

Abstract
In spite of its central roles in cell cycle progression, senescence, and aging, knowledge about the posttranslational regulation of P16 (also known as INK4A and MTS1) remains limited. While it has been reported that P16 could be phosphorylated at Ser7, Ser8, Ser140, and Ser152, the corresponding kinases have not been identified yet. Here we report that IKKβ, a primary kinase for IκBα phosphorylation, is involved in P16 phosphorylation. Immunoprecipitation and kinase assays showed that IKKβ specifically binds to P16 and phosphorylates P16 at Ser8 in WI38 cells. Biochemical characterization of phosphomimetic Ser→Glu P16 mutants demonstrated that phosphorylation at Ser8 of P16 brings about a significant loss of its cyclin-dependent kinase (CDK) 4-inhibitory activity while P16 retains structurally and functionally intact upon phosphorylation at Ser7, Ser140, and Ser152. Our results reveal the novel role of IKKβ in P16 phosphorylation and broaden our understanding of the regulation of P16.
Keywords: P16, phosphorylation, IKKβ, CDK4
Introduction
P16, also designated INK4A and MTS1, is one of the most extensively studied proteins in the past decades due to its critical roles in cell cycle progression, cellular senescence, and the development of human cancers. At the G1-to-S transition, P16 specifically inhibits cyclin-dependent kinases (CDK) 4 and 6-mediated phosphorylation of pRb, the retinoblastoma susceptible gene product, thus sequestering the transcription factors of E2Fs in incompetent pRb/E2F complexes and consequently blocking cell cycle progression [1]. It has also been demonstrated that elevated expression of P16 caused by certain oncogenes, DNA damage response, or aging triggers and accelerates cell senescence [2, 3]. Moreover, while genetic inactivation of the p16 gene (CDKN2A) by deletion, methylation, and point mutations has been found in a significant fraction (close to 50%) of all human cancers [4], over-expression of P16 at both mRNA and protein levels is associated with poor prognosis for cancers including neuroblastoma, cervical, ovarian, breast, prostate tumors, and oral cancers [5], indicating that the cellular level of P16 protein is critical for its functioning. In comparison with genetic and transcriptional regulation of the p16 gene, posttranslational regulation of P16 has been understudied. It has been reported that P16 could be phosphorylated in human fibroblast cells at Ser7, Ser8, Ser140, and Ser152 [6], all of which are located at the flexible N- and C-termini and do not directly contact CDK4 [7]. Such phosphorylation is potentially important since mutations involving these four residues have been found in familial and sporadic melanomas [6]. Nonetheless, the kinases responsible for P16 phosphorylation as well as the functional and structural effect upon P16 phosphorylation remain unknown.
Recently, it has been reported that there are striking functional and structural similarities between P16 and IκBα, a well-known inhibitor of NF-κB [8-10]. On one hand, P16 and IκBα compete with each other for binding to CDK4 and NF-κB, and such binding specifically inhibits the activities of both CDK4 and NF-κB [8, 10]. On the other hand, while P16 and IκBα are composed of 4 and 6 ankyrin repeats (ARs), respectively, the CDK4-binding domain of IκBα is located at the four N-terminal ARs, and the structures of these four ARs in P16 and IκBα are almost superimposable, especially in the helical regions where most of contacts with their target proteins are located [9]. More interestingly, both P16 and IκBα have flexible N-termini harboring two phosphorylation sites, Ser7/Ser8 in P16 [6] and Ser32/Ser36 in IκBα [11]. These findings arguably lead to a postulation that P16 and IκBα, especially their N-termini, may be similar in phosphorylation, i.e. kinases involved in IκBα phosphorylation may function in the regulation of P16.
In the present study, we demonstrated that IKKβ, an IκBα-specific kinase [11], physically associates with P16 in vivo, and the resultant phosphorylation at Ser8 of P16 significantly impairs the CDK4-inhibitory activity of P16.
