

Abstract
Objective:
Identification of low-frequency variants is of clinical importance in the identification of preexisting drug resistance. Using ‘ultra-deep’ sequencing, we address the detection of potential resistance to the chemokine (C–C motif) receptor 5 antagonist, maraviroc, due to the pretreatment presence of low levels of chemokine (CXC motif) receptor 4 (CXCR4)-using virus.
Methods:
We present a novel protocol for the phenotyping of HIV based on ‘454’ pyrosequence data and apply this to two large data sets comprised of 104░628 (before treatment, day 1) and 191░637 (after treatment, day 11) reads from the envelope region. We study resistance in the context of the evolutionary history of the intrapatient viral population. Variation was also investigated both within and outside the V3 region, the region associated with the receptor switch.
Results:
CXCR4-using virus can be detected at low frequency prior to maraviroc treatment (~0.5%) and at high frequency after failure of monotherapy (~81%). Inferring an evolutionary tree from the 1674 unique reads that span the V3 region confirms that the CXCR4-using population emerged from low-frequency CXCR4-using variants present before treatment. Changes in the frequency of amino acid residues used at individual sites were found in regions outside the V3 region, indicative of other potential sites associated with receptor usage.
Conclusion:
We have provided a high-resolution snapshot of intrapatient viral variation, prior and after treatment with maraviroc, and detected preexisting CXCR4-using variants present at an extremely low frequency. The evolutionary analysis demonstrates the extent of diversity present at a single time point within an infected individual and the rapid effect of drug pressure on the structure of a viral population.
Keywords: AIDS, chemokine receptors, chemokine (C–C motif) receptor 5, chemokine (CXC motif) receptor 4, coreceptors, HIV, maraviroc, pyrosequencing
Introduction
The emergence of resistance against drugs that directly target HIV has led to the alternative strategy of targeting the host proteins that the virus interacts with. The first such ‘host’ drug to get clinical approval is Pfizer's chemokine (C–C motif) receptor 5 (CCR5) antagonist, maraviroc [1,2]; CCR5 being a coreceptor that HIV uses to enter a target cell. As this drug has no direct impact on strains using the alternative coreceptor, chemokine (CXC motif) receptor 4 (CXCR4), all patients are screened for the presence of these variants prior to treatment [3]. In their absence, maraviroc may then be administered in combination with other antiretroviral drugs. Detecting coreceptor usage accurately is therefore vitally important in relation to the treatment of individual hosts. Currently, coreceptor usage is screened using the phenotypic coreceptor tropism assay, Trofile (Monogram Biosciences, South San Francisco, California, USA) [4]. The aim is to distinguish a viral population that is exclusively CCR5-using (R5) from a viral population harbouring either dual mixed [R5 and exclusively CXCR4-using (X4)] or R5 and dual-tropic viruses [those that can use both CCR5 and CXCR4 (R5X4)]. We refer to both X4 and R5X4 viruses as CXCR4-using.
As coreceptor usage can be reliably predicted based on specific amino acid changes within the V3 loop of HIV's envelope gene, gp160 [5-11], an alternative approach is to predict the phenotype of an individual's viral population based on genotypic data. However, because of the low frequency at which CXCR4-using variants can be present, especially during the early stages of HIV infection [12-15], Sanger-based sequencing is unlikely to be sufficient for reliably detecting their presence [16]. Here, we investigate whether ultra-deep 454 pyrosequencing [17] can be used in conjunction with sequence-based phenotype tests [10,11] for the successful detection of CXCR4-using variants within the viral population. This next generation sequencing technology permits the quantification of the range of variants present within a sample by producing very high numbers of short sequence fragments. As a consequence, it has the potential to detect minor variants of clinical relevance [18].
