

Abstract
Like many other complex human disorders of unknown aetiology, autoimmune-mediated type 1 diabetes may ultimately be controlled via a therapeutic approach that combines multiple agents, each with differing modes of action. The numerous advantages of such a strategy include the ability to minimize toxicities and realize synergies to enhance and prolong efficacy. The recognition that combinations might offer far-reaching benefits, at a time when few single agents have yet proved themselves in well-powered trials, represents a significant challenge to our ability to conceive and implement rational treatment designs. As a first step in this process, the Immune Tolerance Network, in collaboration with the Juvenile Diabetes Research Foundation, convened a Type 1 Diabetes Combination Therapy Assessment Group, the recommendations of which are discussed in this Perspective paper.
Keywords: clinical trials, combination therapies, interventions, type 1 diabetes
Introduction
Type 1 diabetes (T1D), one of the most common autoimmune diseases, results from the progressive destruction of insulin-producing pancreatic β cells by CD4+ and CD8+ T cells.
Typically, it is thought that as few as 15–20% of β cells remain at the time of the first clinical symptoms of T1D [1,2]. Left unchecked, this residual islet cell function/mass is generally short-lived due to continued immune-mediated β cell death [3]. However, the preservation of even this reduced β cell mass has clear therapeutic benefits by enabling tighter control of blood glucose, reducing exogenous insulin requirements and thus reducing the risk of diabetes-related complications [4–6]. As was apparent in a recent study of a monoclonal anti-CD3 antibody [6], individuals with higher pretreatment levels of stimulated C-peptide (i.e. greater remaining endogenous insulin production) benefit most from intervention at this stage. Thus, clinical trials conducted in patients recruited shortly after diagnosis and with significant residual β cell function (often termed ‘tertiary prevention’ or ‘intervention trials’) have become a critical starting-point for assessing immunological therapies. This approach forms part of a wider strategy that would subsequently see efficacious agents investigated for prophylaxis in high-risk individuals. Trials in new-onset patients have several advantages over prevention trials – potential risks are justified more easily when disease is present and studies can be completed in a shorter, 12–24-month time-period using a well-defined end-point, such as maintenance of stimulated C-peptide secretion. As a consequence, there are savings of both cost and time compared to true T1D prevention trials, which may take 5–10 years to complete and require the screening of large numbers of subjects to identify those at the highest risk.
During the past 20 years, several immune interventions for new-onset T1D have been tested clinically. Early attempts involving broadly immunosuppressive agents with proven track records in solid organ transplantation, such as cyclosporin A, azathioprine and prednisolone, failed to produce lasting remission and beneficial effects were limited only to the duration of treatment [4,7–9]. While highlighting the role of immune-mediated islet injury, these studies also demonstrated the inherent tendency of the autoimmune effector response in humans to recur, an issue that is also evident in islet graft failures 4–5 years post-transplantation. However, because of multiple long-term side effects, including secondary cancers and infections [10], continuous immunosuppression is not a viable option for the management of T1D. Therefore, it is critical that immunomodulatory therapies induce tolerance to β cell antigens while minimizing detrimental effects on host defence. Few treatments, such as monoclonal anti-CD3 antibodies [6,11] and anti-CD20 antibodies [12], in addition to islet antigen-specific therapies, have demonstrated this property to date and these will be central to novel combination therapies discussed herein.
Indeed, as of today, two decades of aggressive basic and preclinical research have led to the identification of a large number of rationally designed and much-improved agents that have fewer systemic side effects and target a variety of mechanisms involved in the development of autoimmune disease and the loss of tolerance in T1D. A key feature of several of these agents is the potential to induce tolerogenic effects that outlast generalized suppression of the immune system and are therefore of particular interest for future interventions in T1D. Fc receptor non-binding anti-CD3 monoclonal antibodies (mAbs) show much promise in preliminary trials, as a short course of treatment can delay the post-diagnosis decline in stimulated C-peptide for up to 5 years, with depletion of T cells evident for a limited period of time (< 1 months) [13]. These agents demonstrate clearly that modulation of β cell autoimmunity in humans can be achieved without the need for continuous immunosuppression. A recent trial using anti-CD20 (Rituxan) to target B lymphocytes in patients with recent-onset T1D [12] found that the window between generalized immunosuppression and tolerance towards β cells appears to be smaller than that of anti-CD3. This trial was nevertheless noteworthy because of the well-documented safety profile of B lymphocyte depletion. It is also known that B lymphocyte infiltration is a significant late-stage event in T1D [14]. Thus, as no single agent demonstrates the ability to induce durable disease remission, anti-CD20 therapy could serve as a rapid, anti-inflammatory component of a rational combinational intervention [14,15].
