

Abstract
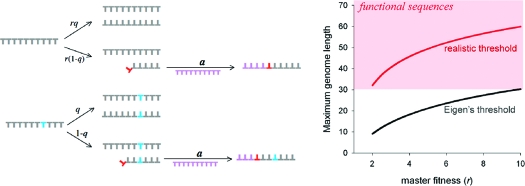
The frequency of errors during genome replication limits the amount of functionally important information that can be passed on from generation to generation. During the origin of life, mutation rates are thought to have been quite high, raising a classic chicken-and-egg paradox: could nonenzymatic replication propagate sequences accurately enough to allow for the emergence of heritable function? Here we show that the theoretical limit on genomic information content may increase substantially as a consequence of dramatically slowed polymerization after mismatches. As a result of postmismatch stalling, accurate copies of a template tend to be completed more rapidly than mutant copies and the accurate copies can therefore begin a second round of replication more quickly. To quantify this effect, we characterized an experimental model of nonenzymatic, template-directed nucleic acid polymerization. We found that most mismatches decrease the rate of primer extension by more than 2 orders of magnitude relative to a matched (Watson−Crick) control. A chemical replication system with this property would be able to propagate sequences long enough to have function. Our study suggests that the emergence of functional sequences during the origin of life would be possible even in the face of the high intrinsic error rates of chemical replication.
Introduction
Biological organisms store information in the sequence of their genomes. The information is propagated during genome replication, but each nucleotide incorporation presents an opportunity for error. At a given mutation rate per base (μ), if the genome is too long, the sequence information will be lost as mutants accumulate (an error “catastrophe”). Therefore, the mutation rate limits the total amount of information that can be carried by a genome. In particular, the maximum genome information is inversely proportional to the mutation rate.(1) Experimental data on mutation rates in RNA viruses, which appear to exist near this limit (the error threshold), also support this relationship.(2) Modern organisms have elaborate machinery for error detection and correction, but the first replicators were presumably very simple and had high error rates. Previous work indicates that nonenzymatic, template-directed nucleic acid polymerization has high error rates (close to 20%), corresponding to a genome of roughly 5 bases,(3) but aptamers, ribozymes, and deoxyribozymes are usually at least 30 bases long.(4) This discrepancy raises a paradox for the emergence of functional sequences during the origin of life. Is nonenzymatic replication accurate enough to propagate functional sequences? Previous proposals to address Eigen’s paradox include a mutualistic hypercycle, a spatially structured environment with cooperating sequences, mutational neutrality, or very high fitness differences.1,5 However, these approaches either invoke special functions or are relatively limited in magnitude.6,7 For example, one analysis of self-cleaving ribozymes found that 25% of bases could be mutated without destroying function, so the physical length of the genome could exceed the informative length by 25%.(6) Here we show that the chemical dynamics inherent in polymerization could offset the error threshold to the extent that sequences long enough to be functional could readily emerge.
The error threshold was first derived by Eigen from the following set of reactions describing replication:
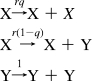 |
In these reactions, X is a “master” sequence that is characterized by higher fitness (r > 1) relative to that of all mutant sequences (Y) and q is the probability of replicating without errors (i.e., q = (1 − μ)L, where μ is the mutation rate per base and L is the number of functionally informative sites). In this classical model,(1) the master sequence can survive only if L is less than a critical L* = (ln r)/μ (Supporting Information). Because L* has a relatively weak logarithmic dependence on r and the prebiotic fitness is thought to have been relatively small, this equation is often approximated as L* ≈ 1/μ (corresponding to r = e) or roughly one error per replication. Beyond this point, the system undergoes a phase transition to a state in which the master sequence disappears and the genomes diffuse randomly through sequence space. L* is often thought of as a physical length, although strictly speaking it is the maximum number of informative sites. In essence, full-length mutant sequences, which are produced both by the replication of mutants and by mutation from the master sequence, grow in number faster than the master sequence when the genome is too long for a given mutation rate. Thus, the mutants outgrow the master sequences because they all consume resources during replication, and in a finite population, the master sequence would eventually disappear.(8) In this simple model, the existence of complementary strands was ignored but the error threshold is similar for complementary replication.(9)
In the classical model, polymerization was assumed to proceed equally fast regardless of whether an error occurred. However, studies of enzymatic polymerization show that if an incorrect nucleotide is incorporated then primer extension stalls after the mutation, presumably because of a suboptimal conformation at the mismatched terminus.(10) Stalling after base pairs are mismatched has been observed for several DNA polymerases, with the ratio of extension rates from a matched versus mismatched terminus (the stalling factor, S) ranging from 10 to 106.(11) Intuitively, this effect might slow the production of inaccurate copies of the master sequence, increasing the effective fidelity and the maximum genome information. However, it was previously unknown whether nonenzymatic polymerization would also slow after mutations. We therefore undertook the determination of mutation rates and stalling factors in a model system for template-directed nonenzymatic polymerization. We used 2′-deoxy-5′phosphorimidazolides (ImpdN) as the activated monomers, DNA templates, and DNA primers terminated by a 3′-amino-2′,3′-dideoxynucleotide.(12) In this system, the rate of a single extension can be determined because the amine reacts much faster than a hydroxyl.3,13 Although other work has focused on 2′-amine analogs, which have properties appropriate for copying long sequences,(14) we chose to focus on a 3′-amine system because it may mimic the biological 3′−5′ linkage more closely. We then calculated the error threshold including the effect of stalling after mutations. Our results indicate that stalling increases the maximum genome information to the extent that functional sequences could have been replicated without enzymes.
Materials and Methods
Preparation of Nucleoside 5′-Phosphoimidazolides
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. The protocol used to synthesize the activated nucleotides (ImpdNs) was based on a previously published method(15). The free acid form of each nucleoside-5′-monophosphate (1.5 mmol) was suspended with imidazole (15 mmol) and 2,2′-dithiodipyridine (4.5 mmol) in 20 mL of a 1:1 mixture of anhydrous dimethyl formamide and anhydrous dimethyl sulfoxide. Subsequently, triethylamine (TEA, 4.5 mmol) and triphenylphosphine (3 mmol) were added and the mixture was stirred at room temperature for ∼4 h. The reaction progress was monitored by thin layer chromatography using a mobile phase of 50% n-butanol and 20% acetic acid in water. The resulting clear, yellow solution was added dropwise to a flask containing a mixture of anhydrous ether (200 mL)/acetone (125 mL)/TEA (15 mL)/anhydrous sodium perchlorate (0.5 g) and precipitated with gentle stirring for 30 min on ice. The resulting precipitate was filtered, washed with 200 mL of a 1:1 mixture of acetone and ether and with 100 mL of anhydrous ether, and dried overnight in vacuum desiccator over phosphorus pentoxide to give the corresponding nucleoside 5′-monophosphate imidazolide sodium salt. The resulting mixture was analyzed by RP-HPLC (Varian, Inc., Palo Alto, CA) using a C18 column (Varian microsorb, 250 × 41 mm2 i.d., 5 μm particle size). The conditions for HPLC were the following: solvent A: 0.025 M TEAB, pH 7.3; solvent B: 70% acetonitrile/water; gradient: isocratic 15% B; flow rate: 15 mL/min; and UV detection: 260 nm. The fractions containing the desired ImpdN were collected and frozen. These were then lyophilized to obtain the solid triethylammonium salt of the imidazolide. All ImpdNs were found to be >93% pure according to analytical HPLC.
Oligonucleotides for Nonenzymatic Polymerization
DNA primers terminated with a 3′-amino-2′,3′-dideoxynucleotide were either radiolabeled or fluorescently tagged for detection and quantification of the reaction products. The primer used to obtain misincorporation rates (AminoG) was synthesized on a dT-CPG column (Glen Research; Sterling, VA). A single 3′-amino-dG residue was added manually using 3′-amino-5′-DMT-dG (RI Chemical Inc.; Orange County, CA) under standard coupling conditions. The remainder of the sequence was synthesized on an Expedite 8900 nucleic acid synthesizer (Millipore; Billerica, MA). After ammonium hydroxide cleavage from the column and deprotection, the oligo was gel purified and then treated with 80% acetic acid overnight to cleave off the terminal phosphoramidate-linked T residue and the hydrolysate was purified by HPLC to isolate the 3′-amino oligo. The correct mass of the oligo was confirmed by matrix-assisted laser desorption ionization−time-of-flight mass spectrometry (MALDI-TOF MS: PerSeptive Biosystems Voyager MALDI-TOF; Framingham, MA). A sample of ∼200 pmol of oligonucleotide was adsorbed on a C18 zip tip. Samples were eluted with 1.5 μL of a matrix solution containing a 2:1 mixture of 52.5 mg/mL 3-hydroxypicolinic acid in 50% acetontrile and 0.1 M ammonium citrate in water. Eluates were directly spotted onto a stainless steel MALDI-TOF plate and analyzed in positive mode. The AminoG primer was end-labeled with a T4 polynucleotide kinase (New England Biolabs; Ipswich, MA) and γ-32-P-ATP (Perkin-Elmer; Waltham, MA) at the 5′-hydroxyl termini of DNA, following an established protocol.(16) This primer was also used for a subset of extension reactions for matched versus mismatched termini.
The three remaining primers (AminoA, AminoT, and AminoC) for these extension reactions were made by reverse synthesis in the W. M. Keck Biotechnology Resource Laboratory at Yale University (New Haven, CT). The synthesis used the following phosphoramidites for the 3′ residue: AminoA: 3′-O-tritylamino-N6-benzoyl- 2′,3′-dideoxyadenosine-5′-cyanoethyl phosphoramidite; AminoC: 3′-O-tritylamino-N4-benzoyl-2′,3′-dideoxycytidine-5′-cyanoethyl phosphoramidite; and AminoT: 3′-tritylamino-3′-deoxythymidine-5′-cyanoethyl phosphoramidite (Metkinen Chemistry; Kuusisto, Finland). These three primers were labeled by Cy3 at their 5′ termini. The primers were purified by anion-exchange chromatography using a 250 × 41.4 mm2 Dionex PA-100 column with a gradient of 0 to 40% B over 20 min followed by an increase to 60% B in 40 min at 15 mL/min (buffer A = 0.01 M NaOH/0.01 M NaCl/H2O; buffer B = 0.01 M NaOH/1.5 M NaCl/H2O). Purification was monitored by UV absorbance at dual wavelengths of 260 and 520 nm. AminoA required further purification by 20% polyacrylamide gel electrophoresis (PAGE using Sequagel (National Diagnostics; Atlanta, GA) on a model V16-2 electrophoresis unit (Labrepco, Horsham; PA) with 20 × 20 cm2 glass plates. The correct mass of these oligos was verified by MALDI-TOF as described above.
The DNA template sequences were synthesized and PAGE purified by Sigma-Aldrich (St. Louis, MO). Primer and template sequences are given below.
primer sequences:
AminoG (“primer G”): 5′ GG GAT TAA TAC GAC TCA CTG-NH2
AminoA (“primer A”): 5′ GG GAT TAA TAC GAC TCA CTA-NH2
AminoT (“primer T”): 5′ GG GAT TAA TAC GAC TCA CTT-NH2
AminoC (“primer C”): 5′ GG GAT TAA TAC GAC TCA CTC-NH2
Template sequences for misincorporation reactions are given below:
MisincorpA: 5′ AGT GAT CTA CAG TGA GTC GTA TTA ATC CC
MisincorpT: 5′ AGT GAT CTT CAG TGA GTC GTA TTA ATC CC
MisincorpG: 5′ AGT GAT CTG CAG TGA GTC GTA TTA ATC CC
MisincorpC: 5′ AGT GAT CTC CAG TGA GTC GTA TTA ATC CC
Template sequences for mismatch extension reactions are given below:
MismatchA: 5′ AGT GAT CTC AAG TGA GTC GTA TTA ATC CC
MismatchT: 5′ AGT GAT CTC TAG TGA GTC GTA TTA ATC CC
MismatchG: 5′ AGT GAT CTC GAG TGA GTC GTA TTA ATC CC
MismatchC: 5′ AGT GAT CTC CAG TGA GTC GTA TTA ATC CC
Primer Extension Reactions and Assay for Nonenzymatic Polymerization
A primer (0.325 μM) and a template (1.3 μM) (1 μL each) were mixed in water, incubated at 95 °C for 5 min, and annealed by cooling to room temperature on a benchtop for 5−7 min. In a typical reaction of 10 μL volume, 1 μL of 1 M Tris (pH 7) and 0.5 μL of 4 M NaCl were added to final concentrations of 100 mM Tris and 200 mM NaCl. For reactions with ImpdA, ImpdC, or ImpdG, the reaction was initiated by the addition of 1 μL of 100 mM ImpdN to a final concentration of 10 mM ImpdN. For reactions involving ImpdT, 1.38 μL of 289 mM stock solution was added to a final concentration of 40 mM ImpdT. The total volume of the reaction was 10 μL. The reaction mixtures were incubated at room temperature, and aliquots were withdrawn during a certain period of time. Time points were obtained by adding 1 μL of the reaction mixture to 9 μL of the loading buffer with 8 M urea, 100 mM EDTA, and 1.3 μM of a competitor DNA with the sequence 5′ GG GAT TAA TAC GAC TCA CTN 3′ where N = A/T/G/C to match the primer employed in the reaction. Time points were heated to 90 °C for 5 min to disrupt primer−template complexes and were run on 20% denaturing PAGE.
The gels were phosphorimaged using a Typhoon TRIO variable-mode imager (Piscataway, NJ), and the scans were analyzed with ImageQuant v5.2 software. The fraction of unreacted primer was calculated by dividing the intensity of the unreacted primer band by the sum of intensities of the unreacted and reacted primer. In some cases, the extended product appeared to be a doublet band that was well separated from the unreacted primer; the doublet intensities were summed. To avoid experimental artifacts late in the reaction, initial rates were estimated by a linear fit to the first several data points.
Calculation of Mutation Rate and Stalling Factor
The frequency of incorporation (ftemplate base:ImpdN) of an ImpdN across a particular template base was calculated by dividing its rate of extension by the sum of the rates of extension for all ImpdN’s of the same primer−template complex (i.e., containing the same template base at the position opposite the incoming nucleotide). The mutation rate for template base N (μN) is the sum of the frequencies of incorrect incorporations (e.g., μA = fA:A + fA:C + fA:G = 1 − fA:T). If the fraction of the genome composed of base N is given by FN, then the average mutation rate of a genome (μave) is Σ(FNμN). For example, a genome composed of equal parts A, C, G, and T would have μave = 0.25(μA + μC + μG + μT), and a genome composed of equal parts of only G and C would have μave = 0.5(μC + μG).
The stalling factor for each mismatch (Stemplate base:primer terminus) was calculated by dividing the rate of extension from the corresponding matched terminus (ktemplate base:primer terminus), which has the same template sequence, by the rate of extension from the mismatched terminus (e.g., SG:A = kG:C/kG:A). The average stalling factor, Save, was calculated by weighting each stalling factor by the frequency of incorporation that leads to that stalled complex (Save = FAΣ(fA:ImpdNSA:N) + FCΣ(fC:ImpdNSC:N) + FGΣ(fG:ImpdNSG:N) + FTΣ(fT:ImpdNST:N)). In other words, the most frequent mutations contribute most to the overall stalling factor because they result in the most frequent mismatched termini. Stalling factors are also weighted by the genome composition because mutations across the most common template base (and the corresponding mismatched termini) would be relatively well represented. In this article, we assume that the genome is equal parts A, C, G, and T for the purpose of the stalling factor calculation (FA = FC = FG = FT = 0.25). The standard deviation of the overall stalling factor and mutation rate, Save and μave, were calculated as the standard deviation of the corresponding values from an initial batch of reactions and a duplicate batch.
Results and Discussion
Mutation Rate of Nonenzymatic Polymerization
We determined the rates of misincorporation in a series of reactions containing a template sequence, a perfectly complementary primer (either radiolabeled or fluorescently tagged), and one ImpdN (A, C, G, or T). Initial experiments showed that the rate of incorporation of T was particularly slow, causing relatively low fidelity when copying across a template base A, so we increased the concentration of ImpdT to 40 mM in our reactions (compared with 10 mM for the other nucleotides). Adjusting the ratio of monomer concentrations has been used previously to improve fidelity in enzymatic reactions.(17) We followed reactions over time to determine apparent first-order rate constants for all possible correct incorporations (4 reactions) and misincorporations (12 reactions) (Figures 1a,b and 2a and Supporting Information).
Figure 1.

Examples of nonenzymatic primer extension over time. (Insets) Denaturing polyacrylamide gel at reaction time points. Exponential curve fits are drawn to guide the eye. (A) Correct incorporation of ImpdT across A. (B) Incorrect incorporation of ImpdG across A. (C) Extension of matched primer terminus (blue). (D) Extension of mismatched primer terminus (blue/red).
Figure 2.

Misincorporation frequencies and stalling factors in nonenzymatic polymerization. (A) Incorporation frequencies of each nucleotide across each base. (B) Stalling factors associated with mismatched termini. Error bars show standard deviations calculated from duplicate sets of reactions. Primer N refers to the primer containing base N at the 3′ terminus.
We found that the average mutation rate (μave) of a genome composed of equal proportions of A, C, G, and T would be 7.6 ± 1.4% in this system. Misincorporations occurred predominantly when copying A and T, so a GC-rich genome would have a lower mutation rate (e.g., for an entirely GC genome, μ ≈ 0.8%; see Methods and Materials for details of the calculation). The absolute rate of incorporation of G and C across their cognate bases was also ∼10 times greater than the rate of incorporation of A and T, consistent with trends from previous work(18) suggesting that hydrogen bonding may also contribute to the reaction rate. Our results differ somewhat from previous estimates of fidelity in a similar system, probably because of differences in ionic conditions and monomer concentrations.(3) According to the original Eigen model, a mutation rate of 7.6% would be too high to sustain a functional genome at low fitness (L* ≈ 13).
Stalling Factor of Nonenzymatic Polymerization
To determine stalling factors for nonenzymatic polymerization, we varied the 3′ terminus of the primer to form either a perfectly matched terminus (4 reactions) or a mismatched terminus (12 reactions) and measured the rate of incorporation of the correct subsequent monomer (Figures 1c,d and 2b and Supporting Information). The overall stalling factor (Save) was calculated as an average weighted by the misincorporation frequency leading to the terminus. (See Methods and Materials for details of the calculation.) Despite the lack of an enzyme, nonenzymatic polymerization showed substantial stalling, with the extension from any mismatch being slower than its matched counterpart by a factor of 20−300 (Save = 124 ± 22).
Modified Error Threshold Including the Effect of Stalling after Mutation
Would the effect of stalling be large enough to permit the nonenzymatic replication of functional sequences? We modified Eigen’s model of replication to include a stalled state after a misincorporation, which progresses to completion at a relatively slow rate. Following Eigen’s model, we assume that the relative fitness of the master sequence is r and resources for replication are available at constant concentration. In the reactions given below, X is the fittest sequence, Z is an incomplete copy in which a mismatched nucleotide was incorporated, and Y is the finished mutant.
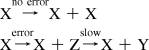 |
Mutant sequences undergo an analogous set of reactions when an error occurs. Strand separation is assumed to occur frequently compared with the relatively slow process of chemical replication (e.g., thermal cycling due to day−night changes or in convection cells (Supporting Information)). The error threshold for the corresponding set of differential equations was determined analytically in the limit of large numbers under the condition that the total density of the system is conserved ([X] + [Y] + [Z] = constant; see Supporting Information for a full description). We obtained a new expression for the maximum genome information corresponding to the condition that [X] > 0 in the stationary state, namely that Ls* = ln[r + μS(r − 1)]/μ (Figure 3a). As with the classical model, Ls* is inversely proportional to μ. As expected, as S increases (i.e., as stalling becomes more pronounced), Ls* also increases. This effect is weighted by μ because the synthesis of new strands is stalled longer if they contain multiple mutations.
Figure 3.

Modified error threshold. (A) Ratio of the maximum genome information including the effect of stalling and strand separation relative to the classical maximum. (B) Maximum genome information for different values of r using experimentally determined values for μave and Save during nonenzymatic polymerization. Curves were calculated for the classical model (black) or with the modified model (red).
Because this limit is always greater than or equal to the original Eigen condition, stalling would be beneficial for a variety of scenarios (i.e., different error rates and stalling factors). We also found that the error threshold was robust to details of the model; a second model in which imperfect copies were more likely to degrade during copying because of their longer copying time (e.g., longer exposure to UV damage or hydrolysis) gave the same error threshold (Supporting Information).
Using our experimentally determined parameters for μave and Save, we calculated the maximum information of a genome undergoing nonenzymatic replication (Figure 3b). Although the classical Eigen model predicts that the mutation rate is too high to propagate a functional sequence, accounting for stalling after errors in polymerization increases the maximum informative length to 39 (at r = e). As with the classical threshold, this length increases with higher fitness (Figure 3b). This result demonstrates that an intrinsic feature of nonenzymatic polymerization could circumvent the Eigen paradox, allowing the propagation of functional sequences before enzymes evolved.
Choice of Experimental Model System
Our studies were carried out with 3′-amino-2′,3′-dideoxynucleotide-terminated primers. Although DNA was probably a relatively late invention in the course of prebiotic evolution, we use this 3′-amine system as an experimentally tractable model of nonenzymatic polymerization. In preliminary experiments, we had attempted to assay misincorporations in the nonenzymatic polymerization of a 2′,3′-hydroxyl system. However, polymerization in the 2′,3′-hydroxyl system was too slow to measure the rate of misincorporation accurately. There are also other unsolved issues with nonenzymatic RNA replication, such as strand separation, leading many to suggest that a different nucleic acid preceded the RNA world.18,19 Another possible experimentally tractable system would use 2′-amino-2′,3′-dideoxynucleotide-terminated primers.(14) Although the 2′-amine system may have superior properties for copying long sequences with the goal of synthesizing a protocell, our goal here was to estimate the error rates associated with the more biological 3′−5′ linkage. In addition to the fairly efficient 3′-amine polymerization observed by Orgel and colleagues,(20) a different 3′-amine system has also been studied by the Richert group,(21) which exhibited very fast reaction rates with nearly quantitative yield, suggesting that a 3′-amine system has the potential to be efficient enough to copy relatively long sequences. It is possible that the 3′-amine system will have a fidelity differing from that of a 3′-hydroxyl system. Our data may not be representative of mutations in the RNA world itself, but our results do demonstrate that a nonenzymatic system exhibits stalling after mutations and that such a system could be capable of propagating sequences long enough to be functional because of this effect.
Conclusions
We have shown that the error catastrophe could be substantially mitigated through the dynamics of replication in which fidelity should not be considered to be a simple constant. Our experimental model system for nonenzymatic, template-directed nucleic acid polymerization demonstrates that stalling can be important even without enzymes. The presence of a mismatched terminus in the nascent sequence stalls extension and effectively decreases the rate of extension of a mutant sequence by more than 2 orders of magnitude. Interestingly, the same features of the prebiotic world that would reduce the maximum genome information in Eigen’s model—low fitness and high mutation rates—also increase the importance of stalling in offsetting the error catastrophe. Thus, nonenzymatic replication could potentially give rise to sequences long enough to be functional despite a high mutation rate. These dynamic effects could still be important after functional sequences emerged, permitting the genome to encode more sequences or longer sequences with higher activity.(22) Furthermore, stalled primer−template complexes could provide a substrate for the evolution of error-correction machinery. Eventually, these effects would become obsolete as the replication machinery evolved greater accuracy and cooperating networks emerged, but early on they could have served to “kick start” the evolution of functional genomes.
Acknowledgments
We thank Jason Schrum, Sylvia Tobe, Michael Lawrence, Ching-Hsuan Tsai, John B. Randolph, Pierre-Alain Monnard, Andrew Murray, David Liu, Johan Paulsson, Eugene Shakhnovich, and Bodo Stern for advice. This work was supported by NIH grant GM068763 to the National Centers of Systems Biology and the Bauer Fellows Program at Harvard University (I.A.C.) and by NSF grant CHE0434507 (J.W.S.). J.W.S. is an Investigator at the Howard Hughes Medical Institute. J.K.I. received a predoctoral fellowship from the Ford Foundation.
Supporting Information Available
Supporting text, figures, and a description of the mathematical analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eigen M. Naturwissenschaften 1971, 58, 465–523. [DOI] [PubMed] [Google Scholar]
- Gago S.; Elena S. F.; Flores R.; Sanjuan R. Science 2009, 323, 1308. [DOI] [PubMed] [Google Scholar]; Drake J. W.; Holland J. J. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 13910–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]; Schuster P. In The Aptamer Handbook; Klussmann S., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp 29−53. [Google Scholar]
- Hagenbuch P.; Kervio E.; Hochgesand A.; Plutowski U.; Richert C. Angew. Chem., Int. Ed. 2005, 44, 6588–6592. [DOI] [PubMed] [Google Scholar]
- Ekland E. H.; Szostak J. W.; Bartel D. P. Science 1995, 269, 364–370. [DOI] [PubMed] [Google Scholar]
- Szathmáry E.; Maynard Smith J. J. Theor. Biol. 1997, 187, 555–571. [DOI] [PubMed] [Google Scholar]; Szabo P.; Scheuring I.; Czaran T.; Szathmary E. Nature 2002, 420, 340–343. [DOI] [PubMed] [Google Scholar]; Altmeyer S.; McCaskill J. S. Phys. Rev. Lett. 2001, 86, 5819–5822. [DOI] [PubMed] [Google Scholar]; Hogeweg P.; Takeuchi N. Origin Life Evol. Biosph. 2003, 33, 375–403. [DOI] [PubMed] [Google Scholar]
- Kun A.; Santos M.; Szathmary E. Nat. Genet. 2005, 37, 1008–1011. [DOI] [PubMed] [Google Scholar]
- Takeuchi N.; Poorthuis P. H.; Hogeweg P. BMC Evol. Biol. 2005, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M.; Schuster P. J. Theor. Biol. 1989, 137, 375–395. [DOI] [PubMed] [Google Scholar]
- Stadler P. F. Math. Biosci. 1991, 107, 83–109. [DOI] [PubMed] [Google Scholar]
- Ichida J. K.; Zou K.; Horhota A.; Yu B.; McLaughlin L. W.; Szostak J. W. J. Am. Chem. Soc. 2005, 127, 2802–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ichida J. K.; Horhota A.; Zou K.; McLaughlin L. W.; Szostak J. W. Nucleic Acids Res. 2005, 33, 5219–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M. M.; Arnheim N.; Goodman M. F. Nucleic Acids Res. 1992, 20, 4567–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]; Perrino F. W.; Loeb L. A. J. Biol. Chem. 1989, 264, 2898–2905. [PubMed] [Google Scholar]; Mendelman L. V.; Petruska J.; Goodman M. F. J. Biol. Chem. 1990, 265, 2338–2346. [PubMed] [Google Scholar]
- Stutz J. A.; Kervio E.; Deck C.; Richert C. Chem. Biodiversity 2007, 4, 784–802. [DOI] [PubMed] [Google Scholar]; Mansy S. S.; Schrum J. P.; Krishnamurthy M.; Tobe S.; Treco D. A.; Szostak J. W. Nature 2008, 454, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel L. E.; Lohrmann R. Acc. Chem. Res. 1974, 7, 368–377. [Google Scholar]
- Schrum J. P.; Ricardo A.; Krishnamurthy M.; Blain J. C.; Szostak J. W. J. Am. Chem. Soc. 2009, 131, 14560–14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrmann R.; Orgel L. E. Tetrahedron 1978, 34, 853–855. [Google Scholar]; Prabahar K. J.; Cole T. D.; Ferris J. P. J. Am. Chem. Soc. 1994, 116, 10914–10920. [DOI] [PubMed] [Google Scholar]
- Chen L.; Rejman D.; Bonnac L.; Pankiewicz K. W.; Patterson S. E. Curr. Protoc. Nucleic Acid Chem. 2005, 13.14.11–−13.14.10. [DOI] [PubMed] [Google Scholar]
- Muller U. F. Cell. Mol. Life Sci. 2006, 63, 1278–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]; Johnston W. K.; Unrau P. J.; Lawrence M. S.; Glasner M. E.; Bartel D. P. Science 2001, 292, 1319–1325. [DOI] [PubMed] [Google Scholar]
- Orgel L. E. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. Science 1999, 284, 2118–2124. [DOI] [PubMed] [Google Scholar]
- Zielinski W. S.; Orgel L. E. Nucleic Acids Res. 1985, 13, 2469–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlingshofer M.; Kervio E.; Lommel T.; Plutowski U.; Hochgesand A.; Richert C. Angew. Chem., Int. Ed. 2008, 47, 6065–6068. [DOI] [PubMed] [Google Scholar]
- Carothers J. M.; Oestreich S. C.; Davis J. H.; Szostak J. W. J. Am. Chem. Soc. 2004, 126, 5130–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.