

Autism spectrum disorders are characterized by abnormal social behavior, deficits in communication, and repetitive behavior. A diverse array of genetic mutations have been linked to autism (1), suggesting that what these behaviorally defined disorders share is not a single genetic lesion, but a disruption of biological processes important in cognitive development. As a result, attempts to understand the etiology of autism spectrum disorders have focused on the neurobiology of related syndromes caused by single-gene mutations that are more easily modeled in the laboratory. One example is mutation of the gene encoding methyl-CpG binding protein 2 (MeCP2), which gives rise to a spectrum of related but distinct postnatal neurodevelopmental disorders including Rett syndrome and autism spectrum disorders (2). Initially identified on the basis of its ability to bind methylated DNA (3), MeCP2 was thought to repress transcription. Mutations disrupting MeCP2 function were thus expected to increase gene expression, perturb neuronal function, and give rise to behavioral disorders such as Rett syndrome. However, the search for genes repressed by MeCP2 has so far identified only a few convincing targets. On page 1224 in this issue, Chahrour et al. (4) demonstrate that MeCP2 may play a more complex role, coordinating either transcriptional repression or activation, depending on the molecular context.
To investigate the effect of MeCP2 gene dosage on transcriptional regulation, Chahrour et al. analyzed gene expression in the hypothalamus of MeCP2-deficient, wild-type, and MeCP2-overexpressing mice. This approach revealed small but important changes in the expression of thousands of genes, many of which are affected in opposite ways with deletion or overexpression of MeCP2. Although these data are consistent with previous studies indicating that many genes may be subtly misregulated in the brains of MeCP2-deficient animals (5), the ability of Chahrour et al. to correlate changes in the expression of particular genes with the level of MeCP2 expression convincingly links these transcriptional changes to the presence or absence of MeCP2. Surprisingly, most of the genes identified in these experiments showed increased expression in the hypothalamus of animals overexpressing MeCP2, and decreased expression in the absence of MeCP2. The study reveals a large number of new genes whose expression depends on MeCP2, and suggests a role for the factor in activating transcription.
A fundamental step toward understanding how mutations in MeCP2 disrupt neuronal development and function is to identify the mechanisms by which MeCP2 regulates transcription. One possibility is that MeCP2 functions as a classical transcription factor that binds directly to regulatory sequences within the promoters of target genes. Repression or, as suggested by Chahrour et al., activation of the targeted genes would depend on the interaction of MeCP2 with molecular complexes that promote or inhibit transcription (see the figure). Alternatively, or perhaps additionally, MeCP2 may play a structural role in the higher-order organization of chromatin architecture (6, 7). Transcriptional repression and activation of individual genes would still be controlled by classical sequence-specific transcription factors, but MeCP2 might influence the chromatin landscape, thus dictating whether regions of genomic DNA are silenced or expressed. Chahrour et al. show that MeCP2 is indeed enriched at the promoters of several genes that are either activated or repressed by MeCP2 in the hypothalamus. These data are consistent with the possibility that MeCP2 directly regulates the expression of at least a subset of the genes identified. However, because MeCP2 is highly expressed in the nucleus and likely binds widely within the genome, further approaches are needed to address whether binding of MeCP2 to specific promoters determines the expression of the targeted genes.
figure. A choice for MeCP2.
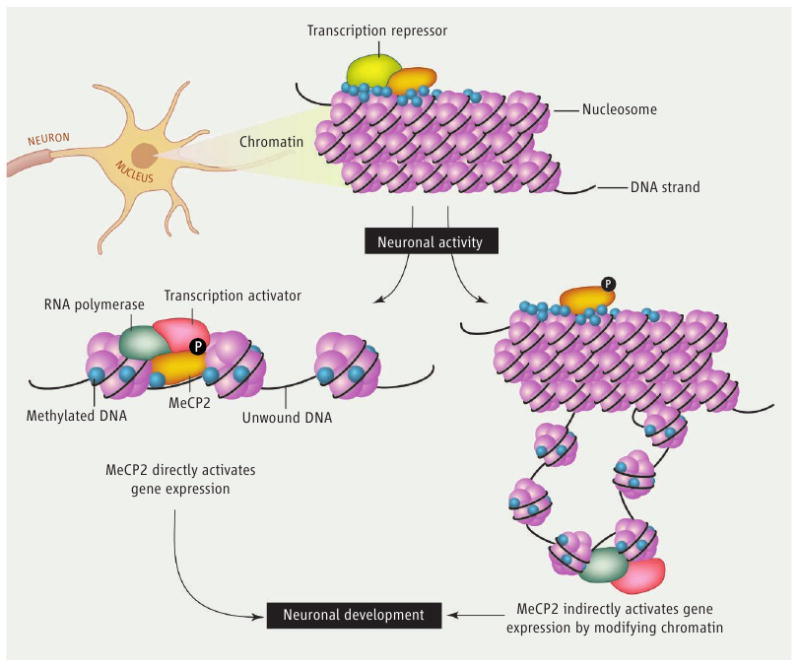
Whether MeCP2 functions as a promoter-specific transcription factor or is involved in coordinating higher-order chromatin structure, both the cellular and molecular context may influence how it modulates transcriptional repression or activation. Neuronal activity may play a central role in determining the transcriptional function of MeCP2.
