

Abstract
The AGBNP2 implicit solvent model, an evolution of the Analytical Generalized Born plus Non-Polar (AGBNP) model we have previously reported, is presented with the aim of modeling hydration effects beyond those described by conventional continuum dielectric representations. A new empirical hydration free energy component based on a procedure to locate and score hydration sites on the solute surface is introduced to model first solvation shell effects, such as hydrogen bonding, which are poorly described by continuum dielectric models. This new component is added to the Generalized Born and non-polar AGBNP terms. Also newly introduced is an analytical Solvent Excluded Volume (SEV) model which improves the solute volume description by reducing the effect of spurious high-dielectric interstitial spaces present in conventional van der Waals representations. The AGBNP2 model is parametrized and tested with respect to experimental hydration free energies of small molecules and the results of explicit solvent simulations. Modeling the granularity of water is one of the main design principles employed for the the first shell solvation function and the SEV model, by requiring that water locations have a minimum available volume based on the size of a water molecule. It is shown that the new volumetric model produces Born radii and surface areas in good agreement with accurate numerical evaluations of these quantities. The results of molecular dynamics simulations of a series of mini-proteins show that the new model produces conformational ensembles in substantially better agreement with reference explicit solvent ensembles than the original AGBNP model with respect to both structural and energetics measures.
1 Introduction
Water plays a fundamental role in virtually all biological processes. The accurate modeling of hydration thermodynamics is therefore essential for studying protein conformational equilibria, aggregation, and binding. Explicit solvent models arguably provide the most detailed and complete description of hydration phenomena.1 They are, however, computationally demanding not only because of the large number of solvent atoms involved, but also because of the need to average over many solvent configurations to obtain meaningful thermodynamic data. Implicit solvent models,2 which are based on the statistical mechanics concept of the solvent potential of mean force,3 have been shown to be useful alternatives to explicit solvation for applications including protein folding and binding,4 and small molecule hydration free energy prediction.5
Modern implicit solvent models6,7 include distinct estimators for the non-polar and electrostatic components of the hydration free energy. The non-polar component corresponds to the free energy of hydration of the uncharged solute while the electrostatic component corresponds to the free energy of turning on the solute partial charges. The latter is typically modeled treating the water solvent as a uniform high-dielectric continuum.8 Methods based on the numerical solution of the Poisson-Boltzmann (PB) equation9 provide a virtually exact representation of the response of the solvent within the dielectric continuum approximation. Recent advances extending dielectric continuum approaches have focused on the development of Generalized Born (GB) models,10 which have been shown to reproduce with good accuracy PB and explicit solvent7,11 results at a fraction of the computational expense. The development of computationally efficient analytical and differentiable GB methods based on pairwise descreening schemes6,12,13 has made possible the integration of GB models in molecular dynamics packages for biological simulations.14–16
The non-polar hydration free energy component accounts for all non-electrostatic solute-solvent interactions as well as hydrophobic interactions,17 which are essential driving forces in biological processes such as protein folding18–21 and binding.22–25 Historically the non-polar hydration free energy has been modeled by empirical surface area models26 which are still widely employed.10,27–35 Surface area models are useful as a first approximation, however qualitative deficiencies have been observed.29,36–41
A few years ago we presented the Analytical Generalized Born plus Non-Polar (AGBNP) implicit solvent model,42 which introduced two key innovations with respect to both the electrostatic and non-polar components. Unlike most implicit solvent models, the AGBNP non-polar hydration free energy model includes distinct estimators for the solute–solvent van der Waals dispersion energy and cavity formation work components. The main advantages of a model based on the cavity/dispersion decomposition of the non-polar solvation free energy stem from its ability to describe both medium range solute–solvent dispersion interactions, which depend on solute composition, as well as effects dominated by short-range hydrophobic interactions, which can be modeled by an accessible surface area term.40 A series of studies highlight the importance of the balance between hydrophobicity and dispersion interactions in regulating the structure of the hydration shell and the strength of interactions between macromolecules.43–45 In AGBNP the work of cavity formation is described by a surface area-dependent model,37,46–48 while the dispersion estimator is based on the integral of van der Waals solute–solvent interactions over the solvent, modeled as a uniform continuum.38 This form of the non-polar estimator had been motivated by a series of earlier studies5,37,49–52 and has since been shown by us38,53–55 and others39–41,56 to be qualitatively superior to models based only on the surface area in reproducing explicit solvent results as well as rationalizing structural and thermodynamical experimental observations.
The electrostatic solvation model in AGBNP is based on the pairwise descreening GB scheme13 whereby the Born radius of each atom is obtained by summing an appropriate descreening function over its neighbors. The main distinction between the AGBNP GB model and conventional pairwise descreening implementations is that in AGBNP the volume scaling factors, which offset the overcounting of regions of space occupied by more than one atom, are computed from the geometry of the molecule rather than being introduced as geometry-independent parameters fit to either experiments or to numerical Poisson-Boltzmann results.14,57–59 The reduction of the number of parameters achieved with this strategy improves the transferability of the model to unusual functional groups often found in ligand molecules, which would otherwise require extensive parametrization.60
Given its characteristics, the AGBNP model has been mainly targeted to applications involving molecular dynamics canonical conformational sampling, and to the study of protein-ligand complexes. Since its inception the model has been employed in the investigation of a wide variety of biomolecular problems ranging from peptide conformational propensity prediction and folding,54,61–63 ensemble-based interpretation of NMR experiments,64,65 protein loop homology modeling,55 ligand-induced conformational changes in proteins,66,67 conformational equilibria of protein-ligand complexes,68,69 protein-ligand binding affinity prediction,70 and structure-based vaccine design.71 The AGBNP model has been re-implemented and adopted with minor modifications by other investigators.72,73 The main elements of the AGBNP non-polar and electrostatic models have been independently validated,39,40,74,75 and have been incorporated in recently proposed hydration free energy models.76,77
In this work we present a new implicit solvent model named AGBNP2 which builds upon the original AGBNP implementation (hereafter referred to as AGBNP1) and improves it with respect to the the description of the solute volume and the treatment of short–range solute–water electrostatic interactions.
Continuum dielectric models assume that the solvent can be described by a linear and uniform dielectric medium.78 This assumption is generally valid at the macroscopic level, however at the molecular level water exhibits significant deviations from this behavior.1 Nonlinear dielectric response, the non-uniform distribution of water molecules, charge asymmetry and electrostriction effects79 are all phenomena originating from the finite size and internal structure of water molecules as well as their specific interactions which are not taken into account by continuum dielectric models. Some of these effects are qualitatively captured by standard classical fixed-charge explicit water models, however others, such as polarization and hydrogen bonding interactions, can be fully modeled only by adopting more complex physical models.80 GB models make further simplifications in addition to the dielectric continuum approximation, thereby compounding the challenge of achieving with GB-based implicit solvent models the level of realism required to reliably model phenomena, such as protein folding and binding, characterized by relatively small free energy changes.
In the face of these challenges a reasonable approach is to adopt an empirical hydration free energy model motivated by physical arguments81 parametrized with respect to experimental hydration free energy data.20 Models of this kind typically score conformations on the basis of the degree of solvent exposure of solute atoms. Historically82 the solvent accessible surface area of the solute has been used for this purpose, while modern implementations suitable for conformational sampling applications often employ computationally convenient volumetric measures.83,84 In this work we take this general approach but we retain the GB model component which we believe is a useful baseline to describe the long–range influence of the water medium. The empirical parametrized component of the model then takes the form of an empirical first solvation shell correction function designed so as to absorb hydration effects not accurately described by the GB model. Specifically, as described below, we employ a short–range analytical hydrogen bonding correction function based on the degree of water occupancy (taking into account the finite size of water molecules) of appropriately chosen hydration sites on the solute surface. The aim of this model is to effectively introduce some explicit solvation features without actually adding water molecules to the system as for example done in hybrid explicit/implicit models.85,86
In this work we also improve the description of the solute volume, which in AGBNP1 is modeled by means of atomic spheres of radius equal to the atomic van der Waals radius. The deficiencies of the van der Waals solute volume model have been recognized.87,88 They stem from the presence of high-dielectric interstitial spaces in the solute interior which are too small to contain discrete water molecules. These spurious high dielectric spaces contribute to the hydration of buried or partially buried atoms causing underestimation of desolvation effects. The volume enclosed by the molecular surface (MS), defined as the surface produced by a solvent spherical probe rolling on the van der Waals surface of the solute,89 represents the region which is inaccessible to water molecules and is often referred as the Solvent Excluded Volume (SEV).90 The SEV, lacking the spurious high-dielectric interstitial spaces, provides a better representation of the low-dielectric region associated with the solute. For this reason accurate Poisson-Boltzmann solvers,9,91,92 have employed the SEV description of the solute region.
Despite its clear advantages, the lack of analytical and computationally efficient representation of the SEV have hampered its deployment in conjunction with GB models for molecular dynamics applications. The GBMV series of models87,93,94 achieve high accuracy relative to numerical Poisson calculations in part by employing the SEV description of the solute volume. The analytical versions of GBMV93,94 describe the SEV volume by means of a continuous and differentiable solute density function which is integrated on a grid to yield atomic Born radii. In this work we present a model for the SEV that preserves the analytical pairwise atomic descreening approach employed in the AGBNP1 model,42 which avoids computations on a grid. We show that this approximate model reproduces some of the key features of the SEV while yielding the same favorable algorithmic scaling of pairwise descreening approaches.
This paper focuses primarily on the description and parametrization of the SEV model and the short range hydrogen bonding function of AGBNP2. In the Methods section we present a brief review of the AGBNP1 model, including the electrostatic and non-polar models, followed by the derivation of the analytical SEV pairwise descreening model and the short range hydrogen bonding function which are new for AGBNP2. In the Results section we validate the AGBNP2 analytical estimates for the Born radii and atomic surface areas using as a reference accurate numerical evaluations of these quantities. This is followed by the parametrization of the hydrogen bonding parameters against experimental hydration free energies of small molecules. This section concludes with a comparison between the structural and energetic properties of a a series of structured peptides (mini-proteins) predicted with the AGBNP2 model to those obtained with explicit solvation. The paper then concludes with a discussion and implications of the results, and with a perspective on future improvements and validation of the AGBNP2 model.
2 Methods
2.1 The Analytical Generalized Born plus Non-Polar Implicit Solvent Model (AGBNP)
In this section we briefly review the formulation of the AGBNP1 implicit solvent model, which forms the basis for the new AGBNP2 model. A full account can be found in the original reference.42 The AGBNP1 hydration free energy ΔGh(1) is defined as
| (1) |
where ΔGelec is the electrostatic contribution to the solvation free energy and ΔGnp includes non-electrostatic contributions. ΔGnp is further decomposed into a cavity hydration free energy ΔGcav and a solute–solvent van der Waals dispersion interaction component ΔGvdW.
2.1.1 Geometrical estimators
Each free energy component in Eq. (1) is ultimately based on an analytical geometrical description of the solute volume modeled as a set of overlapping atomic spheres of radii Ri centered on the atomic positions ri. Hydrogen atoms do not contribute to the solute volume. The solute volume is modeled using the Gaussian overlap approach first proposed by Grant and Pickup.95 In this model the solute volume is computed using the Poincaré formula (also known as the inclusion-exclusion formula) for the volume of the union of a set of intersecting elements
| (2) |
where is the volume of atom i, Vij is the volume of intersection of atoms i and j (second order intersection), Vijk is the volume of intersection of atoms i, j, and k (third order intersection), and so on. The overlap volumes are approximated by the overlap integral, , available in analytical form (see for example Eq. 10 of reference 42), between n Gaussian density functions each corresponding to a solute atom:
| (3) |
where the Gaussian density function for atom i is
| (4) |
where
| (5) |
and
| (6) |
and κ is a dimensionless parameter that regulates the diffuseness of the atomic Gaussian function. In the AGBNP1 formulation κ = 2.227.