454 Life Sciences pyrosequencing [17,19] differs fundamentally from Sanger sequencing. The 454 technology (now owned by Roche Diagnostics, Indianapolis, Indiana, USA) is a sequencing by synthesis method, which makes use of the cyclical delivery of individual reagents containing only one nucleotide and measures their incorporation into the growing DNA strand via pyrophosphate liberation. A nucleotide is incorporated, or not, depending on the complement base on the opposite strand. In regions in which the complement strand contains a homopolymeric stretch (hps), the amount of light emitted by the reaction is directly proportional to the number of bases incorporated. 454's Genome Sequencer system includes software, which, depending on the intensity of the light emitted, determines the quantity of a particular base that is added. These data are reduced to a plot of normalized signal intensities for each nucleotide flow called a flowgram.
Ambiguities in signal intensity, caused by factors such as signal contamination from nearby wells and multiple templates within an individual well, can lead to errors [20]. The majority of pyrosequencing errors manifest as under and overcalls in the sequence reads, whereas miscall errors are much rarer [20]. In a recent study [18] on the application of 454 pyrosequencing to HIV samples, an error rate of 0.98% was estimated that was partitioned into insertion errors (0.73%), deletion errors (0.16%) and mismatches (0.12%). In addition, the error rate within hps of size three or greater was 6.2 times higher than outside of these regions. These errors and the large volumes of data produced by next generation sequencing technologies introduce unique problems for the computational analysis of pyrosequence data [21,22].
In order to determine whether or not this technology can be used to detect low-frequency variants of clinical importance, we have developed a novel protocol for the management and rapid analysis of large quantities of viral pyrosequence data. We apply the protocol to two extremely large 454 data sets containing 104░628 (before treatment, day 1) and 191░637 (after treatment, day 11) reads from an HIV-infected patient treated with maraviroc. Our results demonstrate that CXCR4-using virus is detectable prior to treatment at an extremely low frequency (~0.5%). Furthermore, a phylogenetic analysis of reads completely spanning the V3 region strongly indicates that the CXCR4-using strains that emerge during maraviroc monotherapy do so from a preexisting CXCR4-using viral population that was present prior to treatment.
Methods
Samples
The samples for pyrosequencing were from two time points (days 1 and 11; before and after treatment, respectively) from a patient, A, enrolled in a clinical trial designed to evaluate the effect of short-term monotherapy with maraviroc [3,23]. This was one of three patients (out of 64) in which CXCR4-using virus was detected after 10 days. In one of these patients, C, a sample mix-up had occurred as the coreceptor tropism assay (PhenoSense, related to the Trofile assay) detected CXCR4-using virus prior to treatment. For the other two patients, A and B, the coreceptor tropism assay did not detect CXCR4-using viruses at day 1. For patient A, subsequent screening of 97 functional clones from day 1 revealed two to be dual-tropic demonstrating the presence of CXCR4-using virus prior to treatment. Note, patient A was infected with virus classified as subtype B (see Westby et al. [3] for further details).
Pyrosequencing
RNA extraction and amplification of HIV's envelope gene was first carried out for two patient A samples from days 1 and 11. The amplicons were then subjected to nebulization to generate fragments of approximately 600 nucleotides. These were amplified as in Margulies et al. [17] and sequenced on the Genome Sequencer 20 (Roche Applied Sciences, Indianapolis, Indiana, USA). Standard protocols for the generation of a library of tagged single-stranded DNA molecules were used (see Margulies et al. [17] for details). The standard GS20 pyrosequencing profile uses a sequential flow of each nucleotide in a repeating pattern of TACG. This pattern is repeated for 42 cycles as per the protocol and generates 100 nucleotides of sequence information on average. For the purposes of generating longer sequencing reads, the sequence profile of 42 cycles of nucleotide flows was changed to 100 cycles, which increased the average read length from 100 to 200 nucleotides (Fig. 1, inset). The GS20 has software to recognize high-quality reads and convert the signal (light) into a base call. The software GS20 package was used to generate the sequence files.
Fig. 1. Coverage plots across gp160 for days 1 (blue) and 11 (red) pyrosequencing data sets.