The identification of thousands of genes that are either activated or repressed by MeCP2 raises the intriguing problem of how the transcriptional outcome is determined for a particular MeCP2 target gene. Chahrour et al. approach this question in two ways. First, using mass spectrometry, they identify the transcriptional activator cAMP response element–binding protein (CREB) on chromatin that is associated with MeCP2. Using chromatin immunoprecipitation, the authors verify that CREB is present at the promoter of a gene activated by MeCP2 in the hypothalamus, and is absent from the promoter of a gene repressed by MeCP2. These data suggest that interactions with regulatory complexes present at the promoters of genes bound by MeCP2 may determine whether MeCP2 activates or represses transcription. The authors also find that the promoters of genes activated by MeCP2 are enriched for CpG islands (short regions of DNA enriched in the nucleotides cytosine and guanine) as compared with those genes repressed by MeCP2. Surprisingly, although MeCP2 binds to a similar extent to the regulatory regions of both types of targeted genes, the activated promoters are lightly methylated, whereas the promoters of genes repressed by MeCP2 are more heavily methylated. These data suggest that the function of MeCP2 as an activator or a repressor, rather than the recruitment of MeCP2, may depend on the DNA methylation status of the relevant genomic region.
Consistent with the idea that MeCP2 functions as a context-specific transcriptional modulator, both MeCP2 and CREB are phosphorylated in response to neuronal activation, suggesting that dynamic regulation of these two factors may control activity-dependent gene expression (8, 9). Activity-dependent transcription coordinates the processes of postnatal synaptic development and plasticity that may be disrupted in disorders of cognitive function (10, 11). Indeed, MeCP2-deficient neurons form fewer excitatory synapses than wild-type neurons, whereas MeCP2-overexpressing neurons form more excitatory synapses (12), suggesting that activity-dependent processes of neuronal development are regulated by MeCP2. If so, the products of the genes identified by Chahrour et al. may function in aspects of late neuronal development that depend on neuronal activity, such as synapse formation. Alternatively, genetic mutations that alter MeCP2 protein abundance or function might first affect synapse number through a distinct mechanism, and the broad transcriptional changes seen in the MeCP2-deficient or -overexpressing animals may instead reflect an indirect effect secondary to perturbation of neuronal function. Future work that links the binding of MeCP2 to specific sequences of DNA in regulating particular genes will be required to distinguish between direct and indirect effects on gene expression.
The finding by Chahrour et al. that MeCP2 may have a role beyond simple transcriptional repression comes at a time when the dynamic nature of epigenetic modifications of the genome, including DNA methylation, is being newly appreciated (13, 14). Deeper understanding of the modulatory role of MeCP2 on gene expression will help elucidate the molecular nature of the neuronal dysfunction that underlies neurodevelopmental disorders such as Rett syndrome and other autism spectrum disorders.
Acknowledgments
This work was supported by NIH grant NS048276 (M.E.G.).
Footnotes
A factor implicated in autism spectrum disorders can both suppress and activate the expression of genes involved in neurodevelopment.
References and Notes
- 1.Sebat J, et al. Science. 2007;316:445. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chahrour M, Zoghbi HY. Neuron. 2007;56:422. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Lewis JD, et al. Cell. 1992;69:905. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 4.Chahrour M, et al. Science. 2008;320:1224. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Proc Natl Acad Sci USA. 2002;99:15536. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Georgel PT, et al. J Biol Chem. 2003;278:32181. doi: 10.1074/jbc.M305308200. [DOI] [PubMed] [Google Scholar]
- 7.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Nat Genet. 2005;37:31. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 8.Kornhauser JM, et al. Neuron. 2002;34:221. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Z, et al. Neuron. 2006;52:255. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zoghbi HY. Science. 2003;302:826. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 11.Flavell SW, Greenberg ME. Annu Rev Neurosci. 2008;31 doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao HT, Zoghbi HY, Rosenmund C. Neuron. 2007;56:58. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kangaspeska S, et al. Nature. 2008;452:112. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 14.Metivier R, et al. Nature. 2008;452:45. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]