Gaussian integrals are in principle non-zero for any finite distances between the Gaussian densities. Although not mentioned in reference 42, to reduce computational cost AGBNP1 includes a switching function that reduces to zero the overlap volume between two or more Gaussians when the overlap volume is smaller than a certain value. If is the value of the Gaussian overlap volume between a set of atoms, the corresponding overlap volume V12… used in Eq. (2) is set as
| (7) |
where
| (8) |
| (9) |
where, when using van der Waals atomic radii, v1 = 0.1 and v2 = 1 Å3, and for the augmented radii used in the surface area model (see below), v1 = 0.2 and v2 = 2 Å3. This scheme sets to zero Gaussian overlap volumes smaller than v1, leaves volumes above v2 unchanged and smoothly reduces volumes in between these two limits. It drastically reduces the number of overlap volumes that need to be calculated since the fact that an n-body overlap volume V12…n between n atoms is zero guarantees that all of the n+1-body overlap volumes corresponding to the same set of atoms plus one additional atom are also zero. (Note below that the formulation of AGBNP2 employs modified values of v1 and v2 to improve the accuracy of surface areas).
The van der Waals surface area Ai of atom i, which is another geometrical descriptor of the model, is based on the derivative ∂V/∂Ri of the solute volume with respect to the radius Ri96
| (10) |
where V is given by Eq. (2) and
| (11) |
with a = 5 Å2, is a filter function which prevents negative values for the surface areas for buried atoms while inducing negligible changes to the surface areas of solvent-exposed atoms.
The model further defines the so-called self-volume of atom i as
| (12) |
which is computed similarly to the solute volume and measures the solute volume that is considered to belong exclusively to this atom. Due to the overlaps with other atoms, the self-volume of an atom is smaller than the van der Waals volume Vi of the atom. The ratio
| (13) |
is a volume scaling factor is used below in the evaluation of the Born radii.
2.1.2 Electrostatic Model
The electrostatic hydration free energy is modeled using a continuous dielectric representation of the water solvent using the Generalized Born (GB) approximation
| (14) |
where
| (15) |
and where εin is the dielectric constant of the interior of the solute, εw is the dielectric constant of the solvent; qi and qj are the charges of atom i and j, and
| (16) |
In Eqs. (14)–(16) Bi denotes the Born radius of atom i which, under the Coulomb field approximation,57 is given by the inverse of the integral over the solvent region of the negative 4th power of the distance function centered on atom i,
| (17) |
In the AGBNP1 model this integral is approximated by a so-called pairwise descreening formula
| (18) |
where Ri is the van der Waals radius of atom i, sji is the volume scaling factor for atom j [Eq. (13)] when atom i is removed from the solute and Qji is the integral (available in analytic form see Appendix B of reference 42) of the function (r − ri)−4 over the volume of the sphere corresponding to solute atom j that lies outside the sphere corresponding to atom i. Eq. (18) applies to all of the atoms i of the solute (hydrogen atoms and heavy atoms) whereas the sum over j includes only heavy atoms. The AGBNP1 estimate for the Born radii Bi are finally computed from the inverse Born radii βi from Eq. (18) after processing them through the function:
| (19) |
where b−1 = 50 Å. The filter function Eq (19) is designed to prevent the occurrence of negative Born radii or Born radii larger than 50 Å. The goal of the filter function is simply to increase the robustness of the algorithm in limiting cases. The filter function has negligible effect for the most commonly observed Born radii smaller than 20 Å.
In the AGBNP1 model the scaling factors sji are approximated by the expression
| (20) |
where sj is given by Eq. (13) and Vij is the two body overlap volume between atoms i and j. Also, in the original AGBNP formulation the computation of the scaling factors and the descreening function in Eq. (18) employed the van der Waals radii for the atoms of the solute and the associated Gaussian densities. These two aspects have been modified in the new formulation (AGBNP2) as described below.
2.1.3 Non-Polar Model
The non-polar hydration free energy is decomposed into the cavity hydration free energy ΔGcav and the solute–solvent van der Waals dispersion interaction component ΔGvdW:
| (21) |
The cavity component is described by a surface area model37,46–48
| (22) |
where the summation runs over the solute heavy atoms, Ai is the van der Waals surface area of atom i from Eq. (10), and γi is the surface tension parameter assigned to atom i (see Table 1 of reference 42). Surface areas are computed using augmented radii for the atoms of the solute and the associated Gaussian densities. Augmented radii are defined as the the van der Waals radii (Table 1 of reference 42) plus a 0.5 Å offset. The computation of the atomic surface areas in AGBNP2 is mostly unchanged from the original implementation,42 with the exception of the values of the switching function cutoff parameters v1 and v2 of Eq. (7), which in the new model are set as v1 = 0.01 Å3 and v2 = 0.1 Å3. This change was deemed necessary to improve the accuracy of the surface areas which in the new model also affect the Born radii estimates through Eq. (31) below.
The solute-solvent van der Waals free energy term is modeled by the expression
| (23) |
where αi is an adjustable dimensionless parameter on the order of 1 (see Table 1 of reference 42) and 16
| (24) |
where ρw = 0.033428 Å−3 is the number density of water at standard conditions, and σiw and εiw are the OPLS force field97 Lennard-Jones interaction parameters for the interaction of solute atom i with the oxygen atom of the TIP4P water model.98 If σi and εi are the OPLS Lennard-Jones parameters for atom i,
| (25) |
| (26) |
where σw = 3.15365 Å, and εw = 0.155 kcal/mol are the Lennard-Jones parameters of the TIP4P water oxygen. In Eq. (23) Bi is the Born radius of atom i from Eqs. (18) and (19) and Rw = 1.4 Å is a parameter corresponding to the radius of a water molecule.
2.2 The AGBNP2 Implicit Solvent Model
The AGBNP2 hydration free energy ΔGh(2) is defined as
| (27) |
where ΔGelec and ΔGnp have the same form as in the AGBNP1 model [Eqs. (14) and (21)–(23), respectively]. The only major difference is the pairwise descreening model for the Born radii that in AGBNP2 is based on the solvent excluding volume described below rather than the van der Waals volume as in AGBNP1. ΔGhb, described in Section 2.2.2, is a novel term for AGBNP2 which represents a first solvation shell correction corresponding to the portion of the hydration free energy not completely accounted for by the uniform continuum model for the solvent. We think of this term as mainly incorporating the effect of solute–solvent hydrogen bonding. As described in detail below, the analytical model for ΔGhb is based on measuring and scoring the volume of suitable hydration sites on the solute surface.
2.2.1 Pairwise Descreening Model using the Solvent Excluding Volume
When using van der Waals radii to describe the solute volume, small crevices between atoms (Fig. 1, panel A) are incorrectly considered as high-dielectric solvent regions,93,99,100 leading to underestimation of the Born radii, particularly for buried atoms. The van der Waals volume description implicitly ignores the fact that the finite size of water molecules prevents them to occupy sites that, even though are not within solute atoms, are too small to be occupied by water molecules. Ideally a model for the Born radii would include in the descreening calculation all of the volume excluded from water either because it is occupied by a solute atom or because it is located in an interstitial region inaccessible to water molecules. We denote this volume as the Solvent Excluding Volume (SEV). A realistic description of the SEV is the volume enclosed within the molecular surface89 of the solute obtained by tracing the surface of contact of a sphere with a radius characteristic of a water molecule (typically 1.4 Å) rolling over the van der Waals surface of the solute. The main characteristic of this definition of the SEV (see Fig 2) is that, unlike the van der Waals volume, it lacks small interstitial spaces while it closely resembles the van der Waals volume near the solute-solvent interface. The molecular surface description of the SEV can not be easily implemented into an analytical formulation. In this section we will present an analytical description of the SEV for the purpose of the pairwise descreening computation of the Born radii, as implemented in AGBNP2, that preserves the main characteristics of the molecular surface description of the SEV.
Figure 1.

Schematic diagram illustrating the ideas underpinning the model for the solvent excluded volume descreening. Circles represent atoms of two idealized solutes placed in proximity of each other. The van der Waals description of the molecular volume (panel A) leaves high dielectric interstitial spaces that are too small to fit water molecules. The adoption of enlarged van der Waals radii (B) removes the interstitial spaces but incorrectly excludes solvent from the surface of solvent exposed atoms. The solvent volume subtended by the solvent-exposed surface area is subtracted from the enlarged volume of each atom (C) such that larger atomic descreening volumes are assigned to buried atoms (circled) than exposed atoms (D), leading to the reduction of interstitial spaces while not overly excluding solvent from the surface of solvent exposed atoms.
Figure 2.

Illustration of the relationship between the van der Waals volume and the solvent excluded volume enclosed by the molecular surface.
The main ideas underpinning the SEV model presented here are illustrated in Fig. 1. We start with the van der Waals representation of the solute (model A) which presents an undesirable high dielectric interstitial space between the two groups of atoms. Increasing the atomic radii leads to a representation (model B) in which the interstitial space is removed but that also incorrectly excludes solvent from the surface of solvent exposed atoms. This representation is therefore replaced with one in which the effective volume of each atom in B is reduced by the volume subtended between the solvent-exposed surface of each atom and its van der Waals radius (panel C). This process yields model D in which the effective volume of the most buried atom is larger than those of the solvent-exposed atoms. This SEV model covers the interstitial high-dielectric spaces present in a van der Waals description of the solute volume, while approximately maintaining the correct van der Waals volume description of atoms at the solute surface as in the molecular surface description of the SEV (Fig. 2).
These ideas have been implemented in the AGBNP2 model as follows. The main modification consists of adopting for the pairwise descreening Generalized Born formulation the same augmented van der Waals radii as in the computation of the atomic surface areas. As in the previous model the augmented atomic radii, , are set as
| (28) |
where Ri is the van der Waals radius of the atom and ΔR = 0.5 Å is the offset. The augmented radii are used in the same way as in the AGBNP1 formulation to define the atomic spheres and the associated Gaussian densities [Eqs. (3)–(6)]. Henceforth in this work all of the quantities (atomic volumes, self volumes, etc.) are understood to be computed with the augmented atomic radii, unless otherwise specified. In AGBNP2 the form of the expression for the inverse Born radii [Eq. (18)] is unchanged however the expressions for the volume scaling factors sji and the evaluation of the descreening function Qji are modified as follows to introduce the augmented atomic radii and the reduction of the atomic self volumes in proportion to the solvent accessible surface areas as discussed above.
The pairwise volume scaling factors sji, that is the volume scaling factor for atom j when atom i is removed from the solute, are set as
| (29) |
where sj (defined below) is the volume scaling factor for atom j analogous to Eq. (13) computed with all the atoms present, and the quantity
| (30) |
subtracts from the expression for the self-volume of atom j all those overlap volumes involving both atoms i and j.
Two differences with respect to the original AGBNP1 formulation are introduced. The first is that sj is computed from the self-volume after subtracting from it the volume of the region subtended by the solvent-exposed surface in between the enlarged and van der Waals atomic spheres of atom j, according to the expression
| (31) |
where Aj is the surface area of atom j from Eq. (10) Referring to Fig. 3 the volume of the subtended region is djAj as in Eq. (31) with
Figure 3.
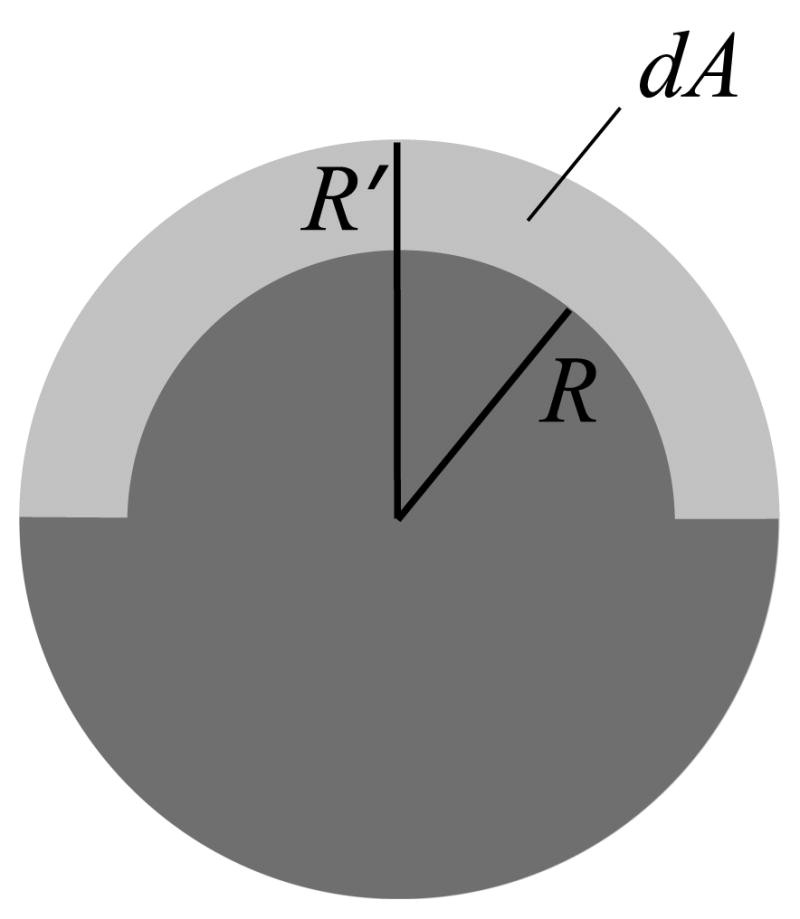
Graphical construction showing the volume subtracted from the atomic self-volume to obtain the surface-area corrected atomic self-volume. R is the van der Waals radius of the atom, R′ = R + ΔR is the enlarged atomic radius. dA is the volume of the region (light gray) subtended by the solvent-exposed surface area in between the enlarged and van der Waals atomic spheres.
| (32) |
The other difference concerns the term which in the AGBNP1 formulation is approximated by the 2-body overlap volume Vij [see Eq. (13)]; the first term in the right hand side of Eq. (30). This approximation is found to lack sufficient accuracy for the the present formulation given the relative increase in size of all overlap volumes. Therefore in AGBNP2 is computed including in Eq. (30) all non-zero overlap volumes after the application of the switching function from Eq. (7).