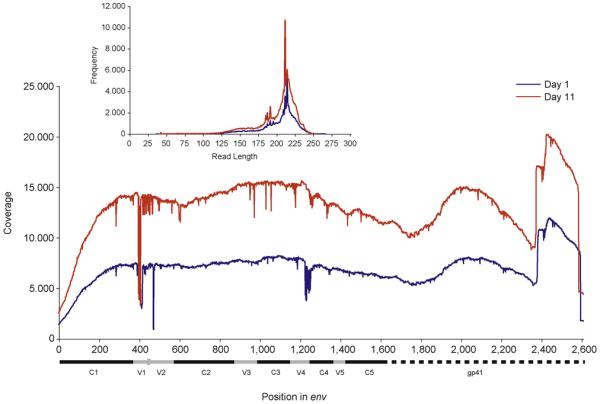
The genomic regions are depicted below the x-axis; the characterized conserved and variable regions (coordinates from LANL HIV Sequence Database) are shown in black and grey, respectively. The inset is a frequency plot of read lengths. All low-quality reads identified by BLAST have been removed.
Read alignment
For each data set in order to locate the direction and genomic region that each read was to be aligned against, a modified BLAST [24] search was used to compare each read with a template sequence. Template sequences were constructed for the days 1 and 11 data by aligning the 12 available clonal sequences from each time point [3] using Muscle (Kimmen Sjolander's Group, Berkeley, California, USA)[25] and constructing a consensus sequence for each. Reads producing an exceptionally poor match to the reference templates were removed from the data set. Match quality was based on the word cover density across the template. This was the frequency of read words being matched to regions of the template sequence, in which words were all subsequences of length five that could be generated from the individual read being processed [26]. The minimum threshold permitted within a matching region was selected from a plot of cover density against frequency for a subset of 1,000 random reads (data not shown). The Smith–Waterman algorithm [27] was then used to more accurately align each read to the genomic region identified by BLAST. The parameters for the alignment were gap opening (−4), gap extension (−2), match (+2), transition (−1) and transversion (−2). This combination of BLAST and Smith–Waterman algorithm significantly increased the speed at which the reads could be processed. To maintain site compatibility between reads, insertions within the template were not permitted.
Phylogenetic analysis
Reads that span the entire V3 region for both time points were truncated and aligned using Muscle. PhyML (PHYlogenetic inferences using Maximum Likelihood) [28] was used to construct a maximum likelihood tree using the HKY model of nucleotide substitution. The centre of the tree (COT) [29] was calculated and reads that were more divergent than the mean distance to the COT (±1.645 SD) were removed from the complete V3 analysis in order to exclude highly divergent sequences that are likely to be sequencing artefacts not removed during the BLAST step. Bootstrap support values were generated using neighbour joining (1000 replicates) and implemented with CLUSTAL X (Plate-Forme de Bio-Informatique, Illkirch, France)[30].
Detection of chemokine (CXC motif) receptor 4CXCR4-using virus
Genotypic detection of CXCR4-using virus was based on both complete and incomplete translated V3 regions and was performed using the 11/24/25 ‘charge rule' (specifically, the presence of a positively charge amino acid residue at one of the sites 306, 319 or 320 in gp120; sites 11, 24 or 25 of the V3 loop, respectively [11]) and the web position-specific scoring matrix (PSSM) phenotype test [8]. Reads that span the entire V3 region were necessary for the latter test.
Entropy and amino acid frequency analysis
Entropy was calculated for each site in gp160 using the standard Shannon entropy formulae. The difference in entropy was calculated by subtracting the entropy at the second time point from the first. The closer the entropies are at the two time points, the closer this value will be to zero, whereas departures from this indicate changes in the entropy at a site. Significant changes in individual amino acid frequencies at sites were detected by comparing the difference in frequencies (day 1 frequency subtracted from day 11 frequency of the same amino acid) to the overall distribution of differences and significant outliers identified.
Results
In total 104░628 and 191░637 nucleotide sequence reads were generated for the days 1 and 11 samples, respectively. After exclusion of the low-identity reads (identified by BLAST), 91░268 and 171░093 reads remained, corresponding to a removal of 12.7 and 10.7% of the data. This amount of data removal is similar to a previous study [18]. The coverage across all regions of gp160 is extremely high for both time points (Fig. 1). Coverage is lowest across the V1 and V2 regions due to high levels of sequence variation in this region. Importantly, for genotypic detection of X4 and R5X4 (CXCR4-using variants), coverage is extremely high across the V3 region with several thousands of reads available for analysis.