Indeed, a further lesson from the past 20 years is that the immunological defects responsible for T1D are multiple and complex, and are not likely to be addressed with a single agent. It is more probable that multiple pathways will need to be modulated in order to achieve a lasting remission. For example, down-regulation of the inflammatory response, elimination of autoreactive effector and memory T cells, and the induction and long-term maintenance of T and B regulatory cell populations may all be required in varied degrees to induce robust disease remission. Furthermore, given the level of β cell destruction observed at the onset of overt disease, the ideal intervention would be one that not only halts the autoimmune response, but also enhances β cell function or stimulates regeneration.
Drugs that have shown promise either in preclinical or early clinical trials fall into a few general classes: T cell modulators [anti-CD3, anti-thymocyte globulin (ATG)], B cell-depleting agents (anti-CD20), anti-inflammatory molecules [anti-interleukin (IL)-1, anti-tumour necrosis factor (TNF)-α], antigen-specific therapies [insulin, glutamic acid decarboxylase-65 (GAD65), islet autoantigenic peptides [16]] and incretin mimetics (insulinotropic agents, such as exenatide) (see Fig. 1 and also an earlier comprehensive review by Staeva-Vieira [17]). Based on the preclinical and clinical outcomes of studies using these therapies, it has become abundantly clear that the future of interventional therapies for T1D lies in the use of combinations of agents that target multiple biological pathways and thus would synergize to achieve a lasting remission.
Fig. 1.
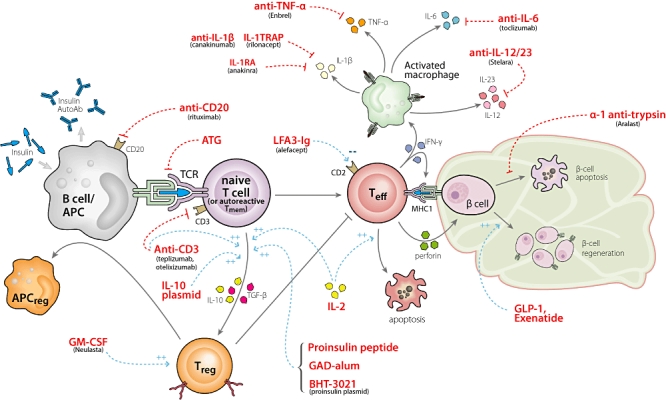
Pathways and opportunities to intervene in type 1 diabetes. This figure shows crucial pathways known to contribute to the pathogenesis of type 1 diabetes and relevant drugs for intervention. A key juncture is the antigen-presenting cell–T cell interaction, where activation of effector T cells can be prevented and generation of regulatory T cells (Tregs) can be enhanced. Another pivotal cell is the β cell itself. Augmentation of β cell mass or function along with prevention of apoptosis may be achievable goals. Most probably a combination of agents dampening inflammation, preventing effector cell activation, enhancing Tregs and augmenting β cell mass will be the ultimate solution for curing type 1 diabetes.
In addition to improved efficacy, specific combinations of agents could be designed to reduce side effects of treatment. The use of agents with different, yet complementary, mechanisms could facilitate dose reductions of drugs known to have toxicities at their conventionally prescribed doses. This could offer considerable advantages in T1D, where the risk : benefit ratio of a new therapy must always be compared with that of daily insulin injections.
Thus, in autumn 2009, the Immune Tolerance Network (ITN), in concert with the Juvenile Diabetes Research Foundation (JDRF), convened a Type 1 Diabetes Combination Therapies Assessment Group to identify and discuss the various challenges and key opportunities for combination therapies in T1D, and develop a framework of potential initiatives that will accelerate their clinical development. A key goal of the discussions was to establish a ranked list of promising combination therapies that will be priority targets for development through these initiatives.