In the AGBNP2 formulation the functional form for the pair descreening function Qji is the same as in the original formulation (see Appendix of reference 42), however in the new formulation this function is evaluated using the van der Waals radius Ri for atom i (the atom being “descreened”) and the augmented radius for atom j (the atom that provides the solvent descreening), rather than using the van der Waals radius for both atoms. So if the pair descreening function is denoted by Q(r, R1, R2), where r is the interatomic distance, R1 the radius of the atom being descreened and R2 the radius that provides descreening, we set in Eq. (18)
| (33) |
The alternative of using enlarged atoms for both atoms and the inclusion of a properly weighted self-descreening term (to take into account the SEV of the atom being descreened) was also tried and judged to be less accurate than Eq. (33) relative to numerical integration.
2.2.2 Short Range Hydrogen Bonding Correction Function
In this section we present the analytical model that implements the short–range hydrogen-bonding correction function for AGBNP2. The model is based on a geometrical procedure, described below, to measure the degree to which a solute atom can interact with hydration sites on the solute surface. The procedure is as follows. A sphere of radius Rs representing a water molecule is placed in a position that provides near optimal interaction with a hydrogen bonding donor or acceptor atom of the solute. The position rs of this water sphere s is function of the positions of two or more parent atoms that compose the functional group including the acceptor/donor atom:
| (34) |
where {rps} represents the positions of the set of parent atoms of the water site s. For instance the water site position in correspondence with a polar hydrogen is:
where rD is the position of the heavy atom donor, rH is the position of the polar hydrogen, and dHB is the distance between the heavy atom donor and the center of the water sphere (see Fig. 4). Similar relationships (see Appendix) are employed to place candidate water spheres in correspondence with hydrogen bonding acceptor atoms of the solute. These relationships are based on the local topology of the hydrogen bonding acceptor group (linear, trigonal, and tetrahedral). This scheme places one or two water spheres in correspondence with each hydrogen bonding acceptor atom (see Table 1).
Figure 4.

Schematic diagram for the placement of the water sphere (w, light gray) corresponding to the hydrogen bonding position relative to the a polar hydrogen (white sphere) of the solute (dark gray). The dashed line traces the direction of the hydrogen-parent heavy atom (circled) bond along which the water sphere is placed. The magnitude of hydrogen bonding correction grows as a function of the volume (light gray) of the water site sphere not occupied by solute atoms.
Table 1.
Optimized surface tension parameters and hydrogen bonding correction parameters for the atom types present in protein molecules. γ is the surface tension parameter, Nw is the number of water spheres and h is the maximum correction corresponding to each atom type [Eq. (37)]. Atom types not listed do not have HB corrections and are assigned γ = 117 cal/mol/Å2
| Atom Type | γ (cal/mol/Å2) | Geometry | Nw | h (kcal/mol) |
|---|---|---|---|---|
| C (aliphatic) | 129 | |||
| C (aromatic) | 120 | |||
| H on N | linear | 1 | −0.25 | |
| H on N (Arg) | linear | 1 | −2.50 | |
| H on O | linear | 1 | −0.40 | |
| H on S | linear | 1 | −0.50 | |
| O (alchol) | 117 | tetrahedral | 2 | −0.40 |
| O (carbonyl) | 117 | trigonal | 2 | −1.25 |
| O (carboxylate) | 40 | trigonal | 2 | −1.80 |
| N (amine) | 117 | tetrahedral | 1 | −2.00 |
| N (aromatic) | 117 | trigonal | 1 | −2.00 |
| S | 117 | tetrahedral | 2 | −0.50 |
The magnitude of the hydrogen bonding correction corresponding to each water sphere is a function of the predicted water occupancy of the location corresponding to the water sphere. In this work the water occupancy is measured by the fraction ws of the volume of the water site sphere that is accessible to water molecules without causing steric clashes with solute atoms [see Fig. (4)]
| (35) |
where is the volume of the water sphere and
| (36) |
is the “free” volume of water site s, obtained by summing over the two-body, three-body, etc. overlap volumes of the water sphere with the solute atoms. Note that the expression of the free volume is the same as the expression for the self-volume [Eq. (12)] except that it lacks the fractional coefficients 1/2, 1/3, etc. The overlap volumes in Eq. (36) are computed using radius Rs for the water site sphere (here set to 1.4 Å) and augmented radii for the solute atoms. Eq. (36) is derived similarly to the expression for the self volumes by removing overlap volumes from the volume of the water sphere rather than evenly distributing them across the atoms participating in the overlap.
Given the water occupancy ws of each water sphere, the expression for the hydrogen bonding correction for the solute is
| (37) |
where hs is the maximum correction energy which depends on the type of solute-water hydrogen bond (see Table 1), and S(w; wa, wb) is a polynomial switching function which is zero for w < wa, one for w > wb, and smoothly (with continuous first derivatives) interpolates from 0 to 1 between wa and wb (see Fig. 5). The expression of S(w; wa, wb) is:
| (38) |
where fw(x) is a switching function given by Eq. (9). In this work we set wa = 0.15 and wb = 0.5. This scheme establishes (see Fig. 5) that no correction is applied if more than 85% of the water sphere volume is not water accessible, whereas maximum correction is applied if 50% or more of the water sphere volume is accessible.
Figure 5.

The switching function S(w; wa, wb) from Eqs. (38) and (9) with wa = 0.15 and wb = 0.5.
2.3 Molecular dynamics of mini-proteins
We conducted molecular dynamics simulations of what we will refer to as mini-proteins (Fig. 6), that is peptides that have been shown to form stable secondary structures in solution: the 23-residue trp-cage peptide of sequence ALQELLGQWLKDGGPSSGRPPPS (PDB id 1RIJ),101 the 28-residue cdp-1 peptide of sequence KPYTARIKGRTFSNEKELRDFLETFTGR (PDB id 1PSV),102 and the 28-residue fsd-1 peptide of sequence QQYTAKIKGRTFRNEKELRDFIEKFKGR (PDB id 1FSD).103 The structure of trp-cage (see Fig. 6) is characterized by a tryptophan sidechain enclosed in a cage formed by a α-helix on one side and a proline-rich loop on the other. The cdp-1 and fsd-1 mini-proteins (Fig. 6) adopt a mixed αβ conformation and are particularly rich in charged residues. The trp-cage mini-protein was chosen because it has been the target of several computational studies.104–107 The cdp-1 and fsd-1 peptides were of interest because they showed in preliminary tests with AGBNP1 solvation significant tendency to deviate from the experimental structures.
Figure 6.

Graphical representations of the NMR structures of the three miniproteins investigated in this work: trp-cage (pdb id: 1RIJ), cdp-1 (pdb id: 1PSV), and fsd-1 (pdb id: 1FSD). In each case the first deposited NMR model is shown. Backbone ribbon is colored from N-terminal (red) to the C-terminal (blue). Charged sidechains are shown in space-filling representation.
Molecular dynamics simulations were conducted for 12ns starting with the first NMR model deposited in the PDB. The temperature was set to 300 K with the Nosé-Hover thermostat,108,109 a MD time-step of 2 fs was employed and covalent bond lengths involving hydrogen atoms were fixed at their equilibrium positions. Backbone motion was restricted by imposing an positional harmonic restraint potential with a force constant of 0.3 kcal/mol/Å2 on the positions of the Cα atoms, which allows for a range of motion of about 5 Å at the simulation temperature. These restraints are sufficiently weak to allow substantial backbone and sidechain motion while preserving overall topology.
Molecular dynamics simulations were conducted with the OPLS-AA potential97,110 with explicit solvation (SPC water model with 2, 450, 3, 110, and 3, 250 water molecules for trp-cage, cdp-1, and fsd-1 respectively) and with both AGBNP1 and AGBNP2 implicit solvation. The DESMOND program111 was used for the explicit solvent simulations and the IMPACT program15 for those with implicit solvation. Identical force field settings were employed in these two programs. The explicit solvent simulations were conducted in the NPT ensemble using the Martyna-Tobias-Klein barostat112 at 1 atm pressure and employed the smooth Particle Mesh Ewald (PME) method113 for the treatment of long range electrostatic interactions with a real space cutoff of 9 Å. Equilibrium averages and energy distributions were obtained by analysis of the latter 10ns of saved trajectories. Convergence was tested by comparing averages obtained using the first and second halves of simulation data. Hydrogen bonds were detected using a minimum hydrogen-acceptor distance of 2.5 Å and a minimum donor angle of 120 degrees.
3 Results
3.1 Accuracy of Born Radii and Surface Areas
The quality of any implicit solvent model depends primarily on the reliability of the physical model on which it is based. The accuracy of the implementation, however, is also a critical aspect for the success of the model in practice. This is true in particular for models, such as AGBNP, based on the Generalized Born formula. It has been pointed out, for instance, that approximations in the integration procedure to obtain the Born radii may actually be of more significance than the physical approximations on which the GB model is based.114 It is therefore important to test that the conformational dependent quantities employed by AGBNP2 are a good approximation to the geometrical parameters that they are supposed to represent. The present AGBNP2 formulation relies mainly on three types of conformational dependent quantities: Born radii [Eq. (18)], solvent accessible surface areas [Eq. (10)], and solvent accessibilities of hydration sites [Eq. (38)]. In this section we analyze the validity of the AGBNP2 analytical estimates for the Born radii and surface areas against accurate numerical results for the same quantities.
We employ the GEPOL program90 to compute numerically atomic solvent accessible surface areas with a solvent probe diameter of 1 Å, the same probe diameter that defines the solute-solvent boundary in the AGBNP model. Fig. 7A shows the comparison between the surface area estimates given by the present formulation of AGBNP and the numerical surface areas produced by GEPOL for a series of native and modeled protein conformations. In Fig. 7B we show the same comparison for the surface areas of the original AGBNP1 model. These representative results show that the present analytical surface area implementation, which as described above employs a weaker switching function for the overlap volumes, produces significantly more accurate atomic surface areas than the original model. These improvements in the computation of the surface areas, introduced mainly to obtain more accurate Born radii through Eq. (31), are also expected to yield more reliable cavity hydration free energies differences.
Figure 7.

Comparisons between numerical and analytical molecular surface areas of the heavy atoms of the crystal structures (1ctf and 1lz1, respectively) of the C-terminal domain of the ribosomal protein L7/L12 (74 aa) and human lysozyme (130 aa), and of four conformations each of the trp-cage, cdp-1, and fsd-1 miniproteins extracted from the corresponding explicit solvent MD trajectories of the same protein conformations as in Fig. 8. (A) Analytical molecular surface areas computed using the present model, and (B), for comparison, analytical surface areas computed using the original model from reference 42.