Performing the charge rule on reads that include at least sites 11, 24 or 25 (of which at lease one had to be positively charged), 30 sequences were predicted to be CXCR4-using on day 1 (before treatment), representing 0.5% of the data (Table 1). In contrast, after 10 days of monotherapy, the charge rule predicts 80.8% of the data to be CXCR4-using (Table 1). This represents a remarkable change in frequency of these variants and must be a direct result of selection against R5 strains because of the presence of CCR5 antagonist.
Table 1.
Coreceptor usage detection for days 1 and 11 pyrosequencing data sets based on the V3 region of env.
| [0,2-3]R5 | [0,4-5]CXCR4-using | |||
|---|---|---|---|---|
|
|
||||
| No. of reads | % of total | No. of reads | % of total | |
| Day 1 | ||||
| ▓Charge rule (sites 11, 24 and 25 present)) | 5859 | 99.5 | 30 | 0.5 |
| ▓Charge rule (complete V3) | 3832 | 99.7 | 11 | 0.3 |
| ▓Composite PSSM (complete V3) | 3839 | 99.9 | 4 | 0.1 |
| Day 11 | ||||
| ▓Charge rule (sites 11, 24 and 25 present) | 2288 | 19.2 | 9660 | 80.8 |
| ▓Charge rule (complete V3) | 1501 | 19.3 | 6277 | 80.7 |
| ▓Composite PSSM (complete V3) | 1537 | 19.8 | 6241 | 80.2 |
CXCR4, chemokine (CXC motif) receptor 4; PSSM, position-specific scoring matrix.
Next, focussing on reads that spanned the entire V3 region, there were 3843 and 7778 sequences from the day 1 and 11 data sets, respectively, representing 570 and 1104 unique nucleotide sequences, respectively. The most frequent complete V3 amino acid sequences (≥10 variants) for days 1 and 11 corresponded to 20 and 48 unique sequences, respectively (Fig. 2a), whereas 11 CXCR4-using viruses were detected at day 1 (Table 1). Figure 2(a) shows the change in the consensus sequence between time points and the shift in relative proportion of R5 to CXCR4-using variants in the patient's viral population; the V3 sequences corresponding to the 11 predicted CXCR4-using viruses at day 1 are also shown.
Fig. 2. Prediction of phenotype based on genotype data.

Sequence-based phenotyping of days 1 and 11 pyrosequence data. (a) The V3 sequences at days 1 and 11 for which 10 or more variants were detected with reference to the consensus clonal sequences (top) for each time point. R5 sequences are shown in green and CXCR4-using sequences in red (predicted based on the charge rule). CXCR4-using V3s detected at day 1 are also shown. The grey boxes indicate sites 11, 24 and 25, and charged amino acids at these sites are in bold. (b) A frequency plot of PSSM scores at days 1 (green) and 11 (red). The light grey box indicates PSSM threshold values for prediction of CXCR4-using and R5 viruses [8]. The circles beneath the x-axis indicate PSSM scores of the day 1 sequences predicted to be CXCR4-using by the composite PSSM test (orange) and charge rule (blue) at sites 11, 24 and 25. CXCR4, chemokine (CXC motif) receptor 4; PSSM, position-specific scoring matrix.
Plotting PSSM scores for the complete V3 data dramatically confirms the charge rule results (Fig. 2b), there being a clear shift in the distribution of scores between days 1 and 11. For the latter time point, the detection of CXCR4-using viruses is very close for the charge rule and PSSM. However, at day 1, only four reads are predicted to be CXCR4-using by the PSSM phenotype test (Table 1). PSSM predicts viruses to be X4 on the basis of scores being higher than a threshold of −2.88 and below this uses the charge rule at sites 11 and 25 (up to a threshold of −6.96) [8]. However, in the day 1 data set, there is a positive charge at site 24 and so these sequences were not classified as CXCR4-using by PSSM. Interestingly, several sites predicted to be CXCR4-using by the charge rule test fall below the −6.96 threshold and so would be classified as R5 by PSSM (Fig. 2b). This emphasises that the utility of any algorithm used to predict a viral tropism phenotype is related to the training data used and the importance of including appropriate information in the prediction, particularly in the presence of dual-tropic data as is the case with this patient [3].
Given the low number of CXCR4-using variants detected at day 1 by the charge rule (Table 1), we checked whether these numbers are above the maximum potential miscall rate for pyrosequencing. The frequency of sequencing errors for HIV data has been previously characterized as 0.0098 (mismatches 0.0012, deletions 0.0016 and insertions 0.0073) [18]. All indels have been conservatively dealt with by our pairwise alignment procedure leaving the 0.0012 error rate in relation to mismatches. With this error rate for mismatches, the maximum number of falsely predicted CXCR4-using variants (combining all three sites 11, 24 and 25) was calculated to be 15, lower than the 30 observed.
The error rate due to mismatches within hps (0.0044), however, has been observed to be 6.2 times higher than the error rate outside of these regions (0.0007) [18]. Thus, to conservatively estimate the error rates, we also take this distinction into account. In the day 1 consensus template, a glycine (GGA) residue present at site 24 does not reside within a hps region. The maximum per nucleotide error rate at this site is therefore taken to be 0.007. Assuming two nonsynonymous positions, the expected number of false positive charged residues arising from a negative residue due to sequencing error is two for this data set. Sites 11 and 25 each have one nucleotide overlapping a hps region. A serine (AGT) is present at site 11, whereas at sites 10 and 9, the presence of a lysine (AAA) and an arginine (AGA) result in the longest hps region found within the V3 loop. At site 25, the residue present is a valine (GAA). The isoleucine (ATA) at site 26 thus results in a hps region of length three. For each of sites 11 and 25 assuming two nonsynonymous positions (one within a hps region and one outside), the expected number of false positive charged residues arising from a negative residue is 12. With all three sites combined a highly conservative estimation of the number of expected falsely predicted CXCR4-using strains is 26, still lower than the observed number. This potential error rate highlights the inherent difficulty in detecting significant variants at low frequency
To investigate the evolutionary origins of the CXCR4-using viruses that have emerged during therapy, a phylogenetic tree was inferred combining the complete V3 data, 3843 and 7778 sequences from days 1 and 11, respectively, collectively corresponding to 1674 unique sequences (Fig. 3a). Interestingly, in this phylogeny, there is strong tendency for variants to cluster together associated with both their time point and predicted coreceptor usage (Fig. 3a). Specifically, the day 1 R5 viruses form clusters towards the top part of the phylogeny and the majority of their day 11 R5 counterparts are relatively closely related to one cluster on the right. In contrast, the majority of CXCR4-using viruses at day 11 form a distinct cluster towards the lower part of the phylogeny (supported by high bootstrap support values, Fig. 3a) that is not closely related to any of the day 1 R5 population. This major CXCR4-using cluster is almost entirely comprised of day 11 sequences, apart from two day 1 CXCR4-using viruses. Mean nucleotide divergence between CXCR4-using viruses from the two time points in this cluster was 4% as compared with 12% for the mean divergence with day 1 CXCR4-using viruses in the top part of the phylogeny. This close proximity of days 1 and 11 CXCR4-using viruses in an evolutionary distinct CXCR4-using cluster – in agreement with Westby et al. [3] – indicates that CXCR4-using variants are at an advantage in the presence of maraviroc due to selection against R5 variants and the pretreatment CXCR4-using variants that gave rise to this distinct cluster were present prior to monotherapy. The position of the clonal sequences from Westby et al. [3] in the phylogeny was consistent with this conclusion (data not shown).
Fig. 3. Evolutionary history of the viral population.

A phylogeny showing CXCR4-using and R5 viruses at days 1 and 11. (a) Colours (see key) indicate predicted coreceptor usage. The two numbers above the main CXCR4 cluster correspond to bootstrap support values out of 1000 replicates, shown for this cluster only. (b) Colours (see key) indicate the frequency of each sequence. The scale bar represents substitutions per site. CXCR4, chemokine (CXC motif) receptor 4.
To investigate viral diversity, Shannon entropy was calculated for each site across gp160 for the two data sets (Fig. 4a and b). For both time points, mean entropy is higher in the variable regions, 0.35 and 0.24, than the conserved regions, 0.16 and 0.15, respectively. In order to compare the shift in entropy between the two time points, the difference was calculated (Fig. 4c). This illustrates that diversity at specific sites changes markedly within just 10 days of the selective environment being altered as a result of drug pressure selecting against R5 viruses (Fig. 2a and b). To identify changes in individual amino acids at these sites, we compared the difference in the per site amino acid frequencies for each amino acid residue. The vast majority of amino acids do not change significantly in frequency (mean for all data −0.0000124). To identify amino acids that have changed significantly, we looked for frequency changes equivalent or greater to that observed at site 11 – the main charge rule site changing within these data (Fig. 2a) – and identified 10 sites (indicated in red in Fig. 4c) of which four (in addition to site 11) are within the V3 region. The other six are potentially associated with CXCR4 use outside of the V3 region. Comparative analysis of multiple data sets will be informative for determining their predictive value.
Fig. 4. Entropy plots.
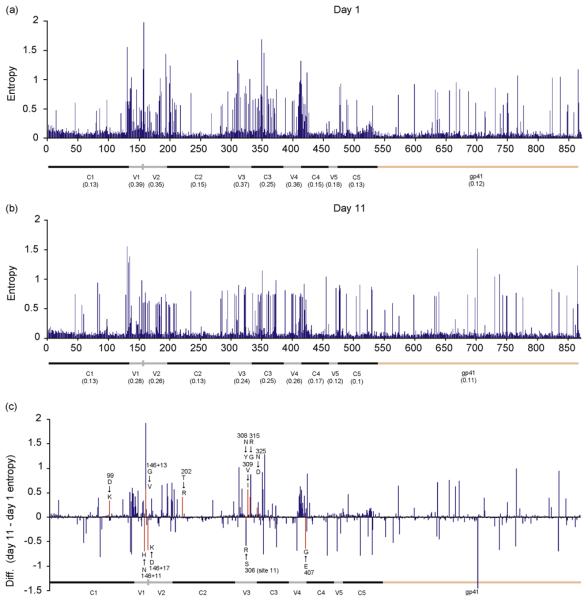
For days 1 (a) and 11(b), including mean entropy values, and difference in entropies (c). Sites at which there is a difference in the frequency of at least one amino acid (equivalent or greater than site 11) are shown in red; the position of these sites relative to HXB2 and major amino changes at that site are indicated. The genomic regions are depicted across the x-axis.
Discussion
We have demonstrated the use of a novel computer-based protocol for the fast and accurate handling of large amounts of short fragment pyrosequence data. Using this protocol, in conjunction with the sequence-based phenotype tests, we detected CXCR4-using variants at extremely low frequency (~0.5%). This minority population is of clinical significance as 10 days after monotherapy with maraviroc, it represents approximately 81% of variants in the population. In the context of viral load, this massive frequency shift in the proportion of CXCR4-using variants detected represents a change of 1.3░×░103░copies/ml on day 1 to 20.5░×░103░copies/ml on day 11. Note, pretreatment viral load (26.6 ░×░104░copies/ml) was calculated from the average of viral load at 38 and 5 days prior to treatment and on day 1; day 11 viral load was significantly lower at 25.6░×░103░copies/ml. Thus, despite the dramatic shift in relative frequencies of CXCR4-using virus, the overall change corresponds to a significant decrease in the patient's viral load.
The evolutionary analysis (Fig. 3a) confirms that these CXCR4-using viruses do not evolve de novo as a result of the drug pressure acting directly on R5 virus, rather they emerge from a preexisting and distinct CXCR4-using population present prior to monotherapy with maraviroc. If the R5 population had evolved on drug to become CXCR4-using, they would have been highly related to day 1 R5 variants. This is not the case. The phylogenetic tree (Fig. 3a) shows that the major day 11 CXCR4-using virus cluster is evolutionary distinct to the day 1 R5 variants. In contrast, the day 11 CXCR4-using virus cluster is closely related to CXCR4-using variants present at day 1 that are highly divergent from the day 1 R5 population. This indicates that the major day 11 CXCR4-using cluster has emerged from preexisting day 1 CXCR4-using variants following maraviroc therapy. This pattern of emergence has also been observed for other patients in whom CXCR4-using virus had been detected after therapy [3,31].
It is important to note that the extent of the divergence in the phylogenetic tree (Fig. 3a) is largely due to the high number of either unique or rare sequences. This is because a few variants predominant in the viral population at both time points (Fig. 3b), whereas only a minority of reads are present once (11 and 10% of sequences at days 1 and 11, respectively). Nonetheless, this amount of variation emerging in only 10 days highlights the extreme mutability of HIV-1.
Collectively, these results demonstrate that 454 pyrosequencing technology combined with a genotypic test has the potential to be used within a clinical setting. Thus, this approach has the potential to provide an alternative to the laboratory-based assay, which is currently a prerequisite for the use of CCR5 antagonists. However, there are a number of considerations for a viable genotypic test. How reproducible are the results given chance variation in amplification, particularly with low copy numbers? How dependable are the results given the additional sequencing errors potentially introduced at the reverse transcriptase and PCR stages? Will the sensitivity of the genotypic tests be sufficient for clinical use [32]? Including additional sites, for example, outside the V3 region [33,34] may improve predictions. How much CXCR4-using virus is acceptable in the context of combination therapy? That patients, despite the presence of CXCR4-using virus, responded to monotherapy [3] indicates that, with appropriate background therapy, patients with low levels of CXCR4-using virus will potentially be appropriate for treatment with CCR5 antagonist. In order to answer these questions and to develop a viable pyrosequencing genotypic test, multiple patient data sets will need to be studied in detail.
In conclusion, we have provided a high-resolution snapshot of intrapatient viral diversity prior and after treatment with the CCR5 antagonist, maraviroc, and detected preexisting CXCR4-using variants of clinical relevance that are present at extremely low frequency. 454 pyrosequencing technology, thus, combined with a V3-based phenotype test can be used to detect low-frequency CXCR4-using viruses.
Acknowledgements
We would like to thank the investigators, study-site staff, the Pfizer maraviroc development team and the patients who participated in the maraviroc studies. Also, thanks to Andrew Rambaut for helpful discussion on the 454 data handling protocol. J.A. is currently funded by Pfizer Global R&D. This work was started while J.A. was a BBSRC funded PhD student. Thanks also to the Wellcome Trust VIP and Apple Research & Technology Support schemes for support.
References
- 1.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum antihuman immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med. 2008;359:1429–1441. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Westby M, Lewis M, Whitcomb J, Youle M, Pozniak AL, James IT, et al. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J Virol. 2006;80:4909–4920. doi: 10.1128/JVI.80.10.4909-4920.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, et al. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob Agents Chemother. 2007;51:566–575. doi: 10.1128/AAC.00853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 6.Shioda T, Levy JA, Cheng-Mayer C. Small amino acid changes in the V3 hypervariable region of gp120 can affect the T-cell-line and macrophage tropism of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1992;89:9434–9438. doi: 10.1073/pnas.89.20.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollakis G, Abebe A, Kliphuis A, Chalaby MI, Bakker M, Mengistu Y, et al. Phenotypic and genotypic comparisons of CCR5- and CXCR4-tropic human immunodeficiency virus type 1 biological clones isolated from subtype C-infected individuals. J Virol. 2004;78:2841–2852. doi: 10.1128/JVI.78.6.2841-2852.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen MA, Li FS, van 't Wout AB, Nickle DC, Shriner D, He HX, et al. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J Virol. 2003;77:13376–13388. doi: 10.1128/JVI.77.24.13376-13388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillai S, Good B, Richman D, Corbeil J. A new perspective on V3 phenotype prediction. AIDS Res Hum Retroviruses. 2003;19:145–149. doi: 10.1089/088922203762688658. [DOI] [PubMed] [Google Scholar]
- 10.Rosen O, Sharon M, Quadt-Akabayov SR, Anglister J. Molecular switch for alternative conformations of the HIV-1 V3 region: implications for phenotype conversion. Proc Natl Acad Sci U S A. 2006;103:13950–13955. doi: 10.1073/pnas.0606312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardozo T, Kimura T, Philpott S, Weiser B, Burger H, Zolla-Pazner S. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res Hum Retroviruses. 2007;23:415–426. doi: 10.1089/aid.2006.0130. [DOI] [PubMed] [Google Scholar]
- 12.Connor RI, Ho DD. Human immunodeficiency virus type 1 variants with increased replicative capacity develop during the asymptomatic stage before disease progression. J Virol. 1994;68:4400–4408. doi: 10.1128/jvi.68.7.4400-4408.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use coreceptor use correlates with disease progression in HIV-1-infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koot M, van Leeuwen R, de Goede RE, Keet IP, Danner S, Eeftinck Schattenkerk JK, et al. Conversion rate towards a syncytium-inducing (SI) phenotype during different stages of human immunodeficiency virus type 1 infection and prognostic value of SI phenotype for survival after AIDS diagnosis. J Infect Dis. 1999;179:254–258. doi: 10.1086/314539. [DOI] [PubMed] [Google Scholar]
- 15.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Low AJ, Dong W, Chan D, Sing T, Swanstrom R, Jensen M, et al. Current V3 genotyping algorithms are inadequate for predicting X4 co-receptor usage in clinical isolates. Aids. 2007;21:F17–F24. doi: 10.1097/QAD.0b013e3282ef81ea. [DOI] [PubMed] [Google Scholar]
- 17.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res. 2007;17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronaghi M, Uhlen M, Nyren P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363– 365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 20.Brockman W, Alvarez P, Young S, Garber M, Giannoukos G, Lee WL, et al. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008;18:763–770. doi: 10.1101/gr.070227.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pop M, Salzberg SL. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008;24:142–149. doi: 10.1016/j.tig.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trombetti GA, Bonnal RJ, Rizzi E, De Bellis G, Milanesi L. Data handling strategies for high throughput pyrosequencers. BMC Bioinformatics. 2007;8(Suppl 1):S22. doi: 10.1186/1471-2105-8-S1-S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–1172. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 24.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durbin R, Eddy S, Krogh A, Mitchison G. Biological sequence analysis: probabilistic models of proteins and nucleic acids. Cambridge University Press; 1998. [Google Scholar]
- 27.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 28.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 29.Nickle DC, Jensen MA, Gottlieb GS, Shriner D, Learn GH, Rodrigo AG, Mullins JI. Consensus and ancestral state HIV vaccines. Science. 2003;299:1515–1518. doi: 10.1126/science.299.5612.1515c. author reply 1515–1518. [DOI] [PubMed] [Google Scholar]
- 30.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis M, Simpson P, Fransen S, Huang W, Whitcomb J, Mosley M, et al. CXCR4-using virus detected in patients receiving maraviroc in the Phase III studies MOTIVATE 1 and 2 originates from a pre-existing minority of CXCR4-using virus. Antiviral Ther. 2007;12:S65–S165. [Google Scholar]
- 32.Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R. Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol. 2007;25:1407–1410. doi: 10.1038/nbt1371. [DOI] [PubMed] [Google Scholar]
- 33.Huang W, Toma J, Fransen S, Stawiski E, Reeves JD, Whitcomb JM, et al. Coreceptor tropism can be influenced by amino acid substitutions in the gp41 transmembrane subunit of human immunodeficiency virus type 1 envelope protein. J Virol. 2008;82:5584–5593. doi: 10.1128/JVI.02676-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pastore C, Nedellec R, Ramos A, Pontow S, Ratner L, Mosier DE. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J Virol. 2006;80:750–758. doi: 10.1128/JVI.80.2.750-758.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]