting the synaptic cell adhesion molecules NRXN and NLGN in individuals with ASD [37]. NRXNs and NLGNs are thought to form a transsynaptic complex stabilized by synaptic scaffolding proteins [5]. NRXNs are presynaptic receptors, whereas NLGNs, the ligands of NRXNs, are present on the postsynaptic side, where they interact with PSD-95 and its binding partners, SAPAP and Shank (Fig. 1). Mice lacking NRXNs or NLGNs show strong deficits in synaptic transmission but not in synapse number, suggesting that NRXNs and NLGNs are not required for the initial formation of synapses but are needed for synaptic function [5]. Moreover, different members of the NLGN family control the functional E/I balance in hippocampal neurons (Fig. 1).
The human genome contains three NRXN genes and four NLGN genes. Several point mutations, distinct translocation events, and different large-scale deletions in NRXN1, NLGN3, and NLGN4 were observed in patients with autism [37]. In addition, several mutations in the gene encoding Shank3 have been found in patients with ASDs. However, Shank3 mutations often are observed in nonsymptomatic siblings, suggesting that Shank3 might only increase the chance of ASDs rather than causing it [5]. Functional studies of the NLGN3-R451C knock-in mice, which mimic a human autistic mutation, showed enhanced spatial learning and impaired social interaction. These mice exhibited a shift of E/I balance by a selective enhancement of GABAA receptor-mediated inhibitory synaptic transmission [38••]. Interestingly, epilepsy, which also is caused by an imbalance between excitatory and inhibitory synaptic transmission, is observed in patients with autism.
These NRXN, NLGN, and Shank3 mutations still concern a limited number of cases (<1% of individuals), but mutations in other synaptic proteins, such as contactin-associated protein-like 2 gene (CNTNAP2) and calcium channel voltage-dependent L-type α1C subunit gene (CACNA1C), were identified in patients with ASD-related syndromes [37]. Moreover, single nucleotide polymorphisms surrounding several prominent ASD candidate loci, including NMDA receptor (GRIN2A), kainate receptor (GRIK2), and cell adhesion molecule cadherin 9/10 (CDH9/10) genes, were identified as risk factors for ASD [39]. Overall, these linkage, association, and functional studies suggest that synaptic dysfunction most likely is the common defect in ASDs.
Mental Retardation: Fragile X Syndrome
FXS, the most common inherited form of mental retardation, affecting approximately 1 in 4,000 males and 1 in 8,000 females, is characterized by reduced intellectual ability, hyperactivity, hypersensitivity to sensory stimuli, anxiety, impaired visuospatial processing, and developmental delay. Thirty percent of children with FXS are diagnosed with autism, and 2% to 5% of autistic children have FXS [40]. A trinucleotide (CGG) repeat expansion, inactivating the FMR1 gene on the X chromosome, leads to the absence of the fragile X mental retardation protein (FMRP), resulting in FXS [41]. The full mutation state is defined as more than 200 CGG repeats in the 5′ untranslated region of the FMR1 gene. Studies in patients and animal models of FXS have identified marked alterations in dendritic spine morphology [2]. Several studies report abnormally long, thin, and immature filopodia-like spines, which are indicative of alterations in synapse development and/or function in FXS.
In normal individuals, FMRP regulates the local processing of a subset of mRNAs—transport, translation, and stability—in response to activation of metabotropic glutamate receptors (mGluRs) [42]. FMRP binds to several dendritically localized mRNAs that encode synaptic proteins, such as PSD-95, activity-regulated cytoskeletal protein, elongation factor 1a, SAPAP3/4, AMPA receptor subunit GluR1/2, and CamKIIα, which is consistent with the observed synapse alterations in FXS [42]. Remarkably, many of the synapse and behavioral deficits are rescued by reduced mGluR5 function [35]. Experimental evidence shows that AMPA receptor internalization triggered by mGluR5 stimulation is increased in FMRP knockout mice. It is tempting to speculate that FMRP inhibits the translation of mRNAs that are important for AMPA receptor trafficking, which might explain the observed increase in LTD and excess of filopodia-like spines in FXS. The potential link between the dysregulation PSD-95 mRNA in FXS and LTD, is of especially great interest [43•].
Neurodegenerative Disease: Alzheimer’s Disease
AD, the most common cause of dementia, is characterized by early memory deficits, followed by the gradual decline of cognitive and intellectual functions. Generally, it is diagnosed in people over 65 years of age, although early-onset AD may occur [33]. In familial AD, autosomal dominant mutations have been identified in the APP gene as well as in genes encoding for presenilin 1 and 2, components of the proteolytic-γ-secretase complex. AD is characterized by loss of neurons and synapses in the hippocampus, cerebral cortex, and subcortical regions, as well as Aβ plaques and neurofibrillary lesions. The major protein component of plaques is the polypeptide Aβ derived from proteolytic cleavage of APP. Consistently, mutations linked to early-onset familial AD lead to increased Aβ production. Importantly, Aβ isolated directly from human AD brains can cause impaired synaptic plasticity and memory in rodents [44•]. Synaptic activity and chronic sleep restriction significantly increase the amount of Aβ in brain interstitial fluid and plaque formation in APP transgenic mice [45]. Mechanistically, Aβ is thought to mediate toxicity by triggering signaling cascades on the postsynaptic membrane. Neuronal culture experiments revealed that surface APP is internalized by clathrin-mediated endocytosis and that proteolytic Aβ cleavage occurs in the endosomal pathway in the axon. Subsequently, Aβ recycles to the cell surface, where it is released into the extracellular fluid and triggers signaling cascades on the postsynaptic membrane, sharing remarkable similarities with LTD, including increased synaptic AMPA receptor endocytosis and dendritic spine loss [46]. Recent studies showed that inhibition of endocytosis reduces APP internalization and immediately lowers Aβ levels in vivo [47•]. Thus, it is tempting to speculate that in its early stages, AD is a disorder of synaptic receptor trafficking and synapse dysfunction.
Addiction
Drug addiction, a chronic neuropsychiatric pathologic condition caused, in part, by powerful and long-lasting memories of the drug experience, has genetic, psychosocial, and environmental dimensions. Generally, it is thought that synaptic plasticity mechanisms normally used to reinforce associated behavior become pathologic in patients with drug addiction. Several in vivo studies indicate that addictive drugs, such as cocaine, nicotine, and ethanol, influence synaptic plasticity mechanisms in brain circuits involved in reinforcement and reward processing [48]. Although modifications at the synaptic level are complex, the current model suggests that addictive drugs might induce LTP in the reward system. For instance, administration of cocaine elicits NMDA-dependent LTP at excitatory synapses in the dopamine system [8]. Consistently, changes in dendritic spine and synaptic proteins have been described after chronic drug administration. An increase in dendritic spine density and larger spine heads have been observed in the striatum and prefrontal cortex of rats after 45 min of acute cocaine administration to those withdrawn earlier from chronic cocaine exposure [48]. Recent data show that several synaptic mechanisms, such as AMPA receptor trafficking, mGluR signaling, and spine actin dynamics, are affected after chronic drug administration [8]. Interestingly, exposure to cocaine generates inactive or “silent” synapses [49•]. Reversing or preventing drug-induced synaptic modifications might provide treatments for drug addiction.
Conclusions
In recent years, genetic linkage studies have identified several synaptic genes contributing to neuropsychiatric disorders [6, 37, 41, 50]. At the same time, basic neurobiological research has led to a better understanding of the molecular composition, structure, and function of synapses (Fig. 1) [11]. Novel pathways upstream of the synapse have been discovered in which failure of the cellular machinery leads to synaptic dysfunction and neuropsychiatric phenotypes. Small non-coding microRNAs that repress the translation of target mRNAs are emerging as important pathophysiologic mechanisms for neurologic and psychiatric disease. Abnormal regulation of protein turnover, chromatin remodeling, and genomic imprinting also are suggested to result in synapse pathology [50]. For instance, the gene responsible for Angelman syndrome encodes for ubiquitin-protein ligase E3A (UBE3A) and marks synaptic proteins for degradation. The gene causing Rett syndrome encodes for X-linked gene encoding methyl-CpG-binding protein 2 (MECP2) and is thought to modulate the expression of synaptic activity-regulated genes, such as brain-derived neurotrophic factor (BDNF).
In some cases, such as in FXS, the basic neurobiological mechanisms underlying the symptoms are well studied and become more clear, but in other cases, the pathways are only beginning to be elucidated. Loss of function of a single gene in FXS [41] or a limited number of genes in WBS [32] provides a unique opportunity to uncover basic neurobiological mechanisms underlying neuropsychiatric diseases. Unfortunately, this model does not account for the common forms of most neuropsychiatric disorders, which are etiologically heterogeneous and complex and likely determined by the combination of variants and/or defects in multiple genes, each having a small effect. For example, genome-wide association studies recently identified polymorphic variants in genes encoding synaptic proteins as important determinants of the risk of developing ASD [39].
Other studies have implied that the genetic factors between neuropsychiatric disorders may not be as diverse as the clinical manifestations. It is not unlikely that an alteration in the same gene may be associated with different phenotypes in different people: one genetic alteration might cause psychosis in one individual, obsessive-compulsive disorder in another, and autism in yet another. Almost certainly, these neuropsychiatric phenotypes are modified by the genetic background and experiences throughout life. It therefore is very likely that many disorders that involve synapse dysfunction manifest in different ways. Conversely, different mutations can produce similar syndromes. For example, mutations in several different genes encoding for proteins regulating the E/I synaptic balance have been linked to epilepsy [18].
Neuropsychiatric diseases nicely illustrate the importance of synapse-specific molecules for normal synapse composition and plasticity. To gain better insight into how synapse pathology underlies psychiatric and neurologic disease, it is essential to combine basic research with clinical genetic studies. In this way, knowledge about synapse function at the basic level has an immediate and significant impact on clinically relevant issues. Finally, synaptic molecules also are important future targets for protective treatments, to slow disease progression and preserve cognitive and functional abilities by preserving synaptic structure and function.
Acknowledgments
Dr. Hoogenraad is supported by The Netherlands Organization for Scientific Research (NWO-ALW/ECHO), Netherlands Organization for Health Research and Development (ZonMw-VIDI/TOP), European Science Foundation (European Young Investigators [EURYI] Award), EMBO Young Investigators Programme (YIP), and Human Frontier Science Program Career Development Award (HFSP-CDA).
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 2.Pfeiffer BE, Huber KM. The state of synapses in fragile X syndrome. Neuroscientist. 2009;15(5):549–567. doi: 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59(10):929–939. doi: 10.1016/j.biopsych.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Calabresi P, Picconi B, Parnetti L, Di Filippo M. A convergent model for cognitive dysfunctions in Parkinson’s disease: the critical dopamine-acetylcholine synaptic balance. Lancet Neurol. 2006;5(11):974–983. doi: 10.1016/S1474-4422(06)70600-7. [DOI] [PubMed] [Google Scholar]
- 5.Sudhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455(7215):903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 7.Welch JM, Lu J, Rodriguiz RM, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448(7156):894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8(11):844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 9.Blanpied TA, Ehlers MD. Microanatomy of dendritic spines: emerging principles of synaptic pathology in psychiatric and neurological disease. Biol Psychiatry. 2004;55(12):1121–1127. doi: 10.1016/j.biopsych.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 12.Craig AM, Graf ER, Linhoff MW. How to build a central synapse: clues from cell culture. Trends Neurosci. 2006;29(1):8–20. doi: 10.1016/j.tins.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008;31(9):487–494. doi: 10.1016/j.tins.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 15.Jin Y, Garner CC. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol. 2008;24:237–262. doi: 10.1146/annurev.cellbio.23.090506.123417. [DOI] [PubMed] [Google Scholar]
- 16.Waxman EA, Giasson BI. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta. 2009;1792(7):616–624. doi: 10.1016/j.bbadis.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 18.Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9(5):331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462(7274):745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X, Winters C, Azzam R, et al. Organization of the core structure of the postsynaptic density. Proc Natl Acad Sci U S A. 2008;105(11):4453–4458. doi: 10.1073/pnas.0800897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6(6):423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- 22.Renner M, Specht CG, Triller A. Molecular dynamics of postsynaptic receptors and scaffold proteins. Curr Opin Neurobiol. 2008;18(5):532–540. doi: 10.1016/j.conb.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10(9):647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 24.Kasai H, Matsuzaki M, Noguchi J, et al. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26(7):360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- 25.Yoshihara Y, De Roo M, Muller D. Dendritic spine formation and stabilization. Curr Opin Neurobiol. 2009;19(2):146–153. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16(1):95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Wang Z, Edwards JG, Riley N, et al. Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell. 2008;135(3):535–548. doi: 10.1016/j.cell.2008.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlager MA, Hoogenraad CC. Basic mechanisms for recognition and transport of synaptic cargos. Mol Brain. 2009;2(1):25. doi: 10.1186/1756-6606-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaworski J, Kapitein LC, Gouveia SM, et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61(1):85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 30.Hoogenraad CC, Bradke F. Control of neuronal polarity and plasticity—a renaissance for microtubules? Trends Cell Biol. 2009;19(12):669–676. doi: 10.1016/j.tcb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64(1):58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- 32.Hoogenraad CC, Akhmanova A, Galjart N, De Zeeuw CI. LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. Bioessays. 2004;26(2):141–150. doi: 10.1002/bies.10402. [DOI] [PubMed] [Google Scholar]
- 33.Gotz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9(7):532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- 34.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8(6):413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 35.Dolen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008;586(6):1503–1508. doi: 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lord C, Cook EH, Leventhal BL, Amaral DG. Autism spectrum disorders. Neuron. 2000;28(2):355–363. doi: 10.1016/S0896-6273(00)00115-X. [DOI] [PubMed] [Google Scholar]
- 37.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9(5):341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tabuchi K, Blundell J, Etherton MR, et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318(5847):71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagerman RJ, Ono MY, Hagerman PJ. Recent advances in fragile X: a model for autism and neurodegeneration. Curr Opin Psychiatry. 2005;18(5):490–496. doi: 10.1097/01.yco.0000179485.39520.b0. [DOI] [PubMed] [Google Scholar]
- 41.Brouwer JR, Willemsen R, Oostra BA. Microsatellite repeat instability and neurological disease. Bioessays. 2009;31(1):71–83. doi: 10.1002/bies.080122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zalfa F, Eleuteri B, Dickson KS, et al. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10(5):578–587. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52(5):831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cirrito JR, Kang JE, Lee J, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58(1):42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10(8):561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 49.Huang YH, Lin Y, Mu P, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63(1):40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455(7215):912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]