Fig. 8 illustrates on the same set of protein conformations the accuracy of the inverse Born radii, , obtained using the AGBNP2 pairwise descreening method using the SEV model for the solute volume described above [Eq. (18)], by comparing them to accurate estimates obtained by evaluating the integral in Eq. (17) numerically on a grid. The comparison is performed for the inverse Born radii because these, being proportional to GB self-energies, are a more reliable accuracy indicators than the Born radii themselves. The grid for the numerical integration was prepared as previously reported,42 except that the solvent excluding volume (SEV) of the solute was employed here rather than the van der Waals volume. The integration grid over the SEV was obtained by taking advantage of the particular way that the GEPOL algorithm describes the SEV of the solute; GEPOL iteratively places auxiliary spheres of various dimensions in the interstitial spaces between solute atoms in such a way that the van der Waals volume of the solute plus the auxiliary spheres accurately reproduces the SEV of the solute. Therefore in the present application a grid point was considered part of the SEV of the solute if it was contained within any solute atom or any one of the auxiliary spheres placed by GEPOL. The default 1.4 Å solvent probe radius was chosen for the numerical computation of the SEV with the GEPOL program to assess the accuracy of the model with respect to a full representation of the solute solvent excluding volume as in the GBMV series of models.93,94 The results of this validation (Fig. 8) show that the analytical SEV pairwise descreening model described above is able to yield Born radii which are not as affected by the spurious high dielectric interstitial spaces present in the van der Waals volume description of the solute. With the van der Waals volume model (Fig. 8B) the Born radii of the majority of solute atoms start to significantly deviate from the reference values for Born radii larger than about 2.5 Å (B−1 = 0.4 Å−1). Born radii computed with the analytical SEV model instead (Fig. 8A) track the reference values reasonably well further up to about 4 Å (B−1 = 0.25 Å−1). Despite this significant improvement most Born radii are still underestimated by the improved model (and, consequently, the inverse Born radii are overestimated - see Fig. 8), particularly those of non-polar atoms near the hydrophobic core of the larger proteins. These regions tend to be loosely packed and tend to contain interstitial spaces too large to be correctly handled by the present model. Because it mainly involves groups of low polarity this limitation has a small effect on the GB solvation energies. It has however a more significant effect on the van der Waals solute–solvent interaction energy estimates through Eq. (23), which systematically overestimate the magnitude of the interaction for atoms buried in hydrophobic protein core. While the present model in general ameliorates in all respects the original AGBNP model, we are currently exploring ways to address this residual source of inaccuracy.
Figure 8.

Comparisons between numerical and analytical inverse Born radii for the heavy atoms of the same protein conformations as in Fig. 7 (A) Analytical Born radii computed using the present SEV model. (B) Analytical Born radii computed using the van der Waals volume model (reference 42).
3.2 Small Molecule Hydration Free Energies
The validation and parametrization of the hydrogen bonding and cavity correction parameters have been performed based on the agreement between experimental and predicted AGBNP2 hydration free energies of a selected set of small molecules, listed in Table 2, containing the main functional groups present in proteins. This set of molecules includes only small and relatively rigid molecules whose hydration free energies can be reliably estimated using a single low energy representative conformation115 as was done here. Table 2 lists for each molecule the experimental hydration free energy, the AGBNP2 hydration free energy computed without HB corrections and the default γ = 117 cal/mol/Å2 surface tension parameter, denoted by AGBNP2/SEV, as well as the hydration free energy from the AGBNP2 model including the HB correction term and the parameters listed in Table 1. For comparison, the corresponding predictions with the original AGBNP142 model are reported in the Supplementary Material.
Table 2.
Experimental and predicted hydration free energies of a set of small molecules.
| Molecule | Exper.a,b | AGBNP/SEVa,c | AGBNP2a,d |
|---|---|---|---|
| n-ethane | 1.83 | 0.98 | 1.80 |
| n-propane | 1.96 | 0.92 | 1.97 |
| n-butane | 2.08 | 0.88 | 2.14 |
| n-pentane | 2.33 | 0.78 | 2.26 |
| n-hexane | 2.50 | 0.70 | 2.40 |
| cyclo-pentane | 1.20 | 0.34 | 1.63 |
| cyclo-hexane | 1.23 | 0.05 | 1.50 |
| benzene | −0.87 | −1.50 | −1.14 |
| toluene | −0.89 | −1.66 | −0.94 |
| acetone | −3.85 | −1.09 | −3.83 |
| acetophenone | −4.58 | −2.74 | −5.07 |
| ethanol | −5.01 | −4.77 | −5.30 |
| phenol | −6.62 | −4.51 | −5.38 |
| ethandiol | −9.60 | −7.99 | −9.87 |
| acetic acid | −6.70 | −2.73 | −7.05 |
| propionic acid | −6.48 | −2.58 | −6.38 |
| methyl acetate | −3.32 | −0.10 | −3.92 |
| ethyl acetate | −3.10 | −0.02 | −3.60 |
| methyl amine | −4.56 | −2.39 | −4.37 |
| ethyl amine | −4.50 | −2.24 | −3.95 |
| dimethyl amine | −4.29 | −1.95 | −3.21 |
| trimethyl amine | −3.24 | −1.78 | −2.39 |
| acetamide | −9.71 | −6.81 | −10.45 |
| N-methylacetamide | −10.08 | −4.75 | −7.51 |
| pyridine | −4.70 | −3.62 | −5.30 |
| 2-methylpyridine | −4.63 | −2.94 | −4.22 |
| 3-methylpyridine | −4.77 | −2.82 | −4.13 |
| methanethiol | −1.24 | −0.61 | −1.46 |
| ethanethiol | −1.30 | −0.57 | −1.22 |
| Neutral AUEa,e | 1.90 | 0.45 | |
| acetate ion | −79.90 | −77.32 | −87.70 |
| propionate ion | −79.10 | −76.29 | −86.29 |
| methylammonium ion | −71.30 | −73.21 | −73.54 |
| ethylammonium ion | −68.40 | −70.63 | −70.75 |
| methylguanidinium | −62.02f | −57.30 | −69.81 |
| Ions AUEa,g | 2.85 | 5.47 | |
In kcal/mol.
Experimental hydration free energy from reference 116 except where indicated.
AGBNP predicted hydration free energies with the default γ parameter for all atoms types (γ = 117 cal/mol/Å2) and without HB corrections.
AGBNP predicted hydration free energies with optimized parameters listed in Table 1.
Average unsigned error of the AGBNP predictions for the neutral compounds relative to the experiments.
From reference 117.
Average unsigned error of the AGBNP predictions for the ionic compounds relative to the experiments.
Going down the results in Table 2 we notice a number of issues addressed by the new implementation. With the new surface area implementation and without corrections (third column in Table 2) the hydration free energies of the normal alkanes are too small compared to experiments, furthermore, in contrast with the experimental behavior, the predicted hydration free energies incorrectly become more favorable with increasing chain length. A similar behavior is observed for the aromatic hydrocarbons. Clearly this is due to the rate of increase of the positive cavity term with increasing alkane size which is insufficient to offset the solute–solvent van der Waals interaction energy term, which becomes more negative with increasing solute size. We have chosen to address this shortcoming by increasing by 10.2% and 2.5% respectively the effective surface tensions for aliphatic and aromatic carbon atoms rather than decreasing the corresponding α parameters of the van der Waals term since the latter had been previously validated against explicit solvent simulations. We have chosen to limit the increases of the surface tension parameters to aliphatic and aromatic carbon atoms since the results for polar functional groups did not indicate that this change was necessary for the remaining atom types. With this new parametrization we achieve (compare the second and fourth column in Table 2) excellent agreement between the experimental and predicted hydration free energies of the alkanes and aromatic compounds. Note that the AGBNP2 model, regardless of the parametrization, correctly predicts the more favorable hydration free energies of the cyclic alkanes relative to their linear analogous. AGBNP2, thanks to its unique decomposition of the non-polar solvation free energy into unfavorable cavity term and an opposing favorable term, is, to our knowledge, the only analytic implicit solvent implementation capable of describing correctly this feature of the thermodynamics of hydration of hydrophobic solutes.
The AGBNP2 model without corrections markedly underpredicts the magnitudes of the experimental hydration free energies of the compounds containing carbonyl groups (ketones, organic acids, and esters). The hydration free energies of alcohols are also underpredicted but by smaller amounts. Much better agreement with the experimental hydration free energies of these oxygen-containing compounds (see Table 2) is achieved after applying hydrogen bonding corrections with h = −1.25 kcal/mol for the carbonyl oxygen, and h = −0.4 kcal/mol for both the hydrogen and oxygen atoms of the hydroxy group (Table 1). Note that the same parameters employed individually for carbonyl and hydroxy groups in ketones and alcohols are applied to the more complex carboxylic groups of acids and esters as well as amides and carboxylate ions. The thiol group of organic sulfides required similar corrections as the hydroxy group (Tables 1 and 2). The AGBNP2 model without corrections also markedly underpredicted the magnitude of the experimental hydration free energies of amines and amides, and, to a smaller extent, of compounds with nitrogen-containing heterocyclic aromatic rings. The addition of HB corrections of −0.25 kcal/mol for amine hydrogens and h = −2.0 kcal/mol for both amine and aromatic nitrogen atoms yields improvement agreement (Table 2), although the effect of N-methylation is still over-emphasized.
3.3 Mini-protein results
As described in Section 2.3 we have performed restricted MD simulations of a series of so-called mini-proteins (trp-cage, cdp-1, and fsd-1) to study the extent of the agreement between the conformational ensembles generated with the original AGBNP implementation (AGBNP1) and the present implementation (AGBNP2) with respect to explicit solvent-generated ensembles. The results of earlier studies4,54,55 suggest that the AGBNP/OPLS-AA model correctly reproduces for the most part the backbone secondary structure features of protein and peptides. The tests in the present study are therefore focused on sidechain conformations. The backbone atoms were harmonically restricted to remain within approximately 3 Å C-α RMSD of the corresponding NMR experimental models. We structurally analyzed the ensembles in terms of the extent of occurrence of intramolecular interactions.
As shown in Table 3 we measured a significantly higher average number of intramolecular hydrogen bonds and ion pairing in the AGBNP1 ensembles relative to the explicit solvent ensembles for all mini-proteins studied. The largest deviations are observed for cdp-1 and fsd-1, two mini-proteins particularly rich in charged sidechains, with on average nearly twice as many intramolecular hydrogen bonds compared to explicit solvent. Many of the excess intramolecular hydrogen bonds with AGBNP1 involve interactions between polar groups (polar sidechains or the peptide backbone) and the sidechains of charged residues. For example for cdp-1 we observe (see Table 3) approximately 8 hydrogen bonds between polar and charged groups on average compared to nearly none with explicit solvation.
Table 3.
Average number of some types of intramolecular electrostatic interactions in the explicit solvent conformational ensembles, and the ensembles generated from simulations using the AGBNP1 and AGBNP2 effective potentials for the trp-cage, cdp-1, and fsd-1 mini-proteins.
| Mini-protein | Explicit | AGBNP1 | AGBNP2 |
|---|---|---|---|
| Intramolecular Hydrogen Bonds | |||
| trp-cage | 13.5 | 18.3 | 15.3 |
| cdp-1 | 12.6 | 24.5 | 15.4 |
| fsd-1 | 14.1 | 24.6 | 14.3 |
| all | 40.2 | 67.4 | 45.0 |
| Polar–Polar Hydrogen Bonds | |||
| trp-cage | 12.9 | 17.1 | 13.9 |
| cdp-1 | 12.5 | 16.4 | 14.1 |
| fsd-1 | 12.0 | 15.0 | 12.9 |
| all | 37.4 | 48.5 | 40.9 |
| Polar–Charged Hydrogen Bonds | |||
| trp-cage | 0.6 | 1.2 | 1.4 |
| cdp-1 | 0.1 | 8.1 | 1.3 |
| fsd-1 | 2.1 | 9.6 | 1.4 |
| all | 2.8 | 18.9 | 4.1 |
| Ion Pairs | |||
| trp-cage | 0.3 | 1.0 | 1.0 |
| cdp-1 | 2.5 | 2.9 | 2.7 |
| fsd-1 | 1.4 | 4.6 | 4.0 |
| all | 4.2 | 8.5 | 7.7 |
Despite the introduction of empirical surface tension correction to penalize ion pairs,55 AGBNP1 over-predicts ion-pair formation. We found that ion pairing involving arginine were particularly over-stabilized by AGBNP1 as we observed stable ion pairing between arginine and either glutamate or aspartate residues during almost the entire duration of the simulation in virtually all cases in which this was topologically feasible given the imposed backbone restrains. In contrast, with explicit solvation some of the same ion pairs were seen to form and break numerous times indicating a balanced equilibrium between contact and solvent-separated conformations. This balance is not reproduced with implicit solvation which instead strongly favors ion pairing. In any case, the relative stability of ion pairs appeared to depend in subtle ways on the protein environment as for example the two ion pairs between arginine and glutamate of cdp-1 were found to be stable with either explicit solvation or AGBNP1 implicit solvation whereas other Arg-Glu ion pairs in trp-cage and fsd-1 were found to be stable only with implicit solvation.
