

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates many of the biological and toxic effects of halogenated aromatic hydrocarbons (HAHs), polycyclic aromatic hydrocarbons (PAHs), and other structurally diverse ligands. While HAHs are several orders of magnitude more potent in producing AhR-dependent biochemical effects than PAHs or other AhR agonists, only the HAHs have been observed to produce AhR-dependent toxicity in vivo. Here we have characterized the dissociation of a prototypical HAH ligand ([3H] 2,3,7,8-tetrachlorodibenzo-p-dioxin [TCDD]) and PAH-like ligand ([3H] β-naphthoflavone [βNF]) from the guinea pig, hamster, mouse, and rat hepatic cytosolic AhR in order to elucidate the relationship between the apparent ligand-binding affinities and the divergent potency of these chemicals. Both compounds dissociated very slowly from the AhR with the amount of specific binding remaining at 96 h ranging from 53% to 70% for [3H]TCDD and 26% to 85% for [3H] βNF, depending upon the species examined. The rate of ligand dissociation was unaffected by protein concentration or incubation temperature. Preincubation of cytosol with 2,3,7,8-tetrachlorodibenzofuran, carbaryl, or primaquine, prior to the addition of [3H]TCDD, shifted the apparent IC50 of these compounds as competitive AhR ligands by ∼10- to 50-fold. Our results support the need for reassessment of previous AhR ligand-binding affinity calculations and competitive binding analysis since these measurements are not carried out at equilibrium binding conditions. Our studies suggest that AhR binding affinity/occupancy has little effect on the observed differences in the persistence of gene expression by HAHs and PAHs.
Keywords: Ah receptor; 2,3,7,8-tetrachlorodibenzo-p-dioxin; TCDD; β-naphthoflavone
The aryl hydrocarbon receptor (AhR) is a ligand-activated member of the basic-helix–loop-helix PAS domain superfamily of proteins that mediates the biological and toxic effects of halogenated aromatic hydrocarbons (HAHs) (Safe, 1990; Schmidt and Bradfield, 1996). HAHs are a diverse group of widespread, persistent, and toxic environmental contaminants that include the polychlorinated dibenzo-p-dioxins, dibenzofurans, biphenyls, and related compounds (Safe, 1990). The most potent HAH activator of the AhR and AhR-dependent effects is that of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin). Mechanistically, the AhR resides in the cytosol as part of a complex containing two molecules of heat shock protein 90 (hsp90) and other proteins (Petrulis and Perdew, 2002). Upon ligand binding, the ligand:AhR protein complex translocates into the nucleus wherein it is released from its associated proteins and heterodimerizes with the aryl hydrocarbon receptor translocator (ARNT) protein (Hankinson, 1995; Whitlock, 1999). Binding of the ligand:AhR:ARNT complex to its specific DNA recognition site, termed the dioxin response element, stimulates transcription of adjacent downstream genes (Denison et al., 1998a; Schmidt and Bradfield, 1996; Whitlock, 1999).
Interestingly, while a closely related class of compounds, the polycyclic aromatic hydrocarbons (PAHs), can bind to and activate the AhR and produce the same AhR-mediated biological effects as HAHs, these compounds typically bind with a much lower affinity and do not cause AhR-dependent toxicity (Poland and Glover, 1980; Poland and Knutson, 1982). While it has been reported that TCDD and the potent PAH AhR ligand 3-methylcholanthrene (3-MC) were equally efficacious inducers of AhR-dependent CYP1A1 in rat liver, TCDD was ∼30,000 more potent (Poland and Glover, 1974). In addition, it was observed that CYP1A1 gene induction by 3-MC was transient, with its associated enzymatic activity returning to control levels by 8 days, compared to persistent maximal induction of CYP1A1 activity for > 35 days by TCDD.
Subsequent studies demonstrated that in mouse hepatoma (Hepa1c1c7) cells, TCDD was ∼1000-fold more potent as an inducer of CYP1A1 activity as compared to 3-MC despite only having a three to fourfold greater binding affinity for the AhR (Riddick et al., 1994). Other AhR agonists, such as the tryptophan- and indole-derived compounds 6-formylindolo [3,2-b]carbazole and indolo[3,2-b]carbazole are very potent inducers with extremely high affinities for the AhR, but, like 3-MC,theyonly transiently induce AhR-dependent gene expression (Chen et al., 1995; Wei et al., 1998). Reports such as these raise questions as to whether the inability of PAHs to produce AhR-dependent toxicity is related to their binding affinity, metabolic stability, and/or other factor(s).
In addition to binding kinetics studies defining the affinity of various HAHs and PAHs for the AhR, characterization of ligand binding indicated that TCDD and other HAHs bind the receptor essentially irreversibly within the window of time that binding analysis is carried out (Bradfield et al., 1988; Bunce et al., 1988; Farrell et al., 1987; Henry and Gasiewicz, 1993). These results raise questions as to the appropriateness of using traditional equilibrium binding affinity calculations for the AhR. However, the majority of these studies on AhR ligand–binding persistence have focused on rat hepatic cytosolic AhR and on HAHs with essentially no information as to whether similar binding characteristics are observed for other species or AhR ligands. Here, we have further characterized persistent binding of prototypical HAH (TCDD) and PAH-like (β-naphthoflavone [βNF]) ligands to the cytosolic AhR from several species.
Materials and Methods
Materials
[3H]TCDD (18 Ci/mmol) and 2,3,7,8-tetrachlorodibenzofuran (TCDF) were generously provided by Dr Steven Safe (Texas A&M University). [3H] βNF (19 Ci/mmol), manufactured by ChemSyn Laboratories (Lenexa, KS), was a kind gift from Dr Mark Hahn (Woods Hole Oceanographic Institute; Butler et al., 2001). [3H]17β-Estradiol (E2; 28 Ci/mmol) was purchased from NEN Life Science (Boston, MA). Dextran (size 124,000 MW average) and diethylstilbestrol (DES) were from Sigma (St Louis, MO), Norit A charcoal from JT Baker (Phillipsburg, NJ), and BiogelHTP hydroxyapatite (HAP) from BioRad (Hercules, CA). Carbaryl (1-naphthyl N-methylcarbamate) was obtained from Chem Service (Media, PA), and primaquine diphosphate from Aldrich (Milwaukee, WI).