Past and current combination studies
To date, there has been little clinical experience evaluating combinations of immunomodulatory agents for T1D; two published trials yielded disappointing results. A study of exenatide and daclizumab (anti-CD25 MAb; Zenapax, Hoffman-La Roche Ltd, Basel, Switzerland) was designed to examine whether stimulating insulin secretion during blockade of IL-2 signalling in effector T cells would affect endogenous insulin production in patients with long-standing T1D (21·3 ± 10·7 years). It is possible that the study aim was overly ambitious, because neither agent has shown efficacy in this setting. Perhaps not surprisingly, the results showed that the combination of intensified insulin therapy, exenatide and daclizumab did not induce improved function of any remaining β cells [18]. Another combination evaluated by Type 1 Diabetes TrialNet examined two doses of daclizumab combined with daily mycophenolate mofetil (CellCept, Roche) in new-onset patients. This combination failed to show any benefit in terms of maintenance of stimulated C-peptide and was halted due to futility [19]. At present, the Immune Tolerance Network is also piloting a combination therapy targeting the IL-2 axis (IL-2 and Rapamycin; Proleukin and Rapamune/Sirolimus from Prometheus Laboratories Inc., San Diego, CA, USA, and Pfizer, New York, NY, USA, respectively) on the basis of a preclinical report of prevention of spontaneous T1D onset in non-obese diabetic (NOD) mice [20]. The mechanism of action of this combination is believed to involve a shift from T helper type 1 (Th1)- to Th2- and Th3-type cytokine-producing cells due to the selective deletion of autoreactive Th1 cells. The pilot study is testing the safety and efficacy of this combination in individuals diagnosed within the previous 3–48 months. All participants will be treated with Proleukin (administered subcutaneously) for 28 days and Rapamune (taken orally) for 12 weeks. Finally, a study of GAD65 (Diamyd) [21] and sitagliptin (DPP-4 inhibitor; also an incretin mimetic) has been initiated by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), although it is temporarily on hold at the time of writing for logistical reasons.
Challenges in selecting combination therapies
The ideal combination therapy would utilize two or more agents whose mechanisms of action are complementary and have already accumulated many patient-years as T1D monotherapies with well-defined safety profiles in humans. Unfortunately, such agents are currently scarce in T1D, as most potential agents have not yet progressed beyond Phase II trials and therefore have limited safety data. Those in late-state development or already approved for other autoimmune or transplant indications might be more appropriate choices. However, even if such data are available for each individual agent and preclinical data indicate the possibility of synergy in recent-onset T1D models, there remains the possibility of deleterious side effects of the combination (especially in cases of two or more immune modulators). This is a key concern, particularly for regulatory agencies, which may require clear evidence of the safety of the proposed combination itself. Fortunately, in T1D there is no shortage of available animal models, including the widely studied non-obese diabetic (NOD) mouse and infection models that can help predict untoward effects of combination therapies on, for example, anti-viral immunity. Regardless, a cautious approach is warranted, first completing preclinical investigations, then establishing safety in small Phase I clinical studies of combination therapies.
The possibility of unforeseen drug interactions appearing in human testing also presents significant challenges for pharmaceutical and biotech companies interested in evaluating combinations that include one or more of their agents. Again, the majority of the therapeutics of interest in T1D are still in the developmental stage for this or other indications. Those in Phases II or III studies, for example, have already required investment of several hundreds of millions of dollars to get there, and their development is associated with a tightly controlled project plan and time-line. Companies are therefore naturally risk-averse, and the prospect of uncovering new side effects associated with their agent, even as part of a combination therapy, could have a serious impact on development costs and time-lines by complicating and delaying regulatory approval of subsequent studies or even progress to market. Thus, in order to engage industry actively in trials of combination therapies for T1D, the means of mitigating such risks are needed – and clearly, industry participation in such trials is very important for ultimate development.
Given such challenges, the most feasible scenario is to identify promising combinations from the limited list of agents that are already approved, whether for T1D or other indications. Rituximab (Rituxan, Genentech, South San Francisco, CA, USA), a B cell-depleting agent approved for rheumatoid arthritis (RA) and lymphoma therapy, abatacept (Orencia, Bristol-Myers Squibb, New York, NY, USA), a co-stimulatory blocker also approved for RA, and anti-thymocyte globulin (thymoglobulin, Genzyme, Cambridge, MA, USA), a cocktail of rabbit-derived antibodies against human T cells currently approved in transplantation, are among the most promising candidates for combination studies. Clearly, this is a limiting factor, as many exciting opportunities (including antigen-specific approaches) for effective combination therapies lie in the many still-investigational agents. Therefore, while the use of approved therapies should take priority for initial combination studies, a means of reconciling industry concerns with the need for access to non-approved agents is certainly required. While there is no way of eliminating all risk to industry, by emphasizing patient safety through intelligent selection of therapeutics and development of clinical protocols that minimize the chance of harmful interactions the risks can, in many cases, be reduced to acceptable levels to encourage industry participation.
As ever, preclinical and human safety studies will raise additional challenges for investigators, not the least of which is the availability of funding. Thus, if promising combinations of agents in T1D are to reach the clinic in a reasonable time-frame, targeted programmes, funding and infrastructure are required to encourage and support the preclinical efforts that are inevitably required. A clear framework must also be developed that specifies the type and quality of preclinical data, including which animal models are acceptable, as well as toxicology and pharmacodynamic data expectations, that will be required for a combination to meet acceptable safety standards to justify human trials.
Challenges in defining trial end-points
Looking forward, the development of any preventive or interventional strategy in T1D, and certainly one involving combination therapies, would also benefit enormously from the identification of biomarkers that could indicate the re-establishment of β cell-specific tolerance (immune modulators and immune suppressants) or the successful induction of a relevant regulatory T cell response (antigen-specific strategies). The current standard end-point for new-onset studies, the stimulated C-peptide response, is a marker of endogenous insulin secretion and a reliable indicator of clinical benefit [22]. However, within the honeymoon phase typical of new-onset diabetes, C-peptide measures have limited value until several months following treatment and it provides no information (other than by inference) on the state of the immune system. What is needed are additional surrogate end-points based upon beneficial immunological responses that manifest soon after treatment, and which would permit rapid assessment and prioritization of the therapeutic potential of novel combinations and, indeed, any individual T1D therapeutic.
In fact, there has never been a more opportune time for research aimed at uncovering biomarkers in T1D: an ever-growing number of clinical studies of new-onset type 1 diabetes should provide unprecedented access to potentially large numbers of clinical specimens. Relevant clinical laboratory assay developments, along with recent developments in high-throughput technologies, now provide the means to assay large numbers of specimens rapidly and affordably. One challenge facing biomarker studies, however, is the lack of defined standards, not only among laboratory protocols for the various assays but also in handling and preparation of clinical specimens, which can have considerable influence on assay results [23]. Another challenge is our lack of knowledge as to how much individual T cell responses fluctuate over time in a given individual – subjects are usually tested only a few times per year, but effector T cell and regulatory T cell (Treg) activities might change multiple times during this period. Indeed, a recent study published by Diabetes TrialNet's Mechanistic Outcomes Committee showed that, while assays measuring overall T cell reactivity against islet autoantigens could distinguish between patients with T1D and healthy controls relatively reliably, those assays that measured individual epitope-specific responses detected variable responses over time [24]. The last challenge is that, as yet, we have no solid data that indicate how T cell responses would be expected to change in a beneficial way in one individual following re-establishment of tolerance to β cells. Animal models tell us what to expect, but do not always correspond to the human case [25]. Thus, precise tracking during clinical interventions is required to develop reliable correlations between T cell responses and clinical outcomes.
The potential benefits of biomarkers of tolerance in T1D are many [26]. They could speed clinical assessments by providing surrogate end-points, permit more robust analysis of trial data through stratification of patients and facilitate personalized medicine by informing treatment decisions. Such benefits argue strongly for the creation of a coordinated biomarker discovery effort that, by establishing common procedures across all new-onset trials, permits comparison of data obtained in trials of varying agents and ultimately the identification of robust immunological markers of disease state and immune tolerance.
The ITN has been working actively to advance such a goal for the past decade by integrating a biomarker discovery programme into each of its clinical trials. As part of this effort, the ITN has adapted and validated a number of cellular and genetic assays for use on clinical specimens, and established and standardized associated handling procedures and laboratory protocols. Similarly, biomarker discovery is integrated into trials conducted by Type 1 Diabetes TrialNet and often accompanied by open Requests for Application (RFA) in the relevant area. Through this process, for example, several biomarker discovery programmes have been commissioned in relation to the Phase II study of GAD65-Alum injection. JDRF has also made a significant investment in T1D biomarker discovery efforts. Clearly, there would be significant benefits to harmonize the efforts of these and other groups into a community-wide biomarker discovery programme that could extend integrated mechanistic investigations to all, even industry-sponsored studies. In the meantime, the ITN, TrialNet and JDRF continue their support for biomarker discovery in T1D and additional National Institutes of Health (NIH)-led initiatives such as the recent RFA for ‘Research on Biosamples From Selected Diabetes Clinical Studies’[27] are encouraging signs that there is a growing recognition of the importance of biomarker research in T1D.
Challenges going forward: defining a roadmap for combination therapies
In light of these discussion points, it can be concluded that there are a number of important opportunities available that will facilitate the clinical translation of combination therapies in T1D. First, there appears to be a strong enthusiasm within the academic community for the development of combination studies and willingness within JDRF, ITN, NIH, and possibly other agencies, to dedicate funding and resources to this effort. Secondly, numerous monotherapy studies in T1D will be completed over the next 1–2 years and will provide safety and efficacy data that will assist the efforts in obtaining regulatory approval and guide the selection of promising combinations. Based on these considerations, the ITN–JDRF Type 1 Diabetes Combination Therapy Assessment Group has developed the recommendations described below.
Regulatory framework for combinations
The US Food and Drug Administration (FDA) has, in general, been open to the application of combination therapies in T1D, recognizing the need for combining agents to achieve synergies while avoiding unwanted side effects from long-term immunosuppression. It is therefore recommended that a formal dialogue be opened with the FDA and interested parties, seeking to establish clearer and more standardized guidelines for the regulatory assessment of combinations of therapeutics for new-onset T1D. Such guidelines would cover the nature of the preclinical data required by the FDA, criteria to decide whether animal data or human Phase I toxicology studies are required for a particular combination or whether individual monotherapy data will suffice, and appropriate patient populations for a given study based on expected adverse effect profiles, as well as currently accepted end-points. Ultimately, a standardized decision tree approach to achieving regulatory approval could be developed. A similar pathway could be envisaged in Europe and Australia, where clinical trial activity in T1D is equally active, although here assessment by the regulatory authorities is typically performed on a case-by-case basis.
Support for preclinical studies and establishment of a network for preclinical intervention studies in recent-onset diabetes
It is recommended that a panel of investigators with a proven track record of using well-characterized animal models of T1D for disease reversal should be assembled with a mandate to develop a consensus on which animal models should be used and how precisely experiments should be carried out to meet FDA requirements for study approvals. Preclinical studies are carried out ideally at more than one site to circumvent local animal colony-related artefacts. In order to assure uniformity when making comparisons between studies, standard operating procedures should be defined and standardized positive controls (e.g. anti-CD3) should be instituted so that data from multiple laboratories could be obtained and be directly comparable. Such a consortium could consist of geographically diverse laboratories employing the same preclinical models in a standardized manner to examine combination therapies that are recommended by the ITN–JDRF Type 1 Diabetes Combination Therapies Assessment Group. This would allow at least three laboratories to test the same combination therapy independently and simultaneously. In general, all tests should be conducted in models of recent-onset diabetes, wherein the blood glucose values and age of each mouse at inception of the intervention have to be tracked as independent variables that are likely to affect the outcome of the treatment.
To this end the ITN, in co-operation with JDRF, has begun developing a consortium of laboratories to carry out preclinical evaluations of combination therapies in type 1 diabetes. The consortium will consist of ∼6 geographically diverse, independent laboratories that will, in parallel, assess toxicology, pharmacodynamics and efficacy of potential combinations. All laboratory protocols will be standardized and all therapeutics would come from a standardized central source, preferably ‘good manufacturing practice’ (GMP)-grade material. The goal of this initiative is to provide an infrastructure that generates high-quality preclinical data rapidly to stimulate clinical assessments of novel combination therapies in T1D.
Coordinated biomarker effort
It is recommended that the above-mentioned preclinical studies also attempt to identify suitable biomarkers. One major gap is that animal studies notoriously track cells in tissues such as the pancreatic draining lymph node, whereas human studies will naturally have to use peripheral blood. As it is known that there can be substantial homing differences between different lymphoid compartments, it would be optimal to generate peripheral blood data during the preclinical animal studies so that precursor numbers and changes in lymphocyte subsets over time can be estimated more accurately. These efforts should then be aligned with current attempts to identify biomarkers within clinical trials in new-onset T1D, for example, at an annual biomarker meeting of participating entities. These should not only include academic laboratories and clinical trial investigators, but should be expanded to include data from additional industry-sponsored studies.
Establish a network for early-stage clinical studies
There are numerous interested and experienced parties that could be assembled into a network of clinical centres to conduct small, short-duration, early-stage, proof-of-concept studies focused predominantly upon mechanistic outcomes, in order to permit a more rapid assessment of the clinical viability of a novel combination. Combinations that show clear evidence of modulation of the immune system would be prioritized for more comprehensive clinical evaluation with C-peptide preservation as the preferred outcome. JDRF, through its Autoimmunity Centers Consortium [28], is currently assessing the feasibility of establishing such a network.
Recommendations for protocol selection and design
Clearly, combinations that will be supported by industry and can navigate the regulatory process successfully will be those for which there is a compelling argument in terms of both efficacy and safety. In addressing the safety of the combinations, several key strategies can be applied to minimize the risk of harmful interactions between agents.
Limit to two agents
First, combinations should be limited to two agents. Both agents may be immunotherapeutics, or one immunotherapeutic and one drug with an alternate mechanism – one that stimulates β cell regeneration, for instance. For reasons stated above, approved agents (or those nearing approval) would have initial priority for development in combination therapies.
Independent/complementary mechanisms
In the case of two immunotherapeutics, combinations should be selected such that individual agents work via mechanisms that are significantly different, so that safety profiles could be considered as, essentially, independent. For instance, combining an antigen-specific therapy and a non-specific therapy would have a reduced theoretical likelihood of resulting in hitherto unrecognized side effects. Antigen-specific therapies in general are regarded as a safer treatment modality, with fewer systemic risks associated with them, and so should be priority considerations for initial combination trials.
Safety in protocol design
Designing safety into clinical protocols is critical and there are a number of steps that can be taken to reduce the risks of harmful drug interactions. For instance, design of a protocol that uses sequential, rather than simultaneous, treatment would be preferred. Similarly, the dose of one or both of the drugs may be reduced in the combination protocol to increase the safety profile. In designing the protocol, implementation of these strategies can be guided by available pharmacodynamic data on each of the agents.
Next steps
With these considerations in mind, the Assessment Group listed and prioritized combination therapies (Table 1) with the understanding that developments in preclinical (combination safety and efficacy) testing and/or ongoing clinical trials could subsequently affect the relative ranking. The Group indicated a preference for combination therapies with anti-CD3 and either antigen (such as oral insulin, GAD alum, proinsulin peptide or proinsulin DNA) or an IL-1 pathway anti-inflammatory (such as Anakinra or Rilonacept). These combinations were attractive in part because of the early positive clinical results using currently available anti-CD3 therapeutics and the anticipation of their clinical progression. In addition, preclinical data indicate good synergy between several antigenic modalities and anti-CD3 in recent-onset T1D [29–31]. Anti-CD20, as an approved therapeutic, has shown potential for preserving β cell function in a Phase II clinical trial [12] and has also been recommended for consideration as a combination therapy alongside a diabetes autoantigen. In order for any of these combination therapies to move forward, co-operation and support from all involved companies will be required, which in some cases will involve complex legal negotiations that could be aided by specialized task forces [32]. In addition, the academic community, ITN, TrialNet and funding agencies as well as industry would be well served to build a coordinated biomarker effort. All parties involved will have to be open to consider different priorities for combination therapies based on emerging preclinical and clinical data.
Table 1.
Consensus priority ranking of combination immune-based therapies for type 1 diabetes
| Combination |
Commentary on: |
||
|---|---|---|---|
| Drug no. 1 | Drug no. 2 | Evidence for tolerance and other (dis)advantages | Drug availability |
| Anti-CD3 •Teplizumab (MacroGenics/Eli Lilly) •Otelixizumab (TolerX/GSK) | Antigen •Oral insulin •GAD-Alum (Diamyd) •Proinsulin DNA (BHT-3021, Genentech) •Proinsulin peptide | –Extended benefit of anti-CD3 monotherapy in Phase II –Long-term induction of Tregs by anti-CD3 in preclinical models –Synergy of anti-CD3 and antigen demonstrated in preclinical models –Antigen-specific suppression shown in preclinical models for antigens | Availability will require negotiations between companies |
| T cell modulation (anti-CD3) •Teplizumab (MacroGenics/Eli Lilly) •Otelixizumab (TolerX/GSK) | Anti-inflammatory •IL-1RA Anakinra (Amgen) •IL-1 Trap Rilonacept (Regeneron) | –See above | Availability will require negotiations between companies |
| B cell depletion (anti-CD20) •Rituximab (Genentech) | Antigen •ral insulin •GAD-Alum (Diamyd) •Proinsulin DNA (BHT-3021, Genentech) •Proinsulin peptide | –Clinical and preclinical studies on all agents show potential effects as monotherapies (and for antigens see above) | Availability will require negotiations between companies |
| Immune depletion/modulation •Anti-thymocyte globulin (ATG; thymoglobulin, Genzyme) | Immune modulation •GM-CSF (Neulasta, Amgen) | –Encouraging preclinical results of combination in NOD mice | Good for all agents |
| Antigen •GAD-Alum (Diamyd) | Antigen •Oral insulin •Proinsulin DNA •Proinsulin peptide | –Encouraging preclinical results of antigen combinations in NOD mice –Induction of Tregs across diverse genetic backgrounds –Specific combinations not tested preclinically or clinically | All early development phase and will therefore require negotiation |
| T cell modulation (anti-CD3) •See above | Incretin mimetic •Exendin-4 (Exenatide, Amylin/Lily) | –Efficacy of combination in animal models | Availability will require negotiations between companies |
| T cell modulation (anti-CD3) or B cell depletion (anti-CD20) | Anti-inflammatory •Anti-TNF-α (Enbrel, Amgen) | –All agents show success in monotherapy trials and target different pathways –No data on combinations in preclinical setting | Availability will require negotiations between companies |
| Other possible combinations with minimal supporting data: | |||
| Anti-inflammatory •e.g. see above or •anti-IL-6 (Tocilizumab, Roche) | Antigen | ||
| B cell depletion (anti-CD20) | IL-2 pathway blockade •Rapamycin (Rapamune, Wyeth) | ||
| B cell depletion (anti-CD20) | Incretin mimetic | ||
| B cell depletion (anti-CD20) | Anti-inflammatory | ||
| T cell modulation (anti-CD3) | Anti-inflammatory •α-1 anti-trypsin (Aralast, Baxter) | ||
| Immune modulation •Anti-IL-12/23 (Stelara, J&J) | Antigen | ||
| Antigen •CTB-Ins plasmid | Antigen | ||
| Antigen •CTB-Ins plasmid | Immune modulation •IL-10 plasmid | ||
GM-CSF: granulocyte–macrophage colony-stimulating factor; IL: interleukin; NOD: non-obese diabetic; TNF: tumour necrosis factor; Treg: regulated T cell.
It is our hope that outlining the activities of the panel at this stage will broaden participation and commitment among diabetes researchers, clinicians, pharmaceutical companies and regulatory agencies to facilitate the development of combination therapies for the treatment of T1D. Already, the first steps taken in establishing a preclinical laboratory consortium and a network for early-stage clinical trials with mechanistic outcomes, as well as dialogues regarding T1D biobanks, provide a basis for optimism regarding progress in T1D immunotherapeutics going into the next decade.
Acknowledgments
This work was supported by the Juvenile Diabetes Research Foundation and the Immune Tolerance Network (National Institute of Allergy and Infectious Diseases contract # N01 AI15416).
Disclosure
Authors have no disclosures to report.
References
- 1.Bekris LM, Kavanagh TJ, Lernmark A. Targeting type 1 diabetes before and at the clinical onset of disease. Endocr Metab Immune Disord Drug Targets. 2006;6:103–24. doi: 10.2174/187153006776056576. [DOI] [PubMed] [Google Scholar]
- 2.Karlsson FA, Berne C, Bjork E, et al. Beta-cell activity and destruction in type 1 diabetes. Ups J Med Sci. 2000;105:85–95. doi: 10.1517/03009734000000056. [DOI] [PubMed] [Google Scholar]
- 3.Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–8. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- 4.Stiller CR, Dupre J, Gent M, et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science. 1984;223:1362–7. doi: 10.1126/science.6367043. [DOI] [PubMed] [Google Scholar]
- 5.Herold KC, Hagopian W, Auger JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–8. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 6.Keymeulen B, Vandemeulebroucke E, Ziegler AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 7.Bougneres PF, Carel JC, Castano L, et al. Factors associated with early remission of type I diabetes in children treated with cyclosporine. N Engl J Med. 1988;318:663–70. doi: 10.1056/NEJM198803173181103. [DOI] [PubMed] [Google Scholar]
- 8.Bougneres PF, Landais P, Boisson C, et al. Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes. 1990;39:1264–72. doi: 10.2337/diab.39.10.1264. [DOI] [PubMed] [Google Scholar]
- 9.Cook JJ, Hudson I, Harrison LC, et al. Double-blind controlled trial of azathioprine in children with newly diagnosed type I diabetes. Diabetes. 1989;38:779–83. doi: 10.2337/diab.38.6.779. [DOI] [PubMed] [Google Scholar]
- 10.Dantal J, Soulillou JP. Immunosuppressive drugs and the risk of cancer after organ transplantation. N Engl J Med. 2005;352:1371–3. doi: 10.1056/NEJMe058018. [DOI] [PubMed] [Google Scholar]
- 11.Herold KC, Gitelman SE, Masharani U, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54:1763–9. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–52. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herold KC, Gitelman S, Greenbaum C, et al. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol. 2009;132:166–73. doi: 10.1016/j.clim.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spencer J, Peakman M. Post-mortem analysis of islet pathology in type 1 diabetes illuminates the life and death of the beta cell. Clin Exp Immunol. 2009;155:125–7. doi: 10.1111/j.1365-2249.2008.03864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thrower SL, James L, Hall W, et al. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol. 2009;155:156–65. doi: 10.1111/j.1365-2249.2008.03814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staeva-Vieira T, Peakman M, von Herrath M. Translational mini-review series on type 1 diabetes: immune-based therapeutic approaches for type 1 diabetes. Clin Exp Immunol. 2007;148:17–31. doi: 10.1111/j.1365-2249.2007.03328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rother KI, Spain LM, Wesley RA, et al. Effects of exenatide alone and in combination with daclizumab on beta-cell function in long-standing type 1 diabetes. Diabetes Care. 2009;32:2251–7. doi: 10.2337/dc09-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottlieb PA, Quinlan S, Krause-Steinrauf H, et al. for the Type 1 Diabetes TrialNet MMF/DZB Study Group. Failure to preserve beta-cell function with mycophenolate mofetil and daclizumab combined therapy in patients with new onset type 1 diabetes. Diabetes Care. 2010 doi: 10.2337/dc09-1349. Jan 12. [Epub ahead of print] PMID: 20067954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabinovitch A, Suarez-Pinzon WL, Shapiro AM, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes. 2002;51:638–45. doi: 10.2337/diabetes.51.3.638. [DOI] [PubMed] [Google Scholar]
- 21.Ludvigsson J, Faresjo M, Hjorth M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–20. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 22.Greenbaum CJ, Harrison LC. Guidelines for intervention trials in subjects with newly diagnosed type 1 diabetes. Diabetes. 2003;52:1059–65. doi: 10.2337/diabetes.52.5.1059. [DOI] [PubMed] [Google Scholar]
- 23.Asare AL, Kolchinsky SA, Gao Z, et al. Differential gene expression profiles are dependent upon method of peripheral blood collection and RNA isolation. BMC Genomics. 2008;9:474. doi: 10.1186/1471-2164-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herold KC, Brooks-Worrell B, Palmer J, et al. Validity and reproducibility of measurement of islet autoreactivity by T-cell assays in subjects with early type 1 diabetes. Diabetes. 2009;58:2588–95. doi: 10.2337/db09-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roep BO. Are insights gained from NOD mice sufficient to guide clinical translation? Another inconvenient truth. Ann NY Acad Sci. 2007;1103:1–10. doi: 10.1196/annals.1394.018. [DOI] [PubMed] [Google Scholar]
- 26.Roep BO, Peakman M. Surrogate end points in the design of immunotherapy trials: emerging lessons from type 1 diabetes. Nat Rev Immunol. 2010;10:145–52. doi: 10.1038/nri2705. [DOI] [PubMed] [Google Scholar]
- 27.National Institutes of Health (NIH) RFA-OD-10-010 – Recovery Act Limited Competition: Research on Biosamples from Selected Diabetes Clinical Studies (RC4), Bethesda (MD) 2010.
- 28.Juvenile Diabetes Research Foundation International. JDRF Autoimmunity Centers Consortium (ACC) New York, 2010. Available at: http://www.jdrf.org/index.cfm?page_id=111042 (accessed.
- 29.Bresson D, Togher L, Rodrigo E, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–81. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bresson D, Fradkin M, Manenkova Y, Rottembourg D, von Herrath M. Genetic-induced variations in the GAD65 T-cell repertoire governs efficacy of anti-CD3/GAD65 combination therapy in new-onset type 1 diabetes. Mol Ther. 2009;18:18. doi: 10.1038/mt.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherry NA, Chen W, Kushner JA, et al. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology. 2007;148:5136–44. doi: 10.1210/en.2007-0358. [DOI] [PubMed] [Google Scholar]
- 32.von Herrath M, Chan A. How can we improve the translational landscape for a faster cure of type 1 diabetes? J Clin Invest. 2009;119:1061–5. doi: 10.1172/JCI37593. [DOI] [PMC free article] [PubMed] [Google Scholar]