This analysis generally confirms quantitatively a series of past observations made in our laboratory indicating that the original AGBNP implementation tends to be biased towards conformations with excessive intramolecular electrostatic interactions, at the expense of more hydrated conformations in which polar groups are oriented so as to interact with the water solvent. During the process of development of the modifications to address these problems, we found it useful to rescore with varying AGBNP formulations and parametrizations the mini-protein conformational ensembles obtained with AGBNP1 and explicit solvation, rather than performing simulations with each new parametrizations. An example of this analysis is shown in the first row of plots of Fig. 9, which compare the probability distributions of the AGBNP1 effective potential energies over the conformational ensembles generated with AGBNP1 implicit solvation and with explicit solvation. These results clearly show that the AGBNP1/OPLS-AA effective potential disfavor conformations from the explicit solvent ensemble relative to those generated with implicit solvation. The AGBNP1 energy scores of the explicit solvent ensembles of all mini-proteins are shifted towards higher energies than those of the AGBNP1 ensemble, indicating that conformations present in the explicit solvent ensemble would be rarely visited when performing conformational sampling with the AGBNP1/OPLS-AA potential. AGBNP1/OPLS-AA assigns a substantial energetic penalty (see Fig. 9A–C) to the explicit solvent ensemble relative to the AGBNP1 ensemble (on average 3.3, 4.4, and 5.7 kcal/mol per residue for, respectively, the trp-cage, cdp-1, and fsd-1 mini-proteins). This energetic penalty, being significantly larger than thermal energy, rules out the possibility that conformational entropy effects could offset it to such an extent so as to equalize the relative free energies of the two ensembles. Detailed analysis of the energy scores shows that, as expected, the AGBNP1 implicit solvent ensemble is mainly favored by more favorable electrostatic Coulomb interaction energies due to its greater number of intramolecular electrostatic contacts relative to the explicit solvent ensemble (see above). Conversely, the AGBNP1 solvation model does not assign sufficiently favorable hydration free energy to the more solvent-exposed conformations obtained in explicit solvent so as to make them competitive with the more compact conformations of the AGBNP1 ensemble.
Figure 9.

Potential energy distributions of the conformational ensembles for the the trp-cage (first column, panels A, D), cdp-1 (second column, panels B, E), and fsd-1 (third column, panels C, F) mini-proteins obtained using the the AGBNP1/OPLS-AA (first row, panels A, B, and C; full line) and AGBNP2/OPLS-AA (second row, panels D, E, F; full line) effective potentials and explicit solvation (dashed line). The distributions are shown as a function of the energy gap per residue (Δu) relative to the mean effective potential energy of the implicit solvent ensemble distribution.
Similar energetic scoring analysis with the AGBNP2 model, see Fig. 1 of the Supplementary Material, with and without hydrogen bonding to solvent corrections showed that the introduction of the SEV model for the solute volume significantly reduced the energetic gap between the explicit solvent and AGBNP1 conformational ensembles, and that the introduction of the hydrogen bonding corrections further favor the explicit solvent ensemble. We proceeded to vary the AGBNP2 parameters to achieve the best possible scoring of the explicit solvent ensembles relative to the AGBNP1 ensembles while maintaining an acceptable level of agreement with the small molecule experimental hydration free energies. This procedure eventually yielded the parameters listed in Table 1, which produces small molecule hydration free energies in good agreement with the experiments (Table 2), as well as energy distributions for the three mini-proteins that, while still favoring the AGBNP1 ensembles, displayed energy gaps between the explicit and AGBNP1 implicit solvation ensembles comparable to thermal energy and smaller than the spread of the energy distributions.
The energy scoring experiments on the explicit solvent and AGBNP1 ensembles described above were very useful for tuning the formulation of the AGBNP2 model without requiring running lengthy MD simulations. They do not, however, guarantee that the conformational ensembles generated with the AGBNP2 solvation model will more closely match the explicit solvent ensembles than those generated with AGBNP1. This is because the new solvation model could introduce new energy minima not encountered with AGBNP1 or explicit solvation that would be visited only by performing conformational sampling with AGBNP2. To validate the model in this respect we obtained MD trajectories with the AGBNP2 implicit solvent model and compared the corresponding probability distributions of the effective energy with those of the explicit solvent ensembles similarly as above. The results for the three mini-proteins, shown in Fig. 9 panels D–F, indicate that the AGBNP2-generated ensembles display significantly smaller bias (mean energy gaps per residue of 2.0, 2.1, and 2.5 kcal/mol for, respectively, the trp-cage, cdp-1, and fsd-1 mini-proteins) than AGBNP1 (Fig. 9, panels A–C) which yielded energy gaps of 3.3, 4.4, and 5.7 kcal/mol per residue respectively. This observation shows that AGBNP2 produces conformational ensembles with energy distributions that more closely match on average that of the explicit solvent ensemble without producing unphysical minima that deviate significantly from it.
We have analyzed structural features of the conformational ensembles obtained with the AGBNP1 and AGBNP2 models to establish the degree of improvement achieved with the new model with respect to intramolecular interactions. The salient results of this analysis are shown in Table 3. This table reports for each mini-protein the average number of intramolecular hydrogen bonds and ion pairs. The number of hydrogen bonds is further decomposed into those involving only polar groups (including the backbone) and those involving a polar groups and the sidechain of a charged residue (arginine, lysine, aspartate, and glutamate). As noted above it is apparent from this data that the AGBNP1 model produces conformations with too many hydrogen bonds and ion pairs. The majority of the excess hydrogen bonds with AGBNP1 involve residue sidechains. Similarly, too many ion pairs are observed in the AGBNP1 ensemble particularly for the fsd-1 mini-protein (4.6 ion pairs on average with AGBNP1 compared to only 1.4 in explicit solvent). The AGBNP2 ensembles, in comparison, yield considerably fewer intramolecular hydrogen bonds. For instance, the average number of hydrogen bonds for cdp-1 is reduced from 24.5 with AGBNP1 to 15.4 with AGBNP2, which is to be compared with 12.6 in explicit solvent. With AGBNP2 the number of polar–polar hydrogen bonds is generally in good agreement with explicit solvation. However the greatest improvement is observed with polar–charged interactions. For example the number of polar–charged hydrogen bonds of fsd-1 is reduced by almost ten fold in going from AGBNP1 to AGBNP2 to reach good agreement with explicit solvation. Importantly, a significant fraction of the excess polar–charged interactions observed with AGBNP1 corrected by AGBNP2 are interactions between the peptide backbone and charged sidechains that would otherwise interfere with the formation of secondary structures.
With AGBNP2 we observe small but promising improvements in terms of ion pair formation. The average number of ion pairs of cdp-1 consistently agrees between all three solvation models, and the only possible ion pair in trp-cage is more stable in both implicit solvent formulations than in explicit solvent (it occurs in virtually all implicit solvent conformations compared to only 30% of the conformations in explicit solvent). However the average number of ion pairs for fsd-1 is reduced from 4.6 with AGBNP1 to 4.0 with AGBNP2. We observe good agreement between the number of ion pairs involving lysine with either AGBNP1 or AGBNP2 and explicit solvation. However ion pairs involving arginine are generally more stable with implicit solvation than with explicit solvation. The agreement in the number of ion pairs with cdp-1 is due to the fact that for this mini-protein the two possible ion pairs involving arginine result stable with explicit solvation as well as with implicit solvation. For the other two mini-proteins however, ion pairs involving arginine that are marginally stable with explicit solvation are found to be significantly more stable with implicit solvation, although less so with AGBNP2 solvation.
4 Discussion
Modern implicit solvent models for biomolecular simulations are generally based on the uniform dielectric continuum representation of the solvent which is accurately modeled by the Poisson-Boltzmann (PB) equation.9 Generalized Born (GB) models,10 which approximate the PB formalism, are applicable to molecular dynamics thanks to their low computational complexity. GB models have reached a high level of accuracy compared to PB following the introduction of more realistic solute volume descriptions87,100 and of higher order corrections to the Coulomb field approximation.118–120 However at the molecular level water is sometimes poorly described by uniform continuum models. Even the best GB models have been found to deviate considerably from, for example, explicit solvent benchmarks.121,127 The non-linear and asymmetric dielectric response of water stems primarily by the finite extent and internal structure of water molecules.1 The modeling of effects due to water granularity is important for the proper description of molecular association equilibria. Integral equation methods122 provide an accurate implicit solvation description from first principles, however, despite recent progress,123 they are not yet applicable to molecular dynamics of biomolecules. The primary aim of the present study has been to formulate an analytical and computational efficient implicit solvent model incorporating solvation effects beyond those inherent in standard continuum dielectric models and, by so doing, achieving an improved description of solute conformational equilibria.
In this work we have developed the AGBNP2 implicit solvent model which is based on an empirical (but physically-motivated) first solvation shell correction function parametrized against experimental hydration free energies of small molecules and the results of explicit solvent molecular dynamics simulations of a series of mini-proteins. The correction function favors conformations of the solute in which polar groups are oriented so as to form hydrogen bonds with the surrounding water solvent thereby achieving a more balanced equilibrium with respect to the competing intramolecular hydrogen bond interactions. A key ingredient of the model is an analytical prescription to identify and measure the volume of hydration sites on the solute surface. Hydration sites that are deemed too small to contain a water molecule do not contribute to the solute hydration free energy. Conversely, hydration sites of sufficient size form favorable interactions with nearby polar groups. This model thus incorporates the effects of both water granularity and of non-linear first shell hydration effects.
The GB and non-polar models in the AGBNP2 implicit solvent model provide the linear continuum dielectric model basis of the model as well as a description of non-electrostatic hydration effects.42 In this work the GB and solute–solvent dispersion interaction energy models are further enhanced by replacing the original van der Waals solute volume model with a more realistic solvent excluding volume (SEV) model. The new volume description improves the quality of the Born radii of buried atoms and atoms participating in intramolecular interactions which would otherwise be underestimated due to high dielectric interstitial spaces present with the van der Waals volume description.88 GB models with these characteristics have been previously proposed. The GBMV series of models87,93,94 represent the SEV on a grid which, although accurate, is computationally costly and lacks frame of reference invariance. The pairwise descreening-based GBOBC model120 introduced an empirical rectifying function to increase the Born radii of buried atoms in an averaged, geometry-independent manner. The GBn model100 introduced the neck region between pairs of atoms as additional source of descreening, dampened by empirical parameters to account in an average way for overlaps between neck regions and between solute atoms and neck regions. The approach proposed here to represent the SEV consists of computing the atomic self-volumes, used in the pairwise descreening computation, using enlarged atomic radii so as to cover the interatomic interstitial spaces. The self-volume of each atom is then reduced proportionally to its solvent accessible surface area [see Eq. (31)] to subtract the volume in van der Waals contact with the solvent. We show (Fig. 8) that this model reproduces well Born radii computed from an accurate numerical representation of the SEV, noting that improvements for the Born radii of atoms in loosely packed hydrophobic interior, while significant, are still not optimal. Although approximate, this representation of the SEV maintains the simplicity and computational efficiency of pairwise descreening schemes, while accounting for atomic overlaps in a consistent and parameter-free manner.
The new AGBNP2 model has been formulated to be employed in molecular dynamics conformational sampling applications, which require potential models of low computational complexity and favorable scaling characteristics, and with analytical gradients. These requirements have posed stringent constraints on the design of the model and the choice of the implementation algorithms. In the formulation of AGBNP2 we have reused as much as possible well-established and efficient algorithms developed earlier for the AGBNP1 model. For example the key ingredient of the hydrogen bonding correction function is the free volume of an hydration site, which is computed using a methodology developed for AGBNP1 to compute atomic self-volumes. Similarly the SEV-based pairwise descreening procedure employs atomic surface areas (see Eq. 31), computed as previously described.42 AGBNP2 suffers additional computational cost associated with the SEV-based pairwise descreening procedure and the hydrogen bonding correction function. This handicap however is offset by having only one solute volume model in AGBNP2 rather than two in AGBNP1. AGBNP1 requires two separate invocations of the volume overlaps machinery [Eq. 2] for each of the two volume models it employs, corresponding to the van der Waals atomic radii for the pairwise descreening calculation and enlarged radii for the surface area calculation.42 AGBNP2 instead employs a single volume model for both the pairwise descreening and surface area calculations. A direct CPU timing comparison between the two models can not be reported at this time because the preliminary implementation of the AGBNP2 computer code used in this work lacks key data caching optimizations similar to those already employed in AGBNP1. Given the computational advantages of the new model discussed above we expect to eventually obtain similar or better performance than with AGBNP1.
The AGBNP2 model has been parametrized against experimental hydration free energies of a series of small molecules and with respect to the ability of reproducing energetic and structural signatures of the conformational ensemble of three mini-proteins generated with explicit solvation. These data sources are chosen so as to ensure that the resulting model would be applicable to both hydration free energy estimation and conformation equilibria, which are fundamental characteristics for models aimed at protein-ligand binding affinity estimation. On the other hand, experimental hydration free energies and explicit solvent conformational ensembles are to some extent incompatible with one another given the limitations of even the best fixed charge force fields and explicit solvation models to reproduce experimental hydration free energies of small molecules with high accuracy.41,124,125 Mindful of this issue we did not seek a perfect correspondence with the experimental hydration free energy values. We first obtained parameters by fitting against the small molecule experimental hydration free energies, then adjusted the parameters to improve the agreement with the explicit solvent data making sure that the predicted small molecule hydration free energies remained within a reasonable range relative to the experimental values. In practice this procedure yielded predicted hydration free energies in good agreement with the experimental values with the exception of the carboxylate and guanidinium ions (see Table 2) for which AGBNP2 predicts more favorable hydration free energies than the experiments; a consequence of the large hydrogen bonding corrections necessary to reduce the occurrence of intramolecular electrostatic interactions in the investigated proteins. As discussed further below, limitations in the description of hydration sites adopted for carboxylate and guanidinium ions may be partly the cause of the observed inconsistencies for these functional groups.
The parametrization and quantitative validation of the model, which is the primary focus of this work, has been based on comparing the effective potential energy distributions of implicit solvent conformational ensembles with those of explicit solvent ensembles. We observed that the AGBNP1 solvation model energetically ranked explicit solvent conformations significantly less favorably than implicit solvent conformations. The AGBNP2 model is characterized by smaller energetic bias relative to the explicit solvent ensembles, indicating that conformational sampling with the AGBNP2/OPLS-AA energy function produces conformations that more closely match those obtained with explicit solvation. This result is a direct consequence of employing the more realistic solvent excluding volume description of the solute which yields larger Born radii for buried groups, as well as the hydrogen bonding to solvent corrections, which favor solvent exposed conformations of polar groups. Furthermore, comparison of the energy distributions of the AGBNP2 and explicit solvent ensembles for the three mini-proteins (Fig. 9D–F) shows, in contrast to the AGBNP1 results, that the AGBNP2 bias for the two more charge-rich mini-proteins (cdp-1 and fsd-1) is similar to that of the least charged one (trp-cage). This suggests that the residual energetic bias of the AGBNP2 model is probably related to the non-polar model rather than the electrostatic model. Future studies will address this particular aspect of the model.
The energy scoring studies conducted in this work indicate that AGBNP2 is a significant improvement over AGBNP1. They also show, however, that the new model falls short of consistently scoring explicit solvent conformations similarly to implicit solvent conformations. Although an optimal match between energy distributions is a necessary condition for complete agreement between implicit and explicit solvation results, it is unrealistic to expect to reach this ultimate goal at the present level of model simplification. Increasing the magnitude of the hydrogen bonding corrections can improve the agreement between the explicit and implicit solvation energy distributions, albeit at the expense of the quality of the predicted small molecule hydration free energies. It seems likely that the no parametrization of the current model would yield both good relative conformational free energies and hydration free energies. Future work will pursue the modeling of additional physical and geometrical features, such us the use of variable dielectric approaches to model polarization effects,126 necessary to improve the agreement between implicit and explicit solvation energy distributions. The energy gap between the implicit solvent and explicit solvent energy distributions used here is, we believe, a meaningful measure of model quality and could serve as a useful general validation tool to compare the accuracy of implicit solvent models.
The excessive number of intramolecular electrostatic interactions involving charged groups has been one of the most noticeable shortcoming of GB-based implicit solvent models.127 To correct this tendency we have in the past adopted in the AGBNP1 model ad-hoc strategies aimed at either destabilizing electrostatic intramolecular interactions54 or, alternatively, stabilizing the competing solvent-separated conformations.55 This work follows the latter approach using a more robust and physically-motivated framework based on locating and scoring hydration sites on the solute surface as well as adopting a more realistic volume model. Structural characterization of the conformational ensembles has shown that AGBNP2 produces significantly fewer intramolecular interactions than AGBNP1 reaching good agreement with explicit solvent results. The reduction of intramolecular interactions has been the greatest for interactions between polar and charged groups. We believe that the excessive tendency toward the formation of intramolecular interactions to be the root cause of the high melting temperatures of structured peptides64 predicted with AGBNP1. Given the reduction of intramolecular interactions achieved with AGBNP2, we expect the new model to yield more reasonable peptide melting temperatures; a result which we hope to report in future publications.
Less visible improvements have been obtained for ion pairs involving arginine sidechains which remain more stable with implicit solvation than explicit solvation. However, significantly, with AGBNP2 we observed a more dynamical equilibrium between ion-paired and solvent-separated conformations of arginine sidechains that was not observed with AGBNP1. This result is promising because it indicates that the AGBNP2 solvation model, although still favoring ion paired conformations, produces a more balanced equilibrium, which is instead almost completely shifted towards contact conformations with AGBNP1. Nevertheless it is apparent that the AGBNP treatment of the guanidinium group of arginine is not as good as for other groups. This limitation appears to be shared with other functional groups containing sp2-hybridized nitrogen atoms as evidenced for example by the relatively lower quality of the hydration free energy predictions for amides and nitrogen-containing aromatic compounds (Table 2). Similar implicit solvent overstabilization solvation of arginine-containing ion pairs has been observed by Yu et al.85 in their comparison of Surface Generalized Born (SGB) and SPC explicit solvation with the OPLS-AA force field. Despite quantitative differences, the explicit solvent studies (with the TIP3P water model) of Masunov and Lazaridis128 and Hassan,129 using the CHARMM force field, and that of Mandell at al.,130 using the OPLS-AA force field, have confirmed that arginine forms the strongest ion pairing interactions, especially in the bidentate coplanar conformation. These observations are consistent with the present explicit solvent results using OPLS-AA and the SPC water model, where we find that most of the ion pairs of the mini-proteins were found to involve arginine sidechains. In contrast to our present implicit solvent results, however, the work of Masunov and Lazaridis128 indicated that the GB-based implicit solvent model that they analyzed14 produced potentials of mean force for arginine-containing ion pairs in general agreement with explicit solvation.
To rationalize the present implicit solvent results concerning ion pair formation, it has been instructive to analyze the potentials of mean force (PMF) of ion pair association with the AGBNP model. As an example, Fig. 10 shows the PMF for the association of propyl-guanidinium (arginine sidechain analog) and ethyl-acetate (aspartate and glutamate analog) in a bidentate coplanar conformation (similar to the arrangement used previously85,128–130) for various AGBNP implementations. The corresponding explicit solvent PMF obtained by Mandell et al.130 is also shown in Fig. 10 for comparison. The original AGBNP1 parametrization42 clearly leads to an overly stable salt bridge with the contact conformation scored at approximately −19 kcal/mol relative to the separated conformation, compared with −8.5 kcal/mol with explicit solvation. The AGBNP1 parametrization analyzed here, which includes an empirical surface area correction to reduce the occurrence of ion pairs,55 yields a contact free energy (−11 kcal/mol) in much better agreement with explicit solvation, although the shape of the PMF is not properly reproduced. The present AGBNP2 model without hydrogen bonding corrections (labeled “AGBNP2-SEV” in Fig. 10) yields a PMF intermediate between the original and corrected AGBNP1 parametrization. The AGBNP2 model with hydrogen bonding corrections yields the PMF with the closest similarity with the one obtained in explicit solvent. Not only the contact free energy (−6.5 kcal/mol) is in good agreement with the explicit solvent result, but, importantly, it also reproduces the solvation barrier of the PMF at 5 Å separation, corresponding to the distance below which there is insufficient space for a water layer between the ions.
Figure 10.

Potential of mean force of ion pair formation between propyl-guanidinium and ethyl-acetate in the coplanar orientation with AGBNP implicit solvation (A) and explicit solvation (B; reference 130). In (A) “AGBNP1 (orig.)” refers to the original AGBNP1 parametrization,42 “AGBNP1” refers to the AGBNP1 model used in this work which includes a surface tension parameter correction for the carboxylate group aimed at reducing the occurrence of ion pairs,55 “AGBNP2” refers to the current model and “AGBNP2-SEV” the current model without hydrogen bonding and surface tension corrections. The potentials of mean force are plotted with respect to the distance between the atoms of the protein sidechain analogs equivalent to the Cζ of arginine and the Cγ of aspartate.
It is in this range of distances that the greatest discrepancies are observed between PMF’s with explicit solvation and some GB-based implicit solvation models85,128 that do not model effects due to the finite size of water molecules. Both the hydrogen bonding correction and the SEV volume description employed in AGBNP2, which are designed to take into account the granularity of the water solvent – the hydrogen bonding correction through the minimum free volume of water sites [Eq. (37)] and the SEV model through the water radius offset [Eq. (28)] – make it possible to reproduce the solvation barrier typical of molecular association processes in water.
It is notable in the PMF results shown in Fig. 10 the lack of a free energy maximum with the AGBNP2/SEV model (AGBNP2 without HB corrections), which would be expected on the basis of results with the GBMV model, indicating that a SEV treatment of the GB model leads to a higher and much broader PMF maximum relative to explicit solvent.88 There are two possible factors contributing to this discrepancy. The first is that the OPLS-AA force field used in this work seems to consistently produce stronger ionic interactions than the CHARMM force field (on which the GBMV model is based) as suggested by the relatively small free energies of salt bridge formation obtained with CHARMM-based implicit solvent models88,128 relative to OPLS-AA based ones (see for example reference 85 and the present results with AGBNP1). Because the shape of the PMF at intermediate separations is determined by a delicate balance between attractive electrostatic interactions and repulsive desolvation forces, stronger electrostatic interactions with OPLS-AA are potentially responsible in part for a missing or smaller PMF maximum. The lack of the PMF maximum with AGBNP2/SEV is most likely also due to the reduced radius offset used in AGBNP42 used to construct the SEV. The small probe radius leads to a smaller reduction, as compared to a full SEV treatment, of the high dielectric volume surrounding the ionic groups as they approach each other. The consequence is a smaller rate of increase of the desolvation penalty which in turn leads to a smaller or absent PMF maximum.
Increasing the magnitude of the AGBNP radius offset is not feasible as we observed that the Gaussian overlap approximation for the overlap volumes [Eq. 10 of reference 42] breaks down for atomic radii much larger than the van der Waals radii. On the other hand, as the results in Fig. 10 show, the added desolvation provided by the short-range HB function is able to properly correct this deficiency yielding a PMF maximum in good correspondence with explicit solvation. This shows that the HB function as parametrized is likely taking into account not only short-range non-linear hydration effects but also inaccuracies in the GB and non-polar models, as well as approximations in the implementation such as the small probe radius discussed above.
The good correspondence between the AGBNP2 and explicit solvent PMF’s for propyl-guanidinium and ethyl-acetate (Fig. 10) stands in contrast with the residual AGBNP2 over-prediction of arginine salt bridges compared to explicit solvation (Table 3). We observed that in the majority of arginine salt bridges occurring with AGBNP2, the guanidinium and carboxylate groups interact at an angle rather than in the coplanar configuration discussed above. We have confirmed that the PMF of ion pair formation for an angled conformation (not shown) indeed shows a significantly more attractive contact free energy than the coplanar one. This result indicates that the in-plane placement of the hydration sites for the carboxylate groups (see Appendix) does not sufficiently penalize angled ion pair arrangements. This observation is consistent with the need of introducing of an isotropic surface-area based hydration correction for carboxylate groups (the reduced γ parameter for the carboxylate oxygen atoms in Table 1), which showed some advantage in terms of reducing the occurrence of salt bridges. Future work will focus on developing a more general hydration shell description for carbonyl groups and related planar polar groups to address this issue.
5 Conclusions
We have presented the AGBNP2 implicit solvent model, an evolution of the AGBNP1 model we have previously reported, with the aim of incorporating hydration effects beyond the continuum dielectric representation. To this end a new hydration free energy component based on a procedure to locate and score hydration sites on the solute surface is used to model first solvation shell effects, such as hydrogen bonding, which are poorly described by continuum dielectric models. This new component is added to the Generalized Born and non-polar AGBNP models which have been improved with respect to the description of the solute volume description. We have introduced an analytical Solvent Excluding Volume (SEV) model which reduces the effect of artefactual high-dielectric interstitial spaces present in conventional van der Waals representations of the solute volume. The new model is parametrized and tested with respect to experimental hydration free energies and the results of explicit solvent simulations. The modeling of the granularity of water is one of the main principles employed in the design of the empirical first shell solvation function and the SEV model, by requiring that hydration sites have a minimum available volume based on the size of a water molecule. We show that the new volumetric model produces Born radii and surface areas in good agreement with accurate numerical evaluations. The results of molecular dynamics simulations of a series of mini-proteins show that the new model produces conformational ensembles in much better agreement with reference explicit solvent ensembles than the AGBNP1 model both with respect to structural and energetics measures.
Future development work will focus on improving the modeling of some functional groups, particularly ionic groups involving sp2 nitrogen, which we think are at the basis of the residual excess occurrence of salt bridges, and on the optimization of the AGBNP2 computer code implementation. Future work will also focus on further validation of the model on wide variety of benchmarks including protein homology modeling and peptide folding.
Supplementary Material
Acknowledgments
This work was supported in part by a National Institute of Health grant no. GM30580. The calculations reported in this work have been performed at the BioMaPS High Performance Computing Center at Rutgers University funded in part by the NIH shared instrumentation grant no. 1 S10 RR022375.
A Hydration Site Locations
Fig. 11 shows the location of the hydration sites for the functional groups listed in Table 1. Each hydration site is represented by a sphere of 1.4 Å radius. The distance dHB between the donor or acceptor heavy atom and the center of the hydration site sphere is set to 2.5 Å.
Figure 11.

Diagram illustrating the hydration site locations for each of the functional group geometries used in this work. Linear: hydrogen bond donor; trigonal(1) and trigonal(2): trigonal planar geometries with, respectively, one and two covalent bonds on the acceptor atom; tetrahedral(2) and tetrahedral(3): tetrahedral geometries with, respectively, two and three covalent bonds on the acceptor atom. Representative molecular structures are shown for each geometry.
There is a single linear geometry for HB donor groups. The corresponding hydration site is placed at a distance dHB from the heavy atom donor along the heavy atom-hydrogen bond.
Acceptor trigonal geometries have one or two hydration sites depending on whether the acceptor atom is bonded to, respectively, two or one other atom. In the former case the water site is placed along the direction given by the sum of the unit vectors corresponding to the sum of the NR1 and NR2 bonds (following the atom labels in Fig. 11). In the latter case the W1 site (see Fig. 11) is placed in the R1CO plane forming an angle of 120° with the CO bond. The W2 site is placed similarly.
Acceptor tetrahedral geometries have one or two hydration sites depending on whether the acceptor atom is bonded, respectively, to three or two other atoms. In the former case the water site is placed along the direction given by the sum of the unit vectors corresponding to the sum NR1, NR2, and NR3 bonds. In the latter case the positions of the W1 and W2 sites are given by
where O is the position of the acceptor atoms, θ = 104.4°, and u1 and u2 are, respectively, the unit vectors corresponding to the OR1 and OR2 bonds.
B Gradients of GB and van der Waals Energies
The component of the gradient of the AGBNP2 van der Waals energy at constant self-volumes is the same as in the AGBNP1 model [see Appendix C of reference 42]. In AGBNP2 the expression for the component of the gradient corresponding to variations in the atomic scaling factors, sij, includes pair corrections at all overlap levels because of the presence of multi-body volumes in . In addition, a new component corresponding to the change in surface areas appears:
| (39) |
| (40) |
| (41) |
Eq. (39) leads to the same expression of the derivative component as in the AGBNP1 model [Eq. (72) in reference 42] (except for the extra elements in the 2-body terms due to the inclusion of the 1/2Vkj correction term). Eq. (40) corresponds to the component of the derivative due to variations in , the volume to be added to the self-volumes of j and k to obtain the sjk and skj scaling factors. In the AGBNP1 model this component included only 2-body overlap volumes; in AGBNP2 this term instead includes all overlap volumes greater than zero. Finally, Eq. (41), where Ak is the surface area of atom k, leads to the component of the derivatives of the GB and vdW terms due to variations of the exposed surface area. The latter two terms are new for AGBNP2.
B.1 Component of derivative from Eq. (40)
From Eq. (63) in reference 42 and Eq. (40) we have
| (42) |
where Wkj has the same expression as in Eq. (69) in reference 42. In working with Eq. (42) it is important to note that, whereas is symmetric with respect to swapping the j and k indexes, Wkj and Wjk are different from each other. Substituting Eq. (30) into Eq. (42) and expanding over symmetric terms we obtain
| (43) |
Eq. (43) is simplified by noting that
| (44) |
Eq. (44) is inserted in Eq. (43) and sums are reduced accordingly, then symmetric terms are collected into single sums by re-indexing the summations, obtaining
| (45) |
The corresponding expression for the gradient of ΔGGB is similar but employs the Uij factors of Eq. (78) of reference 42 rather than Wij.
B.2 Component of derivative from Eq. (41)
Inserting Eq. (41) in Eq. (63) of reference 42 gives
which is the same expression as that for the gradient of ΔGcav (see Appendix A of reference 42) with the replacement
The corresponding expression for the gradient of ΔGGB follows from the substitution:
B.3 Derivatives of HB Correction Energy
From Eq. (37) we have
| (46) |
Inserting Eqs. (35) and (36) in Eq. (46) gives
| (47) |
where
| (48) |
where the first term on the r.h.s. represents the derivative of the overlap volume with respect to the position of atom i keeping the position of the water site s fixed, and the second term reflects the change of overlap volume due to a variation of the position of the water site caused by a shift in position of atom i. The latter term is non-zero only if i is one of the parent atoms of the water site.
References
- 1.Levy RM, Gallicchio E. Annu Rev Phys Chem. 1998;49:531–67. doi: 10.1146/annurev.physchem.49.1.531. [DOI] [PubMed] [Google Scholar]
- 2.Feig M, Brooks C. Curr Op Struct Biol. 2004;14:217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Roux B, Simonson T. Biophysical Chemistry. 1999;78:1–20. doi: 10.1016/s0301-4622(98)00226-9. [DOI] [PubMed] [Google Scholar]
- 4.Felts AK, Andrec M, Gallicchio E, Levy R. Water and Biomolecules - Physical Chemistry of Life Phenomena. Springer Science; 2008. Protein Folding and Binding: Effective Potentials, Replica Exchange Simulations, and Network Models. [Google Scholar]
- 5.Gallicchio E, Zhang LY, Levy RM. J Comp Chem. 2002;23:517–529. doi: 10.1002/jcc.10045. [DOI] [PubMed] [Google Scholar]
- 6.Onufriev A. Annual Reports in Computational Chemistry. 2008;4:125–137. [Google Scholar]
- 7.Chen J, Brooks C, Khandogin J. Curr Opin Struct Biol. 2008;18:140–148. doi: 10.1016/j.sbi.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomasi J, Persico M. Chem Rev. 1994;94:2027–2094. [Google Scholar]
- 9.Baker N. Curr Opin Struct Biol. 2005;15:137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Still WC, Tempczyk A, Hawley RC, Hendrikson T. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 11.Zhang L, Gallicchio E, Friesner RA, Levy RM. J Comp Chem. 2001;22:591–607. [Google Scholar]
- 12.Schaefer M, Froemmel C. J Mol Biol. 1990;216:1045–1066. doi: 10.1016/S0022-2836(99)80019-9. [DOI] [PubMed] [Google Scholar]
- 13.Hawkins GD, Cramer CJ, Truhlar DG. J Phys Chem. 1996;100:19824–19839. [Google Scholar]
- 14.Dominy BN, Brooks CLI. J Phys Chem B. 1999;103:3765–3773. [Google Scholar]
- 15.Banks J, et al. J Comp Chem. 2005;26:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Case D, Cheatham T, Darden T, Gohlke H, Luo R, Merz K, Onufriev A, Simmerling C, Wang B, RJW J Comp Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-naim A. Hydrophobic Interactions. Plenum Press; New York: 1980. [Google Scholar]
- 18.Kauzmann W. Adv in Prot Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 19.Dill KA. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 20.Privalov PL, Makhatadze GI. J Mol Biol. 1993;232:660–679. doi: 10.1006/jmbi.1993.1417. [DOI] [PubMed] [Google Scholar]
- 21.Honig B, Yang AS. Ad Prot Chem. 1995;46:27–58. doi: 10.1016/s0065-3233(08)60331-9. [DOI] [PubMed] [Google Scholar]
- 22.Sturtevant JM. Proc Natl Acad Sci USA. 1977;74:2236–2240. doi: 10.1073/pnas.74.6.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams DH, Searle MS, Mackay JP, Gerhard U, Maplestone RA. Proc Natl Acad Sci USA. 1993;90:1172–78. doi: 10.1073/pnas.90.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Froloff N, Windemuth A, Honig B. Protein Science. 1997;6:1293–1301. doi: 10.1002/pro.5560060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siebert X, Hummer G. Biochemistry. 2002;41:2965–2961. doi: 10.1021/bi0158526. [DOI] [PubMed] [Google Scholar]
- 26.Ooi T, Oobatake M, Nemethy G, Sheraga H. Proc Natl Acad Sci USA. 1987;84:3086–3090. doi: 10.1073/pnas.84.10.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee MR, Duan Y, Kollman PA. Proteins. 2000;39:309–316. [PubMed] [Google Scholar]
- 28.Hünenberger PH, Helms V, Narayana N, Taylor SS, McCammon JA. Biochemistry. 1999;38:2358–2366. doi: 10.1021/bi982064g. [DOI] [PubMed] [Google Scholar]
- 29.Simonson T, Brünger AT. J Phys Chem. 1994;98:4683–4694. [Google Scholar]
- 30.Sitkoff D, Sharp KA, Honig B. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 31.Rapp CS, Friesner RA. Proteins: Struct, Funct, Genet. 1999;35:173–183. [PubMed] [Google Scholar]
- 32.Fogolari F, Esposito G, Viglino P, Molinari H. J Comp Chem. 2001;22:1830–1842. doi: 10.1002/jcc.1134. [DOI] [PubMed] [Google Scholar]
- 33.Pellegrini E, Field MJ. J Phys Chem A. 2002;106:1316–1326. [Google Scholar]
- 34.Curutchet C, Cramer CJ, Truhlar DG, Ruiz-Lòpez MF, Rinaldi D, Orozco M, Luque FJ. J Comp Chem. 2003;24:284–297. doi: 10.1002/jcc.10143. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen W, Ulmschneider J, Tirado-Rives J. J Phys Chem B. 2004;108:16264–16270. [Google Scholar]
- 36.Wallqvist A, Covell DG. J Phys Chem. 1995;99:13118–13125. [Google Scholar]
- 37.Gallicchio E, Kubo MM, Levy RM. J Phys Chem B. 2000;104:6271–6285. [Google Scholar]
- 38.Levy RM, Zhang LY, Gallicchio E, Felts AK. J Am Chem Soc. 2003;25:9523–9530. doi: 10.1021/ja029833a. [DOI] [PubMed] [Google Scholar]
- 39.Wagoner J, Baker N. Proc Natl Acad Sci. 2006;103:8331–8336. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J, Brooks C. Phys Chem Chem Phys. 2008;10:471–481. doi: 10.1039/b714141f. [DOI] [PubMed] [Google Scholar]
- 41.Mobley D, Bayly C, Cooper M, Shirts M, Dill K. J Chem Theory Comput. 2009 doi: 10.1021/ct800409d. Web Article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallicchio E, Levy R. J Comp Chem. 2004;25:479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- 43.Wallqvist A, Gallicchio E, Levy RM. J Phys Chem B. 2001;105:6745–6753. [Google Scholar]
- 44.Huang DM, Chandler D. J Phys Chem B. 2002;106:2047–2053. [Google Scholar]
- 45.Zhou R, Huang X, Margulis C, Berne B. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 46.Pierotti RA. Chemical Reviews. 1976;76:717–26. [Google Scholar]
- 47.Hummer G, Garde S, García AE, Paulaitis ME, Pratt LR. Phys Chem B. 1998;102:10469–82. [Google Scholar]
- 48.Lum K, Chandler D, Weeks JD. J Phys Chem B. 1999;103:4570–4577. [Google Scholar]
- 49.Pitarch J, Moliner V, Pascual-Ahuir JL, Silla A, Tuñón I. J Phys Chem. 1996;100:9955–9959. [Google Scholar]
- 50.Ashbaugh HS, Kaler EW, Paulaitis ME. J Am Chem Soc. 1999;121:9243–9244. [Google Scholar]
- 51.Pitera JW, van Gunsteren WF. J Am Chem Soc. 2001;123:3163–3164. doi: 10.1021/ja0057474. [DOI] [PubMed] [Google Scholar]
- 52.Zacharias M. J Phys Chem A. 2003;107:3000–3004. [Google Scholar]
- 53.Su Y, Gallicchio E. Biophys Chem. 2004;109:251–260. doi: 10.1016/j.bpc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 54.Felts AK, Harano Y, Gallicchio E, Levy RM. Proteins: Structure, Function, and Bioinformatics. 2004;56:310–321. doi: 10.1002/prot.20104. [DOI] [PubMed] [Google Scholar]
- 55.Felts A, Gallicchio E, Chekmarev D, Paris K, Friesner R, Levy R. J Chem Theor Comput. 2008;4:855–858. doi: 10.1021/ct800051k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong F, Wagoner J, Baker N. Phys Chem Chem Phys. 2008;10:4889–4902. doi: 10.1039/b807384h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaefer M, Karplus M. J Phys Chem. 1996;100:1578–1599. [Google Scholar]
- 58.Qiu D, Shenkin PS, Hollinger FP, Still CW. J Phys Chem A. 1997;101:3005–3014. [Google Scholar]
- 59.Tsui V, Case DA. J Am Chem Soc. 2000;122:2489–2498. [Google Scholar]
- 60.Schaefer M, Bartels C, Leclerc F, Karplus M. J Comp Chem. 2001;22:1857–1879. doi: 10.1002/jcc.1137. [DOI] [PubMed] [Google Scholar]
- 61.Chekmarev D, Ishida T, Levy R. J Phys Chem B. 2004;108:19487–19495. [Google Scholar]
- 62.Andrec M, Felts AK, Gallicchio E, Levy RM. Proc Natl Acad Sci USA. 2005;102:6801–6806. doi: 10.1073/pnas.0408970102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gallicchio E, Andrec M, Felts AK, Levy RM. J Phys Chem B. 2005;109:6722–6731. doi: 10.1021/jp045294f. [DOI] [PubMed] [Google Scholar]
- 64.Weinstock D, Narayanan C, Felts AK, Andrec M, Levy R, Wu K, Baum J. J Am Chem Soc. 2007;129:4858–4859. doi: 10.1021/ja0677517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinstock D, Narayanan C, Baum J, Levy R. Protein Science. 2008;17:950–954. doi: 10.1110/ps.073365408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ravindranathan K, Gallicchio E, Levy R. J Mol Biol. 2005;353:196–210. doi: 10.1016/j.jmb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 67.Messina T, Talaga D. Biophys J. 2007;93:579–585. doi: 10.1529/biophysj.107.103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ravindranathan K, Gallicchio E, Friesner RA, McDermott AE, Levy RM. J Am Chem Soc. 2006;128:5786–5791. doi: 10.1021/ja058465i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ravindranathan P, Gallicchio E, McDermott A, Levy R. J Am Chem Soc. 2007;129:474–475. doi: 10.1021/ja0672371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su Y, Gallicchio E, Das K, Arnold E, Levy R. J Chem Theory Comput. 2007;3:256–277. doi: 10.1021/ct600258e. [DOI] [PubMed] [Google Scholar]
- 71.Lapelosa M, Gallicchio E, Ferstandig Arnold G, EA, Levy R. J Mol Biol. 2009;385:675–691. doi: 10.1016/j.jmb.2008.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tjong H, Zhou H. J Phys Chem B. 2007;111:3055–3061. doi: 10.1021/jp066284c. [DOI] [PubMed] [Google Scholar]
- 73.Tjong H, Zhou H. J Chem Phys. 2007;126:195102. doi: 10.1063/1.2735322. [DOI] [PubMed] [Google Scholar]
- 74.Zhu J, Alexov E, Honig B. J Phys Chem B. 2005;109:3008–3022. doi: 10.1021/jp046307s. [DOI] [PubMed] [Google Scholar]
- 75.Fan H, Mark AE, Zhu J, Honig B. Proc Natl Acad Sci. 2005;102:6760–6764. doi: 10.1073/pnas.0408857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grant JA, Pickup B, Sykes MJ, Kitchen C, Nicholls A. Phys Chem Chem Phys. 2007;9:4913–4922. doi: 10.1039/b707574j. [DOI] [PubMed] [Google Scholar]
- 77.Labute P. J Comp Chem. 2008;29:1693–1698. doi: 10.1002/jcc.20933. [DOI] [PubMed] [Google Scholar]
- 78.Levy R, Belhadj M, Kitchen D. J Chem Phys. 1991;95:3627–3633. [Google Scholar]
- 79.Alper H, Levy RM. J Phys Chem. 1990;94:8401–8403. [Google Scholar]
- 80.Morozov A, Kortemme T. Adv Prot Chem. 2005;72:1–38. doi: 10.1016/S0065-3233(05)72001-5. [DOI] [PubMed] [Google Scholar]
- 81.Lazaridis T, Karplus M. Curr Opin Struct Biol. 2000;10:139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 82.Eisenberg D, McLachlan AD. Nature. 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 83.Lazaridis T, Karplus M. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 84.Vitalis A, Pappu R. J Comp Chem. 2009;30:673–699. doi: 10.1002/jcc.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu Z, Jacobson M, Josovitz J, Rapp C, Friesner R. J Phys Chem B. 2004;108:6643–6654. [Google Scholar]
- 86.Okur A, Wickstrom L, Simmerling C. J Chem Theory Comput. 2008;4:488–498. doi: 10.1021/ct7002308. [DOI] [PubMed] [Google Scholar]
- 87.Lee MS, Salsbury FR, Brooks CL., III J Chem Phys. 2002;116:10606. [Google Scholar]
- 88.Swanson JMJ, Mongan J, McCammon JA. J Phys Chem B. 2005;109:14769–14772. doi: 10.1021/jp052883s. [DOI] [PubMed] [Google Scholar]
- 89.Lee B, Richards F. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 90.Pascual-Ahuir JL, Silla E. J Comp Chem. 1990;11:1047–1060. [Google Scholar]
- 91.Cortis CM, Friesner RA. J Comp Chem. 1997;18:1591–1608. [Google Scholar]
- 92.Rocchia W, Sridharan S, Nicholls A, Alexov E, Chiabrera A, Honig B. J Comp Chem. 2002;23:128–137. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- 93.Lee MS, Feig M, Salsbury FR, Jr, Brooks CL., III J Comp Chem. 2003;24:1348–1356. doi: 10.1002/jcc.10272. [DOI] [PubMed] [Google Scholar]
- 94.Chocholousova J, Feig M. J Comp Chem. 2006;27:719–729. doi: 10.1002/jcc.20387. [DOI] [PubMed] [Google Scholar]
- 95.Grant JA, Pickup BT. J Phys Chem. 1995;99:3503–3510. [Google Scholar]
- 96.Kratky KW. J Statist Phys. 1981;25:619–634. [Google Scholar]
- 97.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 98.Jorgensen WL, Madura JD. Mol Phys. 1985;56:1381–1392. [Google Scholar]
- 99.Onufriev A, Bashford D, Case DA. J Phys Chem B. 2000;104:3712–3720. [Google Scholar]
- 100.Mongan J, Simmerling C, McCammon J, Case D, Onufriev A. J Chem Theory Comput. 2007;3:156–169. doi: 10.1021/ct600085e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu Y, Liu Z, Androphy E, Chen J, Baleja J. Biochemistry. 2004;43:7421–7431. doi: 10.1021/bi049552a. [DOI] [PubMed] [Google Scholar]
- 102.Dahiyat B, Sarisky C, Mayo S. J Mol Biol. 1997;273:789–796. doi: 10.1006/jmbi.1997.1341. [DOI] [PubMed] [Google Scholar]
- 103.Dahiyat B, Mayo S. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 104.Snow C, Zagrovic B, Pande V. J Am Chem Soc. 2002;124:14548–14549. doi: 10.1021/ja028604l. [DOI] [PubMed] [Google Scholar]
- 105.Pitera J, Swope W. Proc Natl Acad Sci USA. 2003;100:7587–7592. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou R. Proc Natl Acad Sci USA. 2003;100:13280–13285. doi: 10.1073/pnas.2233312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Paschek D, Hempel S, García A. Proc Natl Acad Sci USA. 2008;105:17754–17759. doi: 10.1073/pnas.0804775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nosè S. J Chem Phys. 1984;81:511–519. [Google Scholar]
- 109.Hoover W. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 110.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 111.Bowers K, Chow E, Xu H, Dror R, Eastwood M, Gregersen B, Klepeis J, Kolossváry I, Moraes M, Sacerdoti F, Salmon J, Shan Y, Shaw D. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); IEEE; Tampa, Florida. 2006. [Google Scholar]
- 112.Martyna G, Tobias D, Klein M. J Comp Phys. 1994;101:4177–4189. [Google Scholar]
- 113.Essman U, Perera L, MLB, Darden T, Lee H, Pedersen L. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 114.Onufriev A, Case DA, Bashford D. J Comp Chem. 2002;23:1297–1304. doi: 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]
- 115.Mobley D, Chodera J, Dill K. J Phys Chem B. 2008;112:938–946. doi: 10.1021/jp0764384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cabani S, Gianni P, Mollica V, Lepori L. J Solut Chem. 1981;10:563–595. [Google Scholar]
- 117.Vorobyov I, Li L, Allen T. J Phys Chem B. 2008;112:9588–9602. doi: 10.1021/jp711492h. [DOI] [PubMed] [Google Scholar]
- 118.Ghosh A, Rapp CS, Friesner RA. J Phys Chem B. 1998;102:10983–10990. [Google Scholar]
- 119.Im W, Lee M, Brooks C. J Comp Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 120.Onufriev A, Bashford D, Case D. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 121.Roe DR, Okur A, Wickstrom L, Hornak V, Simmerling C. J Phys Chem B. 2007;111:1846–1857. doi: 10.1021/jp066831u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yoshida N, Imai T, Phongphanphanee S, Kovalenko A, Hirata F. J Phys Chem B. 2009;113:873–886. doi: 10.1021/jp807068k. [DOI] [PubMed] [Google Scholar]
- 123.Miyata T, Hirata F. J Comp Chem. 2008;29:871–882. doi: 10.1002/jcc.20844. [DOI] [PubMed] [Google Scholar]
- 124.Deng Y, Roux B. J Phys Chem B. 2004;108:16567–16576. [Google Scholar]
- 125.Shirts MR, Pande VS. J Chem Phys. 2005;122:134508. doi: 10.1063/1.1877132. [DOI] [PubMed] [Google Scholar]
- 126.Zhu K, Shirts M, Friesner R. J Chem Theor Comput. 2007;3:2108–2119. doi: 10.1021/ct700166f. [DOI] [PubMed] [Google Scholar]
- 127.Zhou R, Berne B. Proc Natl Acad Sci. 2002;99:12777–12782. doi: 10.1073/pnas.142430099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Masunov A, Lazaridis T. J Am Chem Soc. 2003;125:1722–1730. doi: 10.1021/ja025521w. [DOI] [PubMed] [Google Scholar]
- 129.Hassan S. J Phys Chem B. 2004;108:19501–19509. [Google Scholar]
- 130.Mandell DJ, Chorny I, Groban E, Wong S, Levine E, Rapp C, Jacobson M. J Am Chem Soc. 2007;129:820–827. doi: 10.1021/ja063019w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.