Animals and preparation of cytosol
Male Hartley guinea pigs (400 g), Sprague–Dawley rats (200 g), C57BL/6 mice (20 g), and Golden Syrian hamsters (125 g) were obtained from Charles River Laboratories (Wilmington, MA). All animals were exposed to 12 h of light:12 h of dark daily and given free access to food and water. Calf uterine tissue was obtained from a local slaughter house (Dixon, CA). Hepatic and uterine cytosol was prepared in HEDG (25mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 1mM ethylenediaminetetraacetic acid, 1mM dithiotreitol, 10% [v/v] glycerol) buffer as previously described (Denison et al., 2002b) and protein concentrations determined by dye-binding (Bradford, 1976). Cytosol was stored at −80°C until use.
AhR binding and dissociation experiments
For standard binding experiments, hepatic cytosol from the indicated species (2 mg protein/ml) was incubated with 2nM [3H]TCDD (a saturating concentration [data not shown]), in the presence or absence of 200nM TCDF, for 2 h at 20°C and [3H]TCDD-specific binding determined by the HAP assay as previously described in detail (Denison et al., 2002b). To insure receptor saturation for incubations with higher protein concentrations (5–10mg/ml), 10nM [3H]TCDD and 1μM TCDF were used. For dissociation experiments, after the initial [3H]TCDD/TCDF incubation, samples (1.25 ml) were transferred onto dextran-coated charcoal (DCC) pellets (1 mg:10 mg in HEDG) and incubated at room temperature for 10 min with periodic vortexing to remove free and loosely bound ligand. The “stripped” supernatants were collected following centrifugation and TCDF added to each incubation to a final concentration of 200nM. The time of readdition of TCDF was defined as the zero time point in these studies. Aliquots (200 μl) were taken at the indicated times and [3H]TCDD-specific binding was determined by HAP binding. Dissociation experiments with βNF were the same as described above except 10nM [3H] βNF (a saturating concentration [data not shown]) and 1μM TCDF were used in the incubations. For preincubation binding experiments, hepatic cytosol was incubated with increasing concentrations of the indicated chemical added either simultaneously with 2nM [3H]TCDD or for 1 h prior to the addition of [3H]TCDD. This was followed by the standard 2-h incubation for binding experiments, for a total incubation time of 3 h at 20°C, and [3H]TCDD-specific binding determined by HAP analysis. IC50s from the resulting data were calculated using a Pseudo-Hill plot (Taylor and Insel, 1990).
Estrogen receptor (ER) dissociation experiments
Calf uterine cytosol (2 mg/ml) was incubated with 2nM [3H]E2 in the presence or absence of 100-fold excess DES for 2 h at 20°C. Samples (500 μl) were transferred onto DCC pellets (1 mg:10 mg in HEDG) and incubated at room temperature for 10 min with periodic vortexing to remove free and loosely bound ligand. The “stripped” supernatant was collected following centrifugation, DES was added to a final concentration of 200nM, and samples were further incubated at 20°C. The time of readdition of DES was defined as the zero time point in these studies. Aliquots (250 μl) were taken at the indicated times, transferred to a DCC pellet (1 mg:10 mg in HEDG), and specific binding of [3H]E2 determined as we have previously described (Lashley et al., 2002).
Results
Thermal stability of occupied versus unoccupied AhR
In order to examine the persistence of AhR ligand binding, we first determined the thermal stability of the AhR over time. Previous studies have reported that unoccupied rat AhR is susceptible to thermal inactivation as revealed by a progressive decrease in the ability of cytosol to specifically bind [3H]TCDD when incubated in the absence of ligand for increasing times at different temperatures (Bunce et al., 1988; Kester and Gasiewicz, 1987). In addition, these studies reported that incubation with the ligand (TCDD) stabilized the AhR against thermal inactivation. In order to obtain information about AhR stability for our dissociation analysis, the stability of occupied and unoccupied AhR was examined at 20°C, the temperature of our standard AhR ligand–binding assay, and at 4°C, a temperature at which the AhR is inherently more stable. To be able to compare our analysis to that of previous studies, aliquots of rat hepatic cytosol incubated at each temperature were taken at the indicated times and mixed with [3H]TCDD in the presence or absence of a 100-fold molar excess of TCDF. After an additional 2 h incubation, [3H]TCDD-specific binding was determined by HAP analysis (Fig. 1). Unoccupied receptor was more susceptible to thermal inactivation at 20°C as compared to 4°C. At 20°C, more than 80% of [3H]TCDD-specific binding to rat hepatic cytosol was lost by 8 h whereas at 4°C, it took 60 h to lose the same amount of binding activity. These results are consistent with previous studies that reported half lives ranging from ∼40 min at 30°C to ∼31.9 h at 5°C (Bunce et al., 1988; Kester and Gasiewicz, 1987).
FIG. 1.
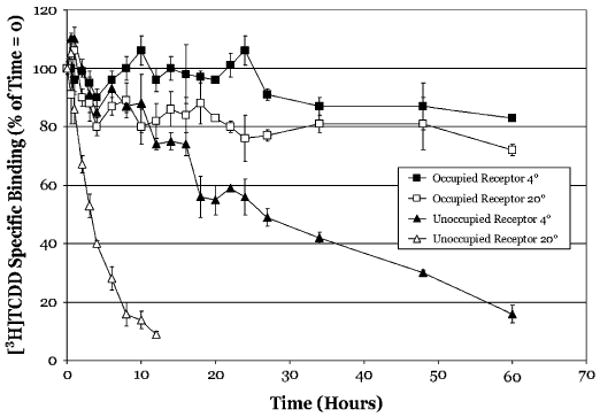
Ligand binding confers thermal stability to Sprague–Dawley rat hepatic cytosolic AhR. Unoccupied receptor: Rat hepatic cytosol (2 mg/ml) was incubated at the indicated temperature and aliquots (500 μl) were removed at the indicated times incubated with 2nM [3H]TCDD in the absence or presence of 200nM TCDF for 2 h. Occupied receptor: Rat hepatic cytosol (2 mg/ml) was incubated with 2nM [3H]TCDD in the absence or presence of 200nM TCDF for 2 h, stripped with DCC to remove free and loosely bound ligand, and then incubated with cold TCDF (to 200nM) for the indicated times at the indicated temperatures. For both unoccupied and occupied receptor incubations, specific binding of [3H]TCDD was determined using the HAP assay and data are presented as the mean ± SD of triplicate incubations.