Materials and methods
Protein Expression and Purification
The cloning, expression, and purification of human P16, IκBα1-214, and Yar 1 have been described previously [8]. Briefly, all P16, IκBα1-276 and Yar 1 proteins including WT and different mutants were expressed in E. coli BL21 Codon Plus (Novagen) as Glutathione-S-transferase (GST)-fusion proteins and purified using reduced Glutathione resin (Sigma). After removal of the GST tag by Thrombin (for P16; Sigma) or Prescission protease (for IκBα1-276 and Yar 1; Amersham), proteins were further purified on a gel filtration column. All P16 mutants were generated using PCR-based site-directed mutagenesis (Stratagene), and were expressed and purified as P16 wild type (WT).
Cell Culturing, Immunoprecipitation (IP) and Western Blot (WB)
U2OS (p16−/−) and WI-38 (p16+/+) cells were purchased from the American Type Culture Collection and cultured in a 5% CO2 humidified atmosphere in Advanced McCoy's 5A and Advanced MEM containing 7% fetal bovine serum (FBS, Invitrogen), respectively. WI-38 cells were used at passage 3–5. Cells were lysed in the non-denaturing lysis buffer (20 mM Tris-HCl, pH 7.4, 0.1% NP-40, 250 mM NaCl, 5 mM EDTA, 20 mM NaF, 2 mM Na3VO4, 1 mM DTT, and 200 μg/ml Sigma P8340 protease inhibitor cocktail). After incubation on ice for 10 minutes, the cells were clarified by centrifugation at 4°C, 20,000g for 15 minutes. The supernatant was then transferred to a clean tube and the protein concentration was determined using a BCA protein assay (Pierce). For protein expression analyses, 50 μg of cell lysates were subjected to SDS-PAGE and western blot to evaluate protein expression using indicated antibodies: IKKβ, sc-56918 (Santa Cruz Biotechnologies); P16, Cat. #554070 (PharMingen); β-actin, sc-56459 (Santa Cruz Biotechnologies). Blots were visualized using the Pico Western Chemiluminescent system (Pierce). For immunoprecipitation (IP) analyses, 400 μg of cell lysates were immunoprecipitated with the afore-mentioned antibodies or a combination of normal mouse serum and rabbit serum (Jackson Immunoresearch Laboratories) [6]. Antibody complexes were captured with 70-100 μl lysis buffer-pretreated protein G-Sepharose (Amersham). Immunoprecipitates were washed three times using the lysis buffer and subjected to further analyses; cell lysates with the removal of immunoprecipitates were used in the in vitro P16 phosphorylation assay as described below. Since there is no endogenous P16 in U2OS, 2 μg of recombinant P16 protein was added into 400 μg of U2OS cell lysate, and after incubation at 4°C for 4 hours, the mixture was subjected to immunoprecipitation using anti-IKKβ antibody as described above.
In vitro Phosphorylation of P16
Reaction mixtures containing 0.1 μg of recombinant IKKβ (Invitrogen), 2.0 μg of P16 proteins, and 5 μCi [γ-32P] ATP in a total volume of 15 μl of the kinase buffer (50 mM HEPES, 10 mM MgCl2, 2.5 mM EGTA, 0.1 mM Na3VO4, 1 mM NaF, 10 mM β-glycerolphosphate, 1 mM DTT, 0.2 mM AEBSF, 2.5 mg/ml leupeptin, and 2.5 mg/ml aprotinin) were incubated at 30°C for 20 minutes. Subsequently, the mixtures were subjected to SDS-PAGE and radio autography. Cell lysate-mediated P16 phosphorylation was evaluated similarly except that each reaction contained 10 μg of U2OS or WI38 cell lysate, or 10 μg of IKKβ-depleted cell lysate, and the incubation at 30°C lasted for 45 minutes. IκBα1-214, truncated IκBα containing Ser32 and Ser36 for IKKβ phosphorylation [8, 11], was used as positive control, while Yar 1, a yeast AR protein of 200 amino acid residues was used as negative control [8].
In vitro Inhibition of P16 on CDK4
The in vitro CDK4 activity assay was performed as previously described [7, 8]. Briefly, each reaction mixture contains about 0.2 μg of recombinant CDK4/cyclin D2 holoenzyme and varying concentrations of P16 in 15 μl of the afore-mentioned kinase buffer. After incubation at 30°C for 30 minutes, 50 ng of GST-Rb791–928 and 5 μCi [γ-32P] ATP were added in the reaction mixture and after incubation at 30°C for another 15 minutes, proteins in the reaction mixture were separated by SDS-PAGE, and the incorporation of 32P into GST-Rb791–928 was quantitatively evaluated using a PhosphorImager (Molecular Dynamics). The IC50 value was defined as the concentration of a kinase inhibitor required to achieve 50% of the maximal inhibition of CDK4 [7], and measurements were performed in triplicate.
Circular Dichroism (CD) Analyses
Samples containing 7.5–10.0 μM proteins in 20 mM sodium borate-40 μM DTT buffer (pH 7.4) were incubated with different amounts of guanidinium chloride (GdnHCl, in a stock solution of 8.5 M) on ice overnight and then equilibrated at 25°C just prior to CD analysis [9]. The rotation at 222 nm was measured on an AVIV far-UV spectropolarimeter using a quartz microcell (Helma) of 0.1 cm light pass length, and the exact concentrations of GdnHCl were determined using the refractive index. For each sample, three scans were averaged, and the free energy of protein denaturation in aqueous condition was obtained on the basis of two-state approximation [9]. Heat-induced unfolding experiments were performed using 5.0 μM proteins with a 1-nm bandwidth and a 10-second response time. Thermal melting spectra were recorded at 222 nm by heating from 5°C to 65°C at the rate of 1°C per minute and a 1°C interval followed by cooling down to 5°C at the same rate. Tm was defined as the temperature at the midpoint of transition [9].
NMR Analyses
NMR samples contained 0.4 mM protein, 5 mM HEPES, 1 mM DTT, 5 μM EDTA in 90% H2O/10% D2O at pH 7.4 [7]. The 1D 1H NMR and 2D 1H-homonulcear NOESY experiments (200 ms mixing time) were performed at 20 °C on a Bruker DMX_600 spectrometer, which was equipped with a 5 mm triple-resonance probe and three-axis gradient coil. Data were processed with XWINNMR 3.5 (Bruker).
Results
IKKβ specifically interacts with P16 and phosphorylates Ser8 of P16
IKKβ is the primary kinase to phosphorylate Ser32 and Ser36 of IκBα thus triggering the release of NF-κB from the inactive NF-κB/IκBα complex [11]. The activation of IKKβ, as a result of inflammatory cytokine signaling, infectious agents, and DNA damage, has been regarded as a predominant pathway in regulating the activity of NF-κB [11]. To explore the potential interaction between IKKβ and P16, we first investigated the potential P16/IKKβ interaction through immunoprecipitations. WI38 (p16+/+) cells were chosen for this study mainly due to the fact that these cells have endogenous P16 as well as P16-phosphorylating activities as demonstrated in previous studies [6]. In contrast, U2OS, a p16-null cell line was used as a control. As shown in Fig. 1A, IKKβ was expressed in both WI38 and U2OS cells while P16 was only present in WI38. In the immunoprecipitation assay using anti-P16 antibody, IKKβ was present only in the immunoprecipitates from WI38 cells (Fig. 1B). Similarly, in immunocipitation using anti-IKKβ antibody, P16 was detected in the immunoprecipitates from WI38, not U2OS (Fig. 1C). These results indicate that P16 and IKKβ interact with each other in WI38 cells. As control, such interaction was not observed in U2OS cells simply due to the absence of endogenous P16. Indeed, as shown in Figure 1C, when exogenous P16 was added to the lysates of U2OS cells, P16 was detected in the immunoprecipitates (using anti-IKKβ), implying that exogenous P16 is able to physically associate with cellular IKKβ.
Fig. 1. P16 physically interacts with IKKβ.
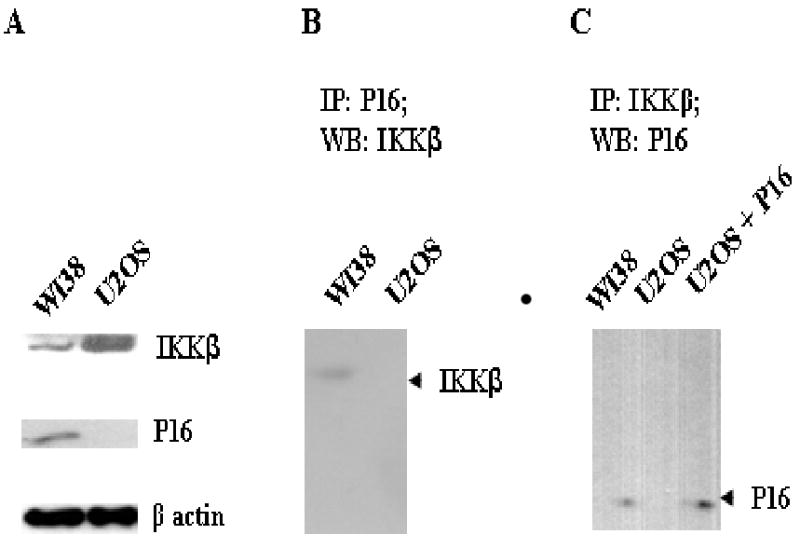
A, Expression of IKKβ and P16 in WI38 and U2OS. 50 μg of cell lysates were subjected to western blotting using corresponding antibodies. β-actin was used as internal control. B and C, Immunoprecipiations to investigate the potential P16/IKKβ interaction. 400 μg of cell lysates from WI-38 and U2OS were immunoprecipitated using indicated antibodies and proteins present in each immunoprecipitate were analyzed by western blotting. In Lane 3 of C, 2 μg of recombinant P16 was added into U2OS cell lysates prior to immunoprecipitation.
We then examined the potential phosphorylation of IKKβ on P16 using an in vitro assay. As shown in Fig. 2A, in a reaction mixture containing recombinant IKKβ and P16, P16 was phosphorylated as the positive control, IκBα1-214, a truncated form of IκBα containing the intact N-terminus and four ARs [8, 11]. In contrast, no phosphorylation was observed on Yar 1, a yeast AR protein [8], indicating that IKKβ is competent in phosphorylating P16 and such phosphorylation is not universal for all AR proteins. Moreover, IKKβ did not phosphorylate P16 S7A/S8A/S140A/S152A mutant, implying that the IKKβ phosphorylation sites are among the previously-identified Ser7, Ser8, Ser140, and Ser152 of P16. Since P16 S7A/S8A/S140A/S152A remains well structured (to be described in the following section), failure in phosphorylating this mutant further supports the notion that IKKβ phosphorylation is specific. We also constructed four P16 mutants, each of which contains only one putative phosphorylation site while the other three serine residues are substituted by alanine, to identify the serine residue(s) for IKKβ phosphorylation. Interestingly, only one mutant, P16 S7A/S140A/S152A was phosphorylated by IKKβ in vitro (Figure 2A). In addition, among P16 S7E, S8E, S140E, and S152E (a group of mutants mimicking serine phosphophorylation as addressed in the following section), P16 S8E was not phosphorylated by IKKβ in vitro (data not shown). Taken together, these results strongly indicate that Ser8 of P16 is the primary target for IKKβ and the removal of this site abolishes IKKβ-mediated phosphorylation of P16.
Fig. 2. In vitro P16 Phosphorylation.
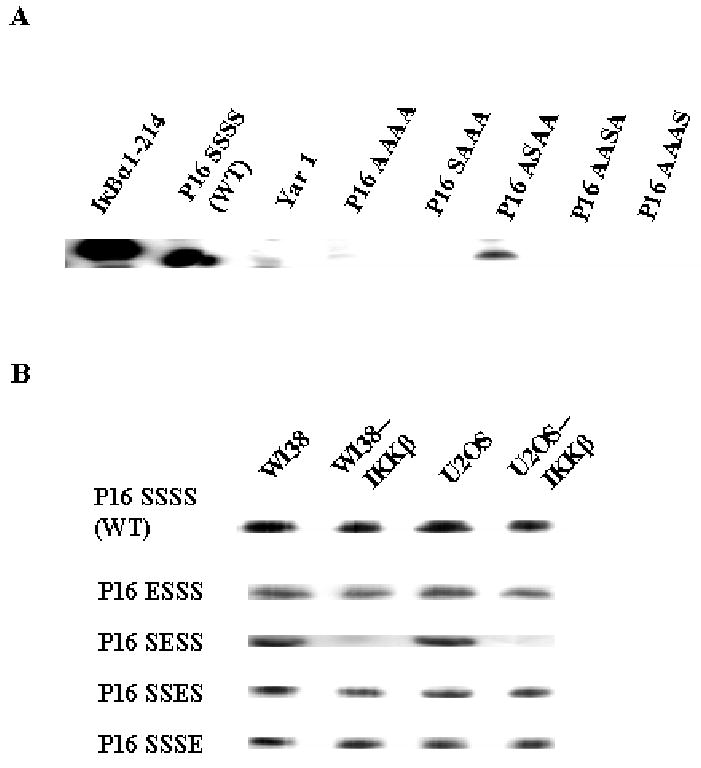
A, P16 phosphorylation by recombinant IKKβ. B, P16 phosphorylation by cell lysates and IKKβ-depleted cell lysates. WI38--IKKβ and U2OS--IKKβ represent IKKβ-depleted WI38 and U2OS lysates, respectively. Each reaction mixture contained 0.1 μg of recombinant IKKβ (A) or 10 μg of cell lysates (B), 2 μg of indicated proteins, and 5 μCi [γ-32P] ATP in a volume of 15 μl. In A, IκBα1-214 and Yar 1 were used as positive and negative controls, respectively. P16 SSSS, P16 WT; P16 AAAA, S7A/S8A/S140A/S152A; P16 SAAA, S8A/S140A/S152A; P16 ASAA, S7A/S140A/S152A; P16 AASA, S7A/S8A/S152A; P16 AAAS, S7A/S8A/S140A.
To further confirm the above findings, we used cell lysates from WI38 and U2OS to phosphorylate the afore-mentioned P16 mutants and wild type (WT). As shown in Fig. 2B, both cell lysates from WI38 and U2OS exhibited P16-phosphorylating activities and such activities were not eliminated as long as at least one serine residue of Ser7, Ser8, Ser140, and Ser152 was present in P16, which is consistent with results in previous studies [6]. However, the depletion of IKKβ from these cell lysates through immunoprecipitation significantly decreased the phosphorylation of P16 S7A/S140A/S152A, indicating that IKKβ is the primary kinase for phosphorylation of P16 Ser8. In the meanwhile, no significant changes were observed in the phosphorylation of P16 S8A/S140A/S152A, S7A/S8A/S152A, and S7A/S8A/S140A upon the depletion of IKKβ, indicating that Ser8 of P16 is the primary phosphorylation site for IKKβ and kinases other than IKKβ are involved in the phosphorylation of Ser7, Ser140, and Ser152 of P16.
IKKβ-mediated phosphorylation of Ser8 of P16 impairs its inhibition to CDK4
Subsequently, we endeavored to assess the structural and functional impacts of P16 phosphorylation by substituting Ser7, Ser8, Ser140, and Ser152 of P16 with glutamates, which provides a rough phosphomimetic [12]. Potential changes in the conformational stability, the core structure, and the CDK4-inhibitory activity of these four single S→E mutants and P16 S7A/S8A/S140A/S152A (as control) were analyzed by Circular dichroism (CD), NMR, and in vitro CDK4 kinase assays, respectively [7]. The results are summarized in Table 1. Interestingly, these five mutants all followed a two-state-transition model in GdnHCl- and heat-induced unfolding (data not shown), and their ΔGdwater and Tm values were comparable to those of P16 WT except that moderately decreased Tm values were observed with S140E and S152E mutants. Furthermore, no significant perturbations to the core structures of these five mutants were observed as evidenced by the fact that the 1D 1H NMR (Fig. 3) and 2D NOESY spectra (data not shown) of these mutants are almost identical to the corresponding NMR spectra of P16 WT [13]. These results strongly suggest that P16 phosphorylation does not bring about significant changes to the conformational stability and the core structure of P16. However, while the IC50 values of P16 S7E (140±17 nM), S140E (114±21 nM), S152E (45±12 nM), and S7A/S8A/S140A/S152A (87±21 nM) are comparable to or moderately different from that of P16 WT (72±15 nM), the IC50 value of S8E (>750 nM) is more than 10-time higher than that of P116 WT, indicating that P16 S8E only retains remnant CDK4-inhibitory activity. Since an alanine substitution at Ser8 as found in P16 S7A/S8A/S140A/S152A does not cause any detectable change in the CDK4-inhibitory activity of P16, the loss of CDK4-inhibitory ability observed in P16 S8E mainly results from the introduction of a negatively-charged side chain in Ser8, implying that phosphorylation of Ser8 largely inactivates P16. The molecular mechanisms underlying this inactivation remain to be further explored. With regard to the dynamic nature of the P16/CDK4 association, it is plausible that phosphorylation at Ser8 of P16 may cause some local conformational changes undetectable by NMR but important in binding kinetics.
Table 1. Characterization of Phosphomimetic Ser→Glu Mutants of P16.
| Protein | IC50 (nM)a |
Core Structureb | ΔGdwater (kcal/mol)c |
D1/2 (M)c |
m (kcal mol-1 M-1)c |
Tm (°C)d |
|---|---|---|---|---|---|---|
| P16 SSSSe (WT) |
72±15 | Retained | 2.32 | 0.72 | 3.23 | 46.0 |
| P16 ESSS (S7E) |
140±17 | Retained | 2.95 | 0.83 | 3.49 | 47.3 |
| P16 SESS (S8E) |
>750f | Retained | 2.14 | 0.70 | 3.05 | 46.1 |
| P16 SSES (S140E) |
114±21 | Retained | 2.49 | 0.79 | 3.07 | 41.7 |
| P16 SSSE (S152E) |
45±12 | Retained | 2.46 | 0.80 | 2.95 | 43.0 |
| P16 AAAA | 87±21 | Retained | 2.82 | 0.76 | 3.73 | 46.3 |
The estimated error in the determination of IC50 is ± 20%, and a 3-fold change in the IC50 values is regarded as biochemically significant [7].
ΔGdwater, D1/2 (the denaturant concentration at the midpoint of transition), and m values were calculated according to a two-state transition model, and the error in ΔGdwater was estimated to be ± 0.5 kcal/mol [9].
The error of Tm was estimated to be ± 0.5 °C [9].
Data cited from [13].
The exact IC50 of P16 S8E was not determined simply because the maximum inhibition of CDK4 was not achieved in the presence of 2.0 μM P16 S8E, the highest concentration used in our assay.
Fig. 3. 1D 1H NMR Spectra showing the amide region as well as upfield aliphatic region of Phosphomimetic P16 Mutants in 2H2O at 600 MHz.
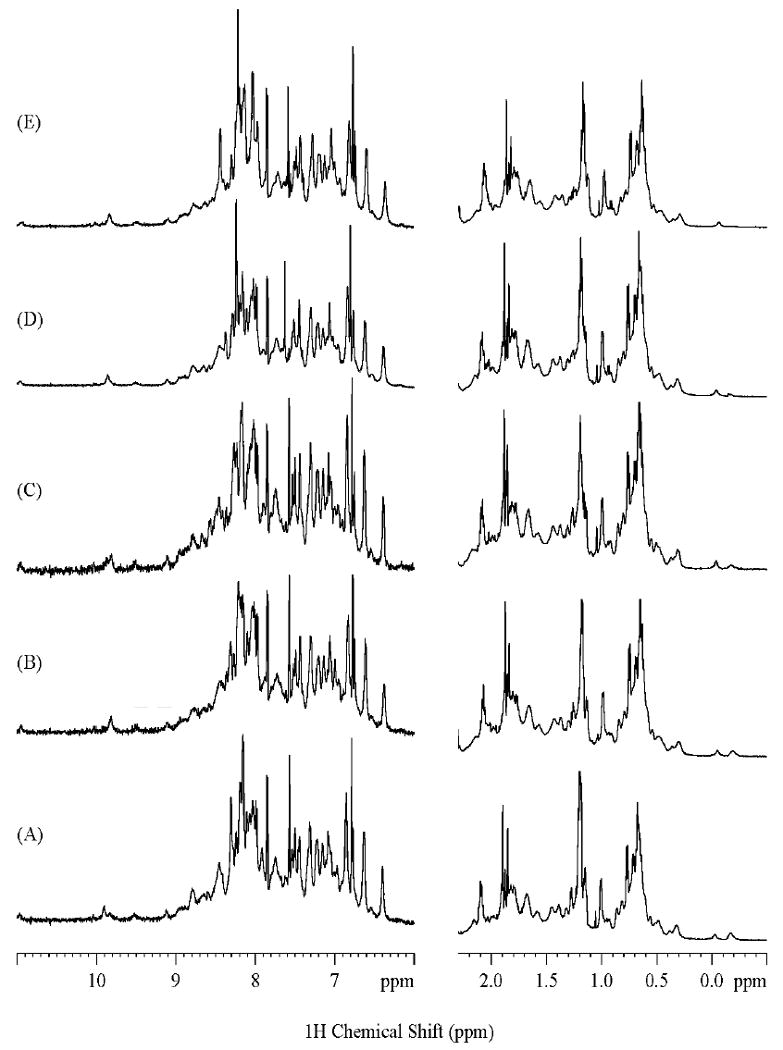
(A), P16 AAAA (as described in Figure 2); (B), P16 ESSS (S7E); (C), P16 SESS (S8E); (D) P16 SSES (S140E); (E), P16 SSSE (S152E). All samples were prepared in 4 mM HEPES- 1mM DTT-5μM EDTA (pH 7.4) in 2H2O, and the NMR experiments were performed at 20 °C.
Discussion
Here, we have shown that IKKβ can upregulate CDK4-mediated phopshorylation of pRb through phosphorylating and inactivating P16, indicating that activation of IKKβ also plays important roles in modulating the pRb pathway. Such resemblance between P16 and IκBα in posttranslational regulation, together with the fact that P16 and IκBα are biochemically “bi-functional” to inhibit both CDK4 and NF-κB [8], strongly supports the notion that there is a crosstalk between the P16/CDK4/pRb pathway and the IκBα/NF-κB pathway. While cancer-related environmental and cellular factors, such as viral infection and genetically toxic chemicals, can modulate both pathways through activating IKKβ, alterations in one pathway may affect the other pathway concomitantly. From this point of view, two prevalent molecular events in human cancers, P16 inactivation and NF-κB activation [4, 11], should be analyzed coordinately.
Out of the four putative phosphorylation sites identified in previous studies [6], IKKβ seemingly only phosphorylates Ser8 of P16. Even for the phosphorylation of Ser8, our results do not rule out the possibility that kinases other than IKKβ are involved. As shown in Figure 2B, there is still a small amount of phosphorylated P16 S7A/S140A/S152A in the reaction mixtures containing IKKβ-depleted cell lysates, which could result from remnant IKKβ after immunoprecipitation or other kinases. Hence, phosphorylation of P16 involves multiple kinases. Moreover, the effects upon phosphorylation at different Ser sites of P16 are different. On one hand, as revealed in our phosphomimetic studies (Table 1), the phosphomimetic substitution at Ser8 of P16 eliminates the majority of its CDK4-inhibitory activity but does not perturb the core structure and the conformational stability. Contrarily, similar substitutions at Ser140 and Ser152 moderately destabilize P16 in heat-induced unfolding while the CDK4-inhibitory activity remains intact. On the other hand, potential changes in biochemical and biophysical properties other than the aforementioned conformational stability, core structure, and CDK4-inhibitory activity of P16, such as its subcellular localization, have not been investigated in our current study. While it has been known that IKKβ-mediated phosphorylation at Ser32 and Ser36 of IκBα facilitates ubiquitylation at Lys21 and Lys22, leading to proteasome-mediated degradation [14], there is evidence implying that phosphorylation of IκBα also may alter its subcellular distribution [15]. Similarly, phosphorylation of P16 may change its localization in the cell thus influencing its association with CDK4/6 due to cellular compartmentalization. This could be true since only phosphorylation at Ser152 has been found in CDK4-bound P16 [6]. In addition, as described earlier, IKKβ phosphorylates P16 S7A/S140A/S152A substantially but fails with P16 S8E, suggesting that phosphorylation at Ser8 is somehow independent of phosphorylation at Ser7, Ser8, and Ser152. Nevertheless, it remains to be further verified whether phosphorylation at Ser7, Ser140, and Ser152 is coordinated.
Acknowledgments
This project was supported by a grant from NIH (CA69472) to J. L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell. 2002;2:103–112. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 2.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Yang D, Liu L, Zheng X. Cyclin-dependent kinase inhibitor p16INK4a and telomerase may co-modulate endothelial progenitor cells senescence. Ageing Res Rev. 2008;7:137–146. doi: 10.1016/j.arr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 5.Lang JC, Borchers J, Danahey D, Smith S, Stover DG, Agrawal A, Malone JP, Schuller DE, Weghorst CM, Holinga AJ, Lingam K, Patel CR, Esham B. Mutational status of overexpressed p16 in head and neck cancer: evidence for germline mutation of p16/p14ARF. Int J Oncol. 2002;21:401–408. doi: 10.3892/ijo.21.2.401. [DOI] [PubMed] [Google Scholar]
- 6.Gump J, Stokoe D, McCormick F. Phosphorylation of p16INK4A correlates with Cdk4 association. J Biol Chem. 2003;278:6619–6622. doi: 10.1074/jbc.C200622200. [DOI] [PubMed] [Google Scholar]
- 7.Byeon IJ, Li J, Ericson K, Selby TL, Tevelev A, Kim HJ, O'Maille P, Tsai MD. Tumor suppressor p16INK4A: determination of solution structure and analyses of its interaction with cyclin-dependent kinase 4. Mol Cell. 1998;1:421–431. doi: 10.1016/s1097-2765(00)80042-8. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Joo S, Tsai MD. An NF-κB-specific inhibitor, IκBα, binds to and inhibits cyclin-dependent kinase 4. Biochemistry. 2003;42:13476–13483. doi: 10.1021/bi035390r. [DOI] [PubMed] [Google Scholar]
- 9.Guo Y, Mahajan A, Yuan C, Joo S, Weghorst CM, Tsai MD, Li J. Comparisions of the conformational stability of cyclin-dependent kinase (CDK) 4-interacting ankyrin repeat (AR) proteins. Biochemistry. 2009;48:4050–4062. doi: 10.1021/bi802247p. [DOI] [PubMed] [Google Scholar]
- 10.Higashitsuji H, Higashitsuji H, Liu Y, Masuda T, Fujita T, Abdel-Aziz HI, Kongkham S, Dawson S, Mayer RJ, Itoh Y, Sakurai T, Itoh K, Fujita J. The oncoprotein gankyrin interacts with RelA and suppresses NF-κB activity. Biochem Biophy Res Commun. 2007;363:879–884. doi: 10.1016/j.bbrc.2007.09.072. [DOI] [PubMed] [Google Scholar]
- 11.Pratt MAC, Tibbo E, Robertson SJ, Jansson D, Hurst K, Perez-Iratxeta C, Lau R, Niu MY. The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population. Oncogene. 2009;28:2710–2722. doi: 10.1038/onc.2009.131. [DOI] [PubMed] [Google Scholar]
- 12.Low C, Homeyer N, Weininger U, Sticht H, Balbach J. Conformational switch upon phosphorylation: human CDK inhibitor p19INK4d between the native and partially folded state. ACS Chem Biol. 2009;4:53–63. doi: 10.1021/cb800219m. [DOI] [PubMed] [Google Scholar]
- 13.Tevelev A, Byeon IJ, Selby T, Ericson K, Kim HJ, Kraynov V, Tsai MD. Tumor suppressor p16INK4A: structural characterization of wild-type and mutant proteins by NMR and circular dichroism. Biochemistry. 1996;35:9475–9487. doi: 10.1021/bi960211+. [DOI] [PubMed] [Google Scholar]
- 14.Jacobs MD, Harrison SC. Structure of an IκBα/NF-κB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 15.Baeuerle PA. IκB-NF-κB structures: At the interface of inflammation control. Cell. 1998;95:729–731. doi: 10.1016/s0092-8674(00)81694-3. [DOI] [PubMed] [Google Scholar]