

Abstract
Background
The success of genome-wide selection (GS) approaches will depend crucially on the availability of efficient and easy-to-use computational tools. Therefore, approaches that can be implemented using mixed models hold particular promise and deserve detailed study. A particular class of mixed models suitable for GS is given by geostatistical mixed models, when genetic distance is treated analogously to spatial distance in geostatistics.
Methods
We consider various spatial mixed models for use in GS. The analyses presented for the QTL-MAS 2009 dataset pay particular attention to the modelling of residual errors as well as of polygenetic effects.
Results
It is shown that geostatistical models are viable alternatives to ridge regression, one of the common approaches to GS. Correlations between genome-wide estimated breeding values and true breeding values were between 0.879 and 0.889. In the example considered, we did not find a large effect of the residual error variance modelling, largely because error variances were very small. A variance components model reflecting the pedigree of the crosses did not provide an improved fit.
Conclusions
We conclude that geostatistical models deserve further study as a tool to GS that is easily implemented in a mixed model package.
Background
Genome-wide selection (GS) is a marker-based method that predicts breeding values on the basis of a large number of molecular markers, which typically cover the entire genome [1]. The idea is to estimate the effects of all genes or chromosomal segments simultaneously and to integrate these estimates in order to predict the total breeding value.
One basic approach for GS is ridge regression (RR) [1]. An interesting alternative to RR is to use spatial models [2] to model genetic correlation among relatives [3].
This study compares RR models and spatial models for estimating genome-wide breeding values for the common dataset provided by the 13th QTL-MAS workshop. The focus is on methods that can be easily implemented using a standard mixed model package with facilities for spatial covariance structures.
Methods
Data
The dataset was simulated as part of the 13th QTL-MAS workshop (see [4] for details). Phenotypes of 1000 of 2025 individuals were recorded at five different times (0, 132, 265, 397 and 530), so there is a series of five repeated measurements for each phenotyped individual. Breeding values for the non-phenotyped individuals were to be predicted for time=600, which constitutes an extrapolation.
Extrapolation for time=600
Careful inspection of the data revealed that a logistic model
would give a reasonable fit to the data, where yit is the trait value of the i-th individual at time t (t = 0, 132, 256, 397, 530 or 600) and αi, βi, γi are parameters pertaining to the i-th individual. Observed data were modelled as yit = E( yit ) + εit, assuming that errors εit are independent and have constant variance. We initially fitted this model separately for each individual. Based on this analysis, we then obtained a pooled residual variance estimate from all individuals as 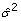 = RSS /2n, where RSS is the pooled residual sum of squares and n is the number of phenotyped individuals. Each individual contributed two error degrees of freedom. Next, nonlinear regressions were re-run for all phenotyped individuals to predict yi600, fixing the residual variance at the pooled estimate. Along with predictions the standard error was determined. The error variance of predicted values was estimated as the square of the standard error. This variance was subsequently regarded as a known within-individual error variance for mixed model analyses.
= RSS /2n, where RSS is the pooled residual sum of squares and n is the number of phenotyped individuals. Each individual contributed two error degrees of freedom. Next, nonlinear regressions were re-run for all phenotyped individuals to predict yi600, fixing the residual variance at the pooled estimate. Along with predictions the standard error was determined. The error variance of predicted values was estimated as the square of the standard error. This variance was subsequently regarded as a known within-individual error variance for mixed model analyses.
Marker scoring
The marker covariate zik for the i-th genotype and the k-th marker for biallelic markers with alleles A1 and A2 was set to 1 for A1A1, -1 for A2A2 and 0 for A1A2 and A2A1. Covariates were stored in a matrix Z = {zik }.
Model
The approach to GS closely follows [2]. Our basic model was
where µ is an intercept, hi is the total genotypic effect of the i-th individual and ei is a residual error. In most cases the total genotypic variance can not be explained by the markers. Thus there is an unexplained part left. We think that this part should be modelled by a polygenic effect. Therefore the total genotypic effect was partitioned into a component explained by the markers (gi) and a polygenic component (vi) not captured by the markers. Thus, the total genotypic effect hi was partitioned as:
[2]. There were two options regarding the model for var(ei), the variance of ei. Either we fixed var(ei) at the squared standard errors of predictions of yi600 obtained from nonlinear regression and assumed heterogeneous residual variance, or we pooled ei with vi and thus implicitly included var(ei) in the variance structure for vi which is defined as var(v) =I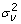 , where
is the variance component for polygenic effects. It is common to model the polygenic effect by the relationship matrix. But in this case we assumed independent polygenic effects, because no pedigree information on the parents was available. It must be stressed that var(ei) is strictly a within-individual error variance component that does not comprise between-individual error components. In the present application, these latter error components are effectively confounded with the polygenic effect vi. By contrast, in plant breeding applications, field replication would allow a separate assessment of between-individual error components. It is conjectured that explicit modelling of such an error component would be of benefit [2], because it is possible that the model captures part of the variance among individuals, which is nongenetic. For this reason it is advisable to generally obtain an independent estimate of error (as in [5]).
, where
is the variance component for polygenic effects. It is common to model the polygenic effect by the relationship matrix. But in this case we assumed independent polygenic effects, because no pedigree information on the parents was available. It must be stressed that var(ei) is strictly a within-individual error variance component that does not comprise between-individual error components. In the present application, these latter error components are effectively confounded with the polygenic effect vi. By contrast, in plant breeding applications, field replication would allow a separate assessment of between-individual error components. It is conjectured that explicit modelling of such an error component would be of benefit [2], because it is possible that the model captures part of the variance among individuals, which is nongenetic. For this reason it is advisable to generally obtain an independent estimate of error (as in [5]).
We considered different models for the variance of g' = (g1, g2,..., gG), conditionally on the markers Z = {zik }. All conditional models were of the form
for some matrix Γ that is a function of Z and
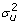 is a variance component. The models that were used are identical to those used in [2]. Under the mixed model for RR the matrix Γ =ZZ' was used and the penalty parameter depends on the variance components through λ2 =
is a variance component. The models that were used are identical to those used in [2]. Under the mixed model for RR the matrix Γ =ZZ' was used and the penalty parameter depends on the variance components through λ2 =
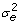 /
, where
is the residual variance [2]. In addition, different spatial models were used. Under these models, the genetic correlation is expressed as
/
, where
is the residual variance [2]. In addition, different spatial models were used. Under these models, the genetic correlation is expressed as
where dii' is the Euclidean distance of genotypes i and i', defined as dii'=||zi – zi' ||, with z'i equal to the i-th row of Z, and f(d) is some monotonically decreasing function of d. There are different options for the function f(d), including those shown in Table 1 [6]. We used all mentioned models and an independent model where
is omitted. It is noteworthy that the quadratic model is equivalent to the RR [2].
Table 1.
Genotypic covariance models of the form Γ = {f(dii')}, where d is the Euclidean distance computed from marker data and θ is a parameter.
| Name | Equation |
|---|---|
| Linear | 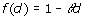 |
| Quadratic | 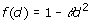 |
| Power | 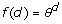 |
| Exponential | 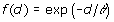 |
| Gaussian | 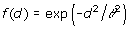 |
| Spherical | 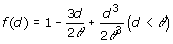 |
We also considered an extended model 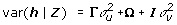 , where Ω represents the covariance due to simple random effects, i.e.,
, where Ω represents the covariance due to simple random effects, i.e., 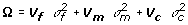 , where
, where 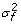 ,
, 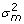 and
and 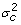 are the variance components for random effects of father and mother of crosses and of the crosses themselves, respectively, and Vf , Vm and Vc are corresponding symmetric matrices of known constants and are the variance component for random effects of the individuals. In this case the polygenic variance is defined as
are the variance components for random effects of father and mother of crosses and of the crosses themselves, respectively, and Vf , Vm and Vc are corresponding symmetric matrices of known constants and are the variance component for random effects of the individuals. In this case the polygenic variance is defined as 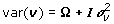 , where v'= v1,v2,...,vG. If Ω = 0 the model is equal to models with independent polygenic effects. When fitting the extended model we did not fix var(ei) at the squared standard errors of predictions of yi600, therefore var(ei) is pooled with .
, where v'= v1,v2,...,vG. If Ω = 0 the model is equal to models with independent polygenic effects. When fitting the extended model we did not fix var(ei) at the squared standard errors of predictions of yi600, therefore var(ei) is pooled with .
For each fitted model we obtained BLUPs of µ + hi corresponding to genome-wide estimated breeding values (GEBV). For non-phenotyped individuals 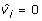 in the case of models with independent polygenic effects and
in the case of models with independent polygenic effects and 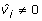 for the extended model when Ω ≠ 0. The Pearson correlation between the GEBV and the fitted values yi600 were calculated. In addition, the Akaike Information Criterion (AIC) was recorded. Small values of AIC indicated a preferable model.
for the extended model when Ω ≠ 0. The Pearson correlation between the GEBV and the fitted values yi600 were calculated. In addition, the Akaike Information Criterion (AIC) was recorded. Small values of AIC indicated a preferable model.
After the 13th QTL-MAS workshop the organizers reported the true breeding values (TBV). The TBV of the non-phenotyped individuals were compared to the GEBV by the Pearson correlation.
Software
The nonlinear regression was done by the NLMIXED procedure of the SAS System, while all mixed models were fitted by the REML method using the MIXED procedure of SAS.
Results
Analysis without fixing the residual variance
First the models were fitted without fixing the residual variance (Table 2). The RR and spatial methods give better fits than the model with independent genotypic effects. The AIC values of the RR and the spatial models are relatively close. The spatial linear, power, exponential and spherical models have a smaller residual variance than the RR/quadratic spatial model and Gaussian spatial model. The latter models show a higher correlation of GEBV with TBV (Table 2). Overall, the RR/quadratic model has the best AIC value, but this model shows a relatively low correlation of GEBV and yi600.
Table 2.
Model fits of different genetic covariance models and Pearson correlation between GEBV and fitted value and between GEBV and true breeding value (TBV).
| Residual | Correlation | ||||
|---|---|---|---|---|---|
| Model for gi | AIC | variance§ | θ | Fitted value$ | TBV# |
| Independent | 6789.1 | 51.89 | |||
| Ridge Regression (RR) | 6418.5 | 28.17 | 0.734 | 0.889 | |
| Spatial models | |||||
| Linear | 6425.8 | 12.00 | 0.974 | 0.880 | |
| Quadratic | 6418.5 | 28.17 | 0.734 | 0.889 | |
| Power | 6428.9 | 12.16 | 0.99 | 0.974 | 0.879 |
| Exponential | 6428.5 | 11.48 | 216.52 | 0.977 | 0.879 |
| Gaussian | 6420.5 | 28.08 | 124.59 | 0.737 | 0.889 |
| Spherical | 6427.8 | 11.96 | 959.97 | 0.974 | 0.880 |
§ The error variance was pooled with that for vi into a single residual variance.
$ The Pearson correlation between GEBV and fitted values (yi600) of the phenotyped individuals.
# The Pearson correlation between GEBV and true breeding values (TBV) of the non- phenotyped individuals.
The fits from the extended models that include Ω are shown in Table 3. The AIC values are a little bit higher than in the models without considering the effects of the parents, the independent model being an exception. The ranking of genotypes remains unaltered. Only when 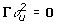 do we find a non-zero variance for mother effects in Ω . Throughout, there is a non-zero estimate for the variance of father effects (), while the variance for cross effects () is zero. The correlations of GEBV with TBV are almost the same as those when Ω was omitted (Table 3).
do we find a non-zero variance for mother effects in Ω . Throughout, there is a non-zero estimate for the variance of father effects (), while the variance for cross effects () is zero. The correlations of GEBV with TBV are almost the same as those when Ω was omitted (Table 3).
Table 3.
Model fits of different genetic covariance models with random effects for father and mother of crosses and for the crosses themselves and Pearson correlation between GEBV and fitted value and between GEBV and true breeding value (TBV).
| Residual | Father& | Mother& | Correlation | ||||
|---|---|---|---|---|---|---|---|
| Model for gi | AIC | variance§ | θ | |
|
Fitted value$ | TBV# |
| Independent | 6605.1 | 40.77 | 7.16 | 6.16 | 0.481 | 0.649 | |
| Ridge Regression (RR) | 6420.2 | 28.18 | 0.61 | 0 | 0.734 | 0.889 | |
| Spatial models | |||||||
| Linear | 6427.2 | 12.17 | 1.08 | 0 | 0.973 | 0.879 | |
| Quadratic | 6420.2 | 28.18 | 0.61 | 0 | 0.734 | 0.889 | |
| Power | 6430.3 | 12.35 | 0.99 | 1.20 | 0 | 0.973 | 0.878 |
| Exponential | 6429.9 | 11.62 | 208.63 | 1.11 | 0 | 0.976 | 0.878 |
| Gaussian | 6422.2 | 28.07 | 118.46 | 0.62 | 0 | 0.737 | 0.889 |
| Spherical | 6429.2 | 12.17 | 802.96 | 1.09 | 0 | 0.973 | 0.879 |
§ The error variance was pooled with that for vi into a single residual variance.
& The estimate of the variance for cross effects was zero in all models.
$ The Pearson correlation between GEBV and fitted values (yi600) of the phenotyped individuals.
# The Pearson correlation between GEBV and true breeding values (TBV) of the non- phenotyped individuals.
Analysis with fixing the residual variance
The results of the models with a fixed residual variance at the squared standard errors of predictions of yi600 obtained from nonlinear regression are shown in Table 4. The AIC values show that the models with an independent estimate of error var(ei) have an equal or nearly equal fit compared to the models without fixing the residual variance (Table 4). The correlations of GEBV with TBV are almost the same as those without fixing the residual variance (Table 4). Overall, the RR/quadratic model had the best AIC value as was the case when var(ei) was not fixed.
Table 4.
Model fits of different genetic covariance models. Residual variance var(ei) fixed at value of squared standard error of yi600 and Pearson correlation between GEBV and fitted value and between GEBV and true breeding value (TBV).
| Polygenic | Correlation | ||||
|---|---|---|---|---|---|
| Model for gi | AIC | variance | θ | Fitted value$ | TBV# |
| Independent | 6789.1 | 51.80 | |||
| Ridge Regression (RR) | 6418.5 | 28.09 | 0.734 | 0.889 | |
| Spatial models | |||||
| Linear | 6425.8 | 11.88 | 0.974 | 0.880 | |
| Quadratic | 6418.5 | 28.10 | 0.734 | 0.889 | |
| Power | 6428.8 | 10.38 | 0.99 | 0.981 | 0.878 |
| Exponential | 6428.2 | 11.17 | 627.76 | 0.977 | 0.879 |
| Gaussian | 6420.5 | 27.98 | 124.10 | 0.737 | 0.889 |
| Spherical | 6427.8 | 11.87 | 959.00 | 0.975 | 0.880 |
$ The Pearson correlation between GEBV and fitted values (yi600) of the phenotyped individuals.
# The Pearson correlation between GEBV and true breeding values (TBV) of the non- phenotyped individuals.
Discussion
There are only minor differences of the AIC values between RR and spatial models, like in [2]. Thus, some of the spatial models are viable alternatives to RR. Among the spatial models, the Gaussian model gave almost the same fit as RR. This can be explained by a Taylor expansion argument. The correlation function under the Gaussian model is exp(– d2 / θ2). When θ is large, then the exponent is close to zero and to first order we have 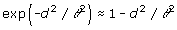 , so the Gaussian model approaches the RR/quadratic model in this case, but when θ is small we expect different fits. It is noteworthy that the Gaussian model is essentially equivalent to reproducing kernel Hilbert spaces regression as proposed by [7] and also to least squares support vector machine (LS-SVM) regression [8]. It may be conjectured that when inheritance is not merely additive it may be of particular importance to model the genetic covariance by some non-linear spatial model such as an exponential or a Gaussian model.
, so the Gaussian model approaches the RR/quadratic model in this case, but when θ is small we expect different fits. It is noteworthy that the Gaussian model is essentially equivalent to reproducing kernel Hilbert spaces regression as proposed by [7] and also to least squares support vector machine (LS-SVM) regression [8]. It may be conjectured that when inheritance is not merely additive it may be of particular importance to model the genetic covariance by some non-linear spatial model such as an exponential or a Gaussian model.
In this study we have used AIC defined as AIC = -2 log(likelihood) + 2 * number of parameters as printed by mixed model packages such as the MIXED procedure of SAS. The models with the lowest AIC values showed the highest correlation between the GEBV and TBV. But the correlation between the model ranks produced by AIC and by the correlation between GEBV and TBV is not perfect. For smoothing methods modifications of the AIC have been proposed (e.g. corrected AIC (AICC: [9]) and different other criteria (e.g. generalized cross-validation (GCV: [10]). The main difference to the AIC is that the complexity of the fitted model is calculated as the trace of the so-called smoother matrix tr(Sλ) described in [11], which relates to the effective degrees of freedom of the fit. It is important to realize that GS may be regarded as a smoothing exercise that replaces observed data (adjusted genotype means) by smoothed fitted values. Thus, model selection criteria developed for smoothing can be a useful extension for selecting a preferable model in GS.
The comparison between GEBV and phenotypes is not a good indictor for accuracy of breeding values, when all individuals are involved in the prediction and no independent validation set is left (Tables 2, 3 and 4). Cross-validation is one option to avoid this problem. The leave-one-out cross-validation procedure is equivalent to the cross-validation criterion, which is related to other selection criteria (AIC, AICC and GCV) [12]. Therefore one idea is to replace the cross-validation procedure by model selection criteria, which would entail a considerable saving of computing time.
In the present study the data were simulated without polygenic effects. Nevertheless, it is prudent to cater for the case that the total genotypic variance can not be fully explained by the markers alone. We think that this unexplained part should be modelled by a polygenic effect.
We modelled the polygenic effect as independent. Alternatively, one can assume that the polygenic effect is correlated due to the pedigree. We modelled the pedigree of the crosses by variance components, however, reflecting the pedigree did not provide an improved fit in the present case.
We also think it is important to separate the polygenic effect from error in order to avoid overfitting [2]. In the present study, however, fixing the residual variance did not have much of an effect because essentially we only had a within-individual error variance estimate. This ignored between-individual error variance, which is therefore expected to be confounded with the variance component for polygenic effects (). In plant breeding trials, where replication is available, one can separate polygenic effects from non-genetic between-individual errors. We expect that such separation will be crucial to the success of GS approaches in plant breeding.
Conclusions
Our study has shown that geostatistical models are viable alternatives to RR that deserve further study as a tool to GS. With respect to our analyses for the QTL-MAS 2009 dataset, however, we prefer the RR/quadratic model without fixed residual variance for predicting GEBV.
Competing interests
The authors declare no competing interests.
Authors' contributions
TSS participated in the design of the study, performed all analyses and drafted the paper. HPP conceived the study, and participated in its design, and helped in the final editing of the paper.
Contributor Information
Torben Schulz-Streeck, Email: torben.schulz-streeck@uni-hohenheim.de.
Hans-Peter Piepho, Email: piepho@uni-hohenheim.de.
Acknowledgements
Two anonymous reviewers are thanked for constructive comments.
This article has been published as part of BMC Proceedings Volume 4 Supplement 1, 2009: Proceedings of 13th European workshop on QTL mapping and marker assisted selection.
The full contents of the supplement are available online at http://www.biomedcentral.com/1753-6561/4?issue=S1.
References
- Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho HP. Ridge regression and extensions for genome-wide selection in maize. Crop Science. 2009;49:1165–1176. doi: 10.2135/cropsci2008.10.0595. [DOI] [Google Scholar]
- Piepho HP, Möhring J, Melchinger AE, Büchse A. BLUP for phenotypic selection in plant breeding and variety testing. Euphytica. 2008;161:209–228. doi: 10.1007/s10681-007-9449-8. [DOI] [Google Scholar]
- Coster A, Bastiaansen J, Calus M, Maliepaard C, Bink M. QTLMAS 2009: Simulated Dataset. BMC Proc. 2010;4(Suppl 1):S3. doi: 10.1186/1753-6561-4-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo R, Yu J. Prospects for genomewide selection for quantitative traits in maize. Crop Science. 2007;47:1082–1090. doi: 10.2135/cropsci2006.11.0690. [DOI] [Google Scholar]
- Schabenberger O, Gotway CA. Statistical methods for spatial data analysis. CRC Press; 2005. [Google Scholar]
- Gianola D, van Kaam JBCHM. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics. 2008;178(4):2289–2303. doi: 10.1534/genetics.107.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suykens JAK Gestel TV de Brabanter J de Moor B Vandewalle J Least squares support vector machines. 2002World Scientific Publishers; 11972910 [Google Scholar]
- Hurvich CM, Simonoff JS, Tsai C. Smoothing parameter selection in nonparametric regression using an improved Akaike information criterion. Journal ofthe Royal Statistical Society Series B. 1998;60:271–93. doi: 10.1111/1467-9868.00125. [DOI] [Google Scholar]
- Craven P, Wahba G. Smoothing noisy data with spline functions: Estimating the correct degree of smoothing by the method of generalized cross-validation. Numerische Mathematik. 1979;31:377–403. doi: 10.1007/BF01404567. [DOI] [Google Scholar]
- Ruppert D, Wand MP, Carroll RJ. Semiparametric regression. Cambridge Univ. Press; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie ADR, Tsai CL. Regression and time series model selection. World Scientific; 1998. [Google Scholar]