To confirm the effect of temperature on the dissociation of the [3H]TCDD–AhR complex, rat hepatic cytosol was first incubated for 2 h with [3H]TCDD in the presence or absence of 100-fold molar excess TCDF at 4°C or 20°C. Samples were then transferred to a DCC pellet to remove free and loosely bound ligand, TCDF added to the supernatant (to 200nM), and further incubated at 4°C or 20°C. The time of readdition of TCDF was defined as the time zero point for these studies. Aliquots were taken at the indicated times and [3H]TCDD-specific binding was determined by HAP assay. Similar to previous studies, ligand-bound receptor was significantly more resistant to thermal inactivation than unoccupied receptor (Fig. 1). At 60 h of incubation at 20°C and 4°C, only 30% and 20% of [3H]TCDD-specific binding was lost, respectively. Not only do these results confirm that TCDD binding confers both stability and resistance to thermal inactivation to the rat AhR (with respect to ligand binding), but the minimal loss in [3H]TCDD-specific binding over time also demonstrates that the [3H]TCDD ligand does not readily dissociate from the rat AhR at either temperature.
Persistent binding of [3H]TCDD to hepatic cytosol of various species
TCDD has been reported to bind to the AhR with a relatively high affinity (1pM–10nM, depending on the species and assay employed (Bradfield et al., 1988; Denison et al., 1986a) and for the rat hepatic AhR, TCDD dissociates very slowly with a T1/2 on the order of ∼70 h (Henry and Gasiewicz, 1993). To determine whether this slow rate of ligand dissociation is a characteristic of AhRs from other species, ligand dissociation experiments identical to those described above were carried out using hepatic cytosol from Hartley guinea pigs, Golden Syrian hamsters, C57BL/6 mice, and Sprague–Dawley rats (Fig. 2). Similar to the above results, [3H]TCDD dissociated slowly from the AhR of all the species tested, especially within the 2 h time frame of standard competitive ligand-binding assays. At 3 h (Fig. 2A), [3H]TCDD-specific binding to guinea pig, hamster, mouse, and rat cytosol, was 102 ± 11%, 82 ± 1%, 86 ± 6%, and 92 ± 6% of that at time zero, respectively. In addition, to characterize better the time course of persistent binding (i.e., resistance to ligand dissociation), identical experiments were carried out and specific binding determined at times out to 96 h after the addition of TCDF. At 96 h, [3H]TCDD-specific binding was 70 ± 5%, 64 ± 5%, 53 ± 15%, and 65 ± 11% of that at time zero for the species listed above, respectively (Fig. 2B). Taken together, a small amount of ligand (10–20%) dissociated within 3 h from the mouse, rat, and hamster cytosolic AhR, and the remainder dissociated very slowly throughout the remaining experimental time period. There was no apparent ligand dissociation from the guinea pig AhR during the 3-h incubation period.
FIG. 2.
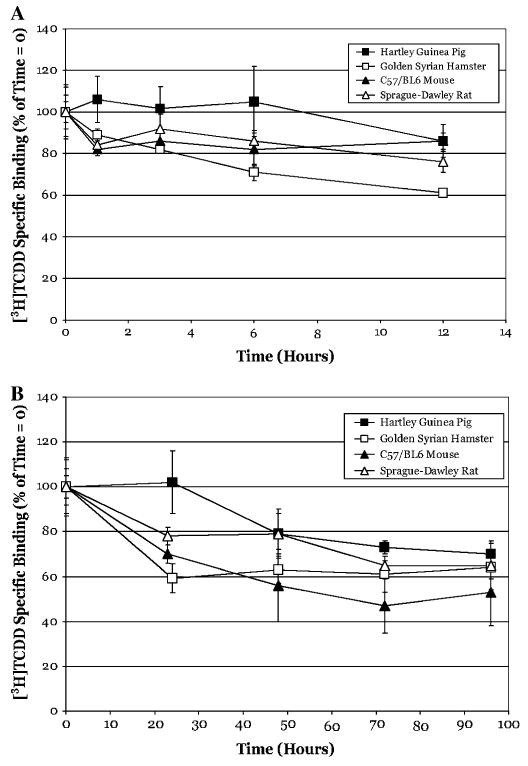
[3H]TCDD does not readily dissociate from the hepatic cytosolic AhR from various species. Hepatic cytosol (2 mg/ml) from the indicated species was incubated with 2nM [3H]TCDD in the absence or presence of 200nM TCDF for 2 h, charcoal stripped to remove free and loosely bound ligand and then further incubated with cold TCDF (to 200nM) at 20°C. Specific binding of [3H]TCDD was determined by the HAP assay at the indicated times out to 12 h (A) or 96 h (B). Data are presented as the mean ± SD of at least quadruple incubations.
While some of the reduction in ligand binding could result from AhR inactivation/denaturation, our results clearly demonstrate the persistence of [3H]TCDD-specific binding to hepatic cytosolic AhR for all of the species studies here. One possible explanation for the slow rate of ligand dissociation could be that the low cytosolic protein concentration typically used in binding experiments (2 mg/ml) may not favor dissociation of extremely hydrophobic ligands, such as TCDD, into the aqueous incubation mixture. To test this possibility, we examined the rate of ligand dissociation from guinea pig hepatic cytosolic AhR (the species with the most persistent ligand binding) in incubations containing increasing cytosolic protein concentration (Fig. 3). These results revealed no significant difference between the protein concentrations examined and the relative rate of ligand dissociation from the AhR for up to 48 h.
FIG. 3.
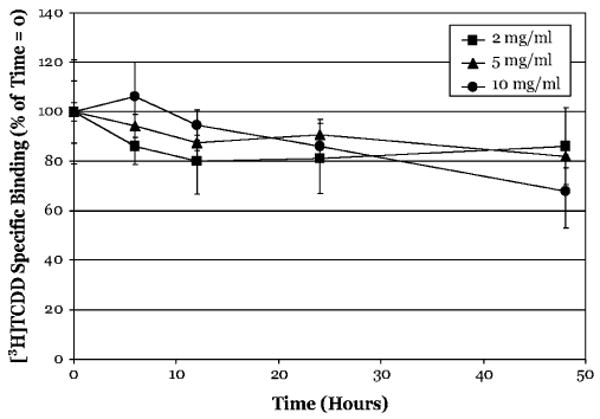
Protein concentration does not affect the persistence of [3H]TCDD binding to Hartley guinea pig hepatic cytosolic AhR. Guinea pig hepatic cytosol of increasing protein concentrations (2–10 mg/ml) was incubated with [3H]TCDD in the absence or presence of 100-fold excess TCDF for 2 h. The final concentration of [3H]TCDD was 2nM for the 2 mg/ml samples, and 10nM for the 5 and 10 mg/ml samples. After 2 h, incubation were charcoal stripped to remove free and loosely bound ligand and then incubated with 200nM TCDF (2 mg/ml samples) or 1μM TCDF (5–10 mg/ml samples) at 20°C. Specific binding of [3H]TCDD was determined by the HAP assay at the indicated times. Data are presented as the mean ± SD of at least triplicate incubations.
Persistent binding of [3H] βNF to hepatic cytosol of various species
The ability of other HAHs, to bind persistently to the AhR has been reported (Brown et al., 1994), but the resistance to dissociation was only inferred by the inability of [3H]TCDD to displace bound, nonradioactive PCB congeners from the AhR. Although a similar indirect approach could be taken with other AhR ligands, the availability of [3H] βNF of high specific activity (Butler et al., 2001) provided an avenue with which to test directly the persistence of binding of PAH-like ligands to the AhR. Accordingly, dissociation experiments, identical to those described above, were performed using [3H] βNF, a prototypical PAH-like ligand for the AhR. Similar to our results with [3H]TCDD, dissociation of [3H] βNF from the AhR was also very slow. At 3 h, < 10% of [3H] βNF was displaced from all three of the species tested (Fig. 4). [3H] βNF dissociation was slowest from the guinea pig AhR and more rapid from the hamster and rat AhRs with 85 ±13%, 60 ± 5%, and 26 ± 25% of specific binding remaining at 96 h for each species, respectively. These results demonstrate that βNF, like TCDD, persistently binds to the hepatic cytosolic AhR from various species, with some species differences in ligand dissociation.
FIG. 4.
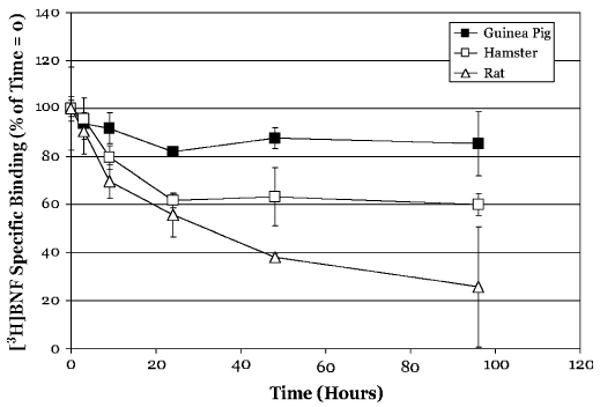
[3H]βNF does not readily dissociate from hepatic cytosolic AhR from various species. Hepatic cytosol (2 mg/ml) from the indicated species was incubated with 10nM [3H] βNF in the absence or presence of 1 μM TCDF for 2 h, charcoal stripped to remove free ligand and then incubated with cold TCDF (1 μM) at 20°C. Specific binding of [3H] βNF was determined by the HAP assay at the indicated times. Data are presented as the mean ± SD of at least triplicate incubations.
It may be questioned as to whether the dissociation we observe at the nonphysiological temperature of 20°C is biologically relevant and as such, we examined the rate of ligand dissociation from guinea pig hepatic cytosolic AhR at 37°C. These experiments revealed that the rate of dissociation for [3H]TCDD at 37°C was similar to that observed at 20°C (compare Figs. 2b and 5). Interestingly, the dissociation of [3H] βNF from the AhR was actually significantly reduced at 37°C as compared to 20°C (compare Figs. 4 and 5). While the reason for the more persistent binding of βNF at the elevated temperature remains to be determined, these results do confirm that the persistent binding results we obtained are not an artifact of the reduced temperature of the binding reaction.
FIG. 5.
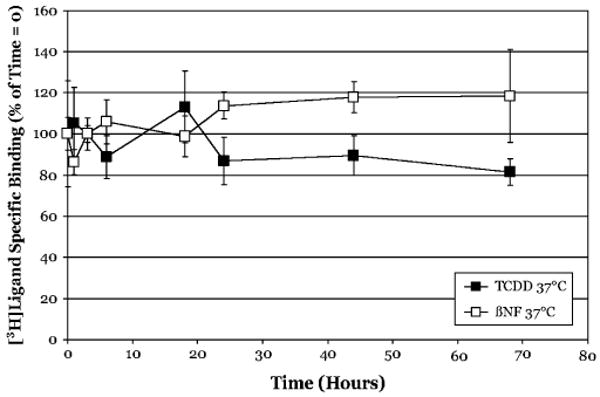
Persistent binding of [3H]TCDD and [3H]βNF to guinea pig hepatic cytosolic AhR is observed at 37°C. Guinea pig hepatic cytosol (2 mg/ml) was incubated with 2nM [3H]TCDD in the absence or presence of 200nM TCDF, or 10nM [3H]βNF in the absence or presence of 1μM TCDF for 2 h, charcoal stripped to remove free and loosely bound ligand and then incubated with TCDF (200nM or 1 μM, respectively, at 37°C). Specific binding of [3H]TCDD and [3H] βNF was determined by the HAP assay at the indicated times. Data are presented as the mean ± SD of at least triplicate incubations.
[3H]E2 dissociates relatively rapidly from the bovine uterine cytosolic ER
The relatively rapid dissociation of ligands from other nuclear receptors has been previously reported, with T1/2 values on the order of 2–4 h for the estrogen and progesterone receptor (Boctor et al., 1983; Ogle, 1986; Wooge et al., 1992). To confirm that the persistence of AhR ligand binding we observed was not simply an artifact of our experimental system, but is a characteristic of the AhR, we examined the dissociation of [3H]E2 from calf uterine ER. Experimental controls were the same as that for the AhR experiments and incubations consisted of uterine cytosol treated with [3H]E2 (2nM) in the presence or absence of 100-fold molar excess unlabeled DES for 2 h at 20°C, followed by charcoal stripping of free and loosely bound ligand and the readdition of unlabeled DES to a final concentration of 200nM. Aliquots were taken at various times and [3H]E2 specific binding determined using the DCC binding assay with the time of readdition of DES defined as the zero time point. In contrast to the persistent binding of ligands to the AhR, dissociation of [3H]E2 from the ER was significantly faster with ∼40% of [3H]E2-specific binding lost by 3 h, ∼60% lost by 6 h, and ∼80% lost by 24 h (Fig. 6). These results are consistent with previously published dissociation rates for [3H]E2 and confirm the validity of the experimental system we used to measure ligand dissociation and the unique ligand-binding persistence of the AhR.
FIG. 6.
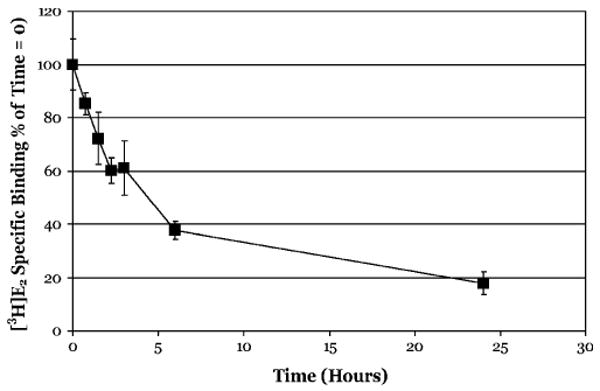
[3H]E2 dissociates relatively rapidly from bovine uterine cytosolic ER. Bovine calf uterine cytosol (2 mg/ml) was incubated with 2nM [3H]E2 in the absence or presence of 200nM DES for 2 h, charcoal stripped to remove free and loosely bound ligand and incubated with cold DES (200nM) at 20°C. Specific binding of [3H]E2 was determined at the indicated time using the DCC method as described under the “Materials and methods” section. Data are presented as the mean ± SD of at least triplicate incubations.
Effect of persistent AhR ligand binding on analysis and interpretation of binding results
Our dissociation experiments indicate that both a high affinity ligand (TCDD) and a lower affinity ligand (βNF) bind the hepatic cytosolic AhR from several species relatively persistently. This has significant implications with regard to analysis of typical AhR competitive ligand-binding experiments and interpretation of their results. The persistent binding of ligands to the AhR suggests that competitive ligand-binding assays may actually represent a “race between ligands to bind first,” with higher affinity ligands binding preferentially if the ligands are at equal concentrations. If this is true, then the actual binding affinity calculated from competitive binding experiments between TCDD and a weaker ligand may result in a significant underestimation of the actual affinity of a competitive ligand for the AhR. To test this, guinea pig hepatic cytosol was incubated with increasing concentrations of TCDF added either simultaneously with [3H]TCDD, as in a typical competitive binding experiment, or allowed to preincubate for 1 h prior to the addition of [3H]TCDD, followed by a 2-h incubation and determination of [3H]TCDD-specific binding. As was expected, the competitive binding curve for TCDF upon 1-h preincubation was shifted to the left by ∼50-fold, indicating a greater apparent binding affinity than suggested by the simultaneous addition experiments (Fig. 7). The calculated IC50s for TCDF were 1.6nM and 44.3pM for the simultaneous addition and preincubation samples, respectively (Table 1). This result raises questions as to whether the apparent relative affinities measured for other ligands using radioligand displacement from the AhR are also significantly greater than currently thought.
FIG. 7.
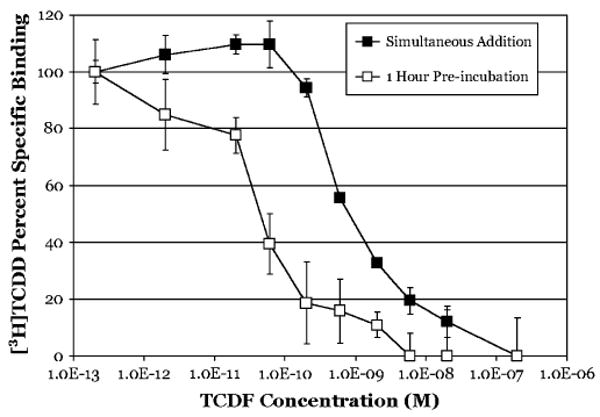
TCDF has a higher apparent binding affinity for guinea pig hepatic cytosolic AhR when incubated prior to [3H]TCDD addition. Simultaneous addition: Guinea pig hepatic cytosol (2 mg/ml) was incubated with 2nM [3H]TCDD in the absence or presence of TCDF at the indicated concentration for 3 h at 20°C. Preincubation: Guinea pig hepatic cytosol (2 mg/ml) was incubated in the absence or presence of the indicated concentration of TCDF for 1 h at 20°C followed by the addition of 2nM [3H]TCDD and further incubation for 2 h. Specific binding of [3H]TCDD was determined by the HAP assay at the indicated times. Data are presented as the mean ± SD of at least triplicate incubations.
TABLE 1. Preincubation Shifts the Apparent IC50 of Ligands for the AhR.
| Compound | IC50 (M) | |
|---|---|---|
| Simultaneous addition | Preincubation | |
| TCDF | 1.6 × 10−9 | 4.4 × 10−11 |
| Carbaryl | 1.5 × 10−5 | 3.1 × 10−6 |
| Primaquine | 3.2 × 10−5 | 4.7 × 10−6 |
Note. Simultaneous addition: Guinea pig hepatic cytosol (2 mg/ml) was incubated with 2nM [3H]TCDD in the absence or presence of increasing concentrations of the indicated compounds for 3 h at 20°C. Preincubation: Guinea pig hepatic cytosol (2 mg/ml) was incubated in the absence or presence of increasing concentrations of the indicated compounds for 1 h at 20°C followed by the addition of 2nM [3H]TCDD and further incubation for 2 h. Specific binding of [3H]TCDD in all incubations was determined by the HAP assay and IC50s calculated from a Pseudo-Hill plot of the competition displacement curves.
Numerous compounds have been described that can activate the AhR yet do not appear to compete with [3H]TCDD for binding to the AhR (Denison and Nagy, 2003; Denison et al., 1999; Fontaine et al., 1999; Ledirac et al., 1997). While we have previously documented that there are some technical issues with regards to the specific binding assay methodologies used in these experiments, the binding persistence we observe here could contribute to the inability of other investigators to demonstrate competitive ligand binding by these so-called “ligand-independent” or nonbinding activators of the AhR. One interpretation is that these chemicals are actually very weak AhR ligands that do not effectively compete with [3H]TCDD for ligand binding. If this is true, then utilizing the preincubation approach demonstrated above should allow us to demonstrate the ability of some of these nonbinding AhR activators to bind effectively to the AhR. In these experiments, three compounds (thiabendazole, carbaryl, and primaquine) were added to a final concentration of 100μM to guinea pig hepatic cytosol either 1 h before, or simultaneously with [3H]TCDD and specific binding of [3H]TCDD determined as described above. Interestingly, using the binding assay method we previously optimized for lower affinity ligands (Denison et al., 1998b), we actually observe that each of these chemicals can displace [3H]TCDD binding to the AhR even when added simultaneously with [3H]TCDD (Fig. 8). Moreover, as expected, preincubation with either carbaryl or thiabendazole at this concentration resulted in a significantly lower amount of [3H]TCDD binding under the experimental conditions used. In contrast, primaquine was equally effective at preventing radio-ligand binding in both incubation formats. Concentration-dependent binding inhibition experiments with carbaryl and primaquine revealed that each compound was a more efficient competitor when added prior to [3H]TCDD with an increase in the calculated IC50 for binding of each compound of ∼10-fold (Table 1). These results also support the use of preincubation in [3H]TCDD-binding experiments to aid in the identification of weak AhR ligands since equilibrium is not reached in a typical experiment due to the very slow dissociation of the radioligand.
FIG. 8.
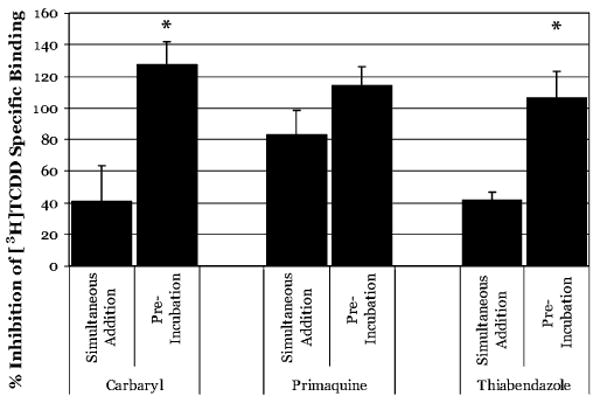
Binding of “ligand-independent” AhR activators to the guinea pig hepatic cytosolic AhR. Simultaneous addition: Guinea pig hepatic cytosol (2 mg/ml) was incubated with 2nM [3H]TCDD in the absence or presence of 100μM of the indicated compounds for 3 h at 20°C. Preincubation: Guinea pig hepatic cytosol (2 mg/ml) was incubated in the absence or presence of 100μM of the indicated compounds for 1 h at 20°C followed by the addition of 2nM [3H]TCDD and further incubation for 2 h. Specific binding of [3H]TCDD determined by the HAP assay. Data are presented as the mean ± SD of at least triplicate incubations. The asterisk (*) identifies those compounds for which significantly more [3H]TCDD-specific binding was inhibited by preincubation as compared to simultaneous addition, as determined by the Student's two-tailed t-test (p < 0.05).
Discussion
In this study, we have demonstrated the persistent binding (i.e., extremely slow dissociation) of [3H]TCDD to the guinea pig, mouse, hamster, and rat hepatic cytosolic AhR. Ligand dissociation from the rat AhR was previously shown to have a biphasic pattern (Bradfield et al., 1988; Henry and Gasiewicz, 1993). This two component dissociation rate was suggested to result from an initial relatively fast dissociation of ligand from nontransformed receptor and a slower rate of dissociation from both the monomeric (ARNT-free) and heterodimeric (ARNT-bound) forms of the receptor. While our results suggest a similar two component dissociation rate for rat, mouse, and hamster AhR, we had an insufficient number of data points during the initial dissociation time to allow rate calculations. It is possible that the differences in [3H]TCDD dissociation rates between the cytosolic AhR of the species tested that occurred primarily during the initial phase of our time course study may be related to species differences in AhR transformation efficiencies. The relative TCDD-induced transformation efficiencies for AhR from these four species in order of highest to lowest is guinea pig > rat > hamster > mouse (Bank et al., 1992; Denison et al., 1991). However, if ligand dissociation was directly related to transformation state of the AhR as suggested for the rat AhR (Henry and Gasiewicz, 1993), then the rank order of ligand dissociation for these four species would be expected to be inversely related to their rank order of transformation efficiency. Consistent with this hypothesis, the greatest degree of transformation and most persistent ligand binding of both [3H]TCDD and [3H] βNF was observed with guinea pig cytosolic AhR, while that of the mouse AhR, which transforms relatively poorly in vitro (< 10%, Bank et al., 1992; and unpublished observations), demonstrated the greatest degree of ligand dissociation in these experiments. This low efficiency of transformation of the mouse AhR has been suggested to be due to the extreme resistance of Hsp90 to dissociate from the mouse AhR, a phenomenon not observed for the AhR from other species (Denison, 1992; Denison and Vella, 1990; Denison et al., 1986b). However, the rate of ligand dissociation from the AhR of the remaining species did not correlate as well with their transformation efficiencies. The lack of a consistent relationship between the degree of AhR transformation and ligand dissociation suggests the involvement of other factors, and/or the existence of unique structural differences among the AhRs of different species which remain to be identified.
Recent studies have demonstrated relatively rapid dissociation of ligand from human AhR expressed in COS cells (Ramadoss and Perdew, 2004, 2005). These results, combined with those reported by Bradfield et al. (1988), suggested that AhR ligand–binding affinity was affected by protein concentration. Accordingly, the lack of TCDD dissociation we observed could result not from persistent ligand binding, but from the extreme lipophilicity of this ligand such that it would remain more tightly bound to the AhR because it is essentially insoluble in the aqueous incubation media. However, several lines of evidence obtained here argue against this possibility. First, if the lack of ligand (TCDD) dissociation was due to its lipophilicity, then it would be expected that the off rate of βNF, a more water soluble compound, would be much faster than that for TCDD. Given that the log Kow for TCDD is 6.80 (Gobas et al., 1988) whereas the calculated log Kow for βNF is 4.68 (Meylan and Howard, 1995), βNF is approximately two orders of magnitude more soluble in water than TCDD. However, our results show that both compounds persistently bind hepatic AhR from several species. In fact, at 37°C, βNF dissociated somewhat slower from guinea pig hepatic cytosolic AhR than TCDD. In addition, increasing the protein concentration of the binding incubation in order to provide more hydrophobic and nonspecific binding sites had no significant effect on [3H]TCDD-binding persistence. While a previous study demonstrated that increasing C57BL/6J cytosolic protein concentrations could alter the apparent dissociation constant (Kd) values determined by Scatchard analysis (Bradfield et al., 1988), their results show that this change was due to an alteration in the association rate constant (k1) and not the apparent dissociation rate constant (k−1), and this would be consistent with our results. In contrast, a recent study reported a protein concentration-dependent dissociation of the photoaffinity AhR ligand 2-azido-3-[(125)I]iodo-7,8-dibromodibenzo-p-dioxin from the human, but not the mouse AhR (Ramadoss and Perdew, 2004). Site-directed mutagenesis, and additional studies using mouse and human AhR chimeras (Ramadoss and Perdew, 2005), indicate that this species-specific difference was attributed to valine 381 of the human AhR (Ala375 in the mouse AhR), an amino acid within the ligand-binding pocket responsible for its 10-fold lower binding affinity (Ema et al., 1994; Ramadoss and Perdew, 2004). While this previously reported lack of ligand dissociation from the mouse AhR is consistent with our results, the relatively rapid dissociation of ligand from the human AhR may reflect a species-specific difference in persistent binding of ligands and may be related to the lower ligand-binding affinity of the human AhR. However, this remains to be determined.
Our studies demonstrate persistent binding of ligand to the AhR in vitro and suggest the ability of the AhR to be persistently activated, however, the AhR is known to be relatively rapidly degraded by the proteasome in vivo and in cells in culture following ligand binding (Pollenz, 1996, 2002; Pollenz and Buggy, 2006; Song and Pollenz, 2002). As might be expected, the dramatic reduction in measurable AhR levels is directly correlated with reductions in the maximal level of induced AhR-dependent gene expression (Song and Pollenz, 2002). While effect of persistent ligand binding to the AhR we observe may be countered to some degree by AhR degradation, it should be noted that these and other studies demonstrate that AhR is not completely eliminated and that ligand- and AhR-dependent gene expression continues, albeit at a reduced level. The ability of the guinea pig AhR to bind to DNA up to 48 hours after ligand addition (data not shown) indicates that the persistently bound ligand (TCDD) maintains the AhR in its transformed DNA binding state. However, given reported differences in the overall rate and amount of ligand-dependent AhR degradation in various cells in culture and in vivo (Pollenz, 1996; Pollenz and Buggy, 2006; Pollenz and Dougherty, 2005), it is likely that persistent ligand binding to and activation of the AhR could contribute to cell-, tissue-, and species-specific differences in AhR-dependent gene expression.
The persistent binding of both TCDD and βNF to the AhR has interesting implications with regard to our understanding the mechanism of AhR-dependent toxic and biological effects of HAH versus non-HAH ligands. While, all AhR agonists induce AhR-dependent gene expression (Denison et al., 1999; Poland and Glover, 1974; Riddick et al., 1994), their potencies can vary by several orders of magnitude and only HAHs have been observed to produce the AhR-dependent battery of toxic effects associated with TCDD exposure. A number of reports have examined the mechanistic reason for this. Possible explanations include differences in binding affinity, binding persistence, AhR transformation and DNA binding, recruitment of transcriptional cofactors, gene expression and/or embolic stability of the ligands. The results of our studies suggest that it is unlikely that the differences in toxicity of HAH and non-HAH AhR agonists are due to their persistence of ligand binding. While affinity may play a role in the initial binding of ligand to AhR, dictating how much AhR is occupied at a given ligand concentration, loss of ligand from the AhR does not appear to be significantly different once the AhR has transformed. The more likely difference is that the less potent PAHs and other compounds are relatively quickly eliminated by both basal and AhR-induced metabolizing enzymes in exposed cells. Once the pool of PAH ligand is reduced, less is available to bind newly synthesized AhR and the induction response is progressively reduced. Interestingly, the ability of PAHs (i.e., BNF and 3-MC) produce persistent induction of CYP1A1 and/or CYP1A1-dependent enzymatic activity (in some cases for up to 45 days) has been reported by several laboratories (Brauze, 2004; Moorthy, 2000 and references therein). While the mechanism for this persistent induction response is not clear, it appears to occur in an AhR independent manner (Moorthy, 2000). In contrast, HAH ligands that are metabolically stable produce persistent gene activation as a result of continual availability for binding and activation of newly synthesized AhR and this can lead to toxic effects. This hypothesis is supported by a study showing prolonged induction of CYP1A1 mRNA expression in human keratinocytes when cotreated with βNF and the CYP1A1 suicide inhibitor 1-ethynylpryene (Berghard et al., 1992). In addition, increased in vivo AhR-dependent embryotoxicity by metabolically labile PAHs in Fundulus heteroclitus has been observed when they are treated in conjunction with a CYP1A1 inhibitor (Wassenberg and Di Giulio, 2004; Wassenberg et al., 2005). These results indicate that PAHs can produce AhR-dependent toxicity if they are not metabolized and eliminated, but instead remain available for AhR binding for an extended period of time.
From an experimental standpoint, our results also question the feasibility of using typical equilibrium binding kinetics approaches for the calculation of accurate AhR ligand–binding affinity. The expression of ligand-binding affinity as a Kd is determined by the relationship between the on and off rates of a ligand for its receptor, k1 and k-1, respectively. In the case of the AhR, accurate determination of a Kd is limited by the fact that there is little to no dissociation (k-1), especially within the time frame of standard binding experiments, typically 2 h. According to the assumptions and equations of receptor occupancy theory, in order to reach equilibrium and accurately determine an equilibrium dissociation constant for a ligand, the incubation time for a competitive binding experiment must continue for at least four to five half lives of dissociation (Taylor and Insel, 1990). Our results clearly demonstrate that this requirement is not met using current AhR competitive binding analysis protocols (1–3 h incubation), and is also not realistically practical for this system. The inappropriateness of deriving Kd values from [3H]TCDD-binding experiments is further demonstrated by the results of our preincubation experiments with TCDF. Previous studies have demonstrated that the order of addition of cold competitor and labeled ligand can affect apparent affinity measurements from competitive displacement of specific binding (Brown et al., 1994; Farrell and Safe, 1987). However, these studies were only carried out at single competitor concentrations. Here we have demonstrated that when cytosol is preincubated with increasing concentrations of TCDF for 1 h prior to the addition of labeled TCDD, the relative affinity estimated from its IC50 is increased by ∼50-fold. Accordingly, since equilibrium is not reached during the incubation time of a standard [3H]TCDD-binding experiment, absolute Kd values cannot be determined. What is actually being measured is a race between ligands competing to be “first to bind” which is based on their relative affinities and concentrations, and could be used to determine an association constant. Useful information can still be obtained from [3H]TCDD-binding experiments for determining relative Kd values for comparisons between ligands providing that they are tested using the exact same experimental protocols. However, values for different compounds cannot be directly compared from studies with different experimental designs, especially with regards to incubation time and order of chemical addition.
Although the results obtained in our studies complicate binding analyses, they also provide an avenue to address the ongoing debate regarding ligand-independent activators of the AhR. A number of reports have described compounds that can stimulate ligand-dependent AhR transformation and DNA binding, and also induce AhR-dependent gene expression, but state that these agonists must activate the AhR indirectly since they fail to displace [3H]TCDD in ligand-binding assays (Delescluse et al., 1998; Fontaine et al., 1999; Gradelet et al., 1997; Ledirac et al., 1997; Lesca et al., 1995). In contrast, other investigators have been able to demonstrate the specific binding of some of these same compounds to the AhR, carbaryl and primaquine being two examples (Backlund and Ingelman-Sundberg, 2004; Denison et al., 1998b). One possible explanation for these differing results is the ligand-binding protocol used in the former studies, particularly with regard to the concentrations of [3H]TCDD and competitor used. Consistent with our previous report, use of a modified competitive binding assay that facilitates the identification of low-affinity AhR ligands (Denison et al., 1998b), as well as preincubating cytosolic with the chemicals in question, has allowed us to demonstrate clearly the ability of carbaryl, primaquine, and thiabendazole to inhibit the binding of [3H]TCDD to the AhR. More importantly, allowing ligand to preincubate with cytosol prior to the addition of [3H]TCDD resulted in a consistent ∼10-fold shift in the IC50 for each of the compounds tested suggesting that this experimental modification of the ligand-binding protocol will be useful in demonstrating the binding of low-affinity ligands to the AhR
The lack of protein structure information on the AhR ligand-binding domain limits our ability to propose an exact mechanism for persistent ligand binding. However, using an available homology model of the AhR ligand–binding domain (Denison et al., 2002a; Procopio et al., 2002; Pandini et al., 2007) we hypothesize that ligand binding induces a conformational change in the receptor that traps the ligand within the binding pocket. The current homology model represents an hsp90-free state of the AhR, and we propose that this model represents the closed, or ligand bound, conformation of the ligand-binding domain. One can envision that in the presence of hsp90, the ligand-binding pocket is in a much more open conformation, and ligand binding within the pocket leads to a structural change that causes the pocket to close over the ligand, effectively trapping it and thus dramatically reducing ligand dissociation. This structural change, and apparent increase in binding affinity, could occur simultaneously with AhR transformation as previously suggested (Henry and Gasiewicz, 1993). While a similar “ligand trap” model has been proposed for steroid hormone receptors, this mechanism does not appear to trap their ligands as effectively as that of the AhR since ligands can more rapidly dissociate from these receptors (Notides et al., 1979; Weichman and Notides, 1977). Like the AhR, ER transformation from an inactive to active state was demonstrated to increase the apparent affinity of ligand binding and is a proposed mechanism for the biphasic dissociation of ligands from the ER (Notides et al., 1979; Weichman and Notides, 1977). We propose a ligand-binding model that is very similar to that of the ER wherein ligand can dissociate from untransformed ligand:receptor complex (LR), but once AhR is converted into its transformed state (LR*), ligand cannot readily dissociate. In this scenario, binding affinity is important in the initial interaction between ligand and receptor (LR), but once binding has induced the necessary conformational change in the AhR (LR*), all ligands, regardless of their initial affinity, would become similarly trapped within the binding pocket and not dissociate from the receptor. It remains to be determined whether ligand-dependent differences in AhR structure and/or interaction with nuclear cofactors exist that also play a role in the differential response and/or responsiveness of a given species or cell to HAHs, PAHs, and other AhR agonists.
Acknowledgments
We wish to thank Dr Steve Safe (Texas A&M University) for the [3H]TCDD, Dr Mark Hahn (Woods Hole Oceanographic Institution) for the [3H] βNF. This work was supported by grants from the National Institutes of Environmental Health Sciences (ESO12498, ESO7685, ES05707) and the California Agricultural Experiment Station.
Footnotes
For Permissions, please email: journals.permissions@oxfordjournals.org
References
- Backlund M, Ingelman-Sundberg M. Different structural requirements of the ligand binding domain of the aryl hydrocarbon receptor for high- and low-affinity ligand binding and receptor activation. Mol Pharmacol. 2004;65:416–425. doi: 10.1124/mol.65.2.416. [DOI] [PubMed] [Google Scholar]
- Bank PA, Yao EF, Phelps CL, Harper PA, Denison MS. Species-specific binding of transformed Ah receptor to a dioxin responsive transcriptional enhancer. Eur J Pharmacol. 1992;228:85–94. doi: 10.1016/0926-6917(92)90016-6. [DOI] [PubMed] [Google Scholar]
- Berghard A, Gradin K, Toftgard R. The stability of dioxin-receptor ligands influences cytochrome P450IA1 expression in human keratinocytes. Carcinogenesis. 1992;13:651–655. doi: 10.1093/carcin/13.4.651. [DOI] [PubMed] [Google Scholar]
- Boctor AM, Band P, Grossman A. Analysis of binding of [3H]Estradiol to the cytosol fraction of rat pancreas: Comparison with sites in the cytosol of uterus. Endocrinology. 1983;113:453–462. doi: 10.1210/endo-113-2-453. [DOI] [PubMed] [Google Scholar]
- Bradfield CA, Kende AS, Poland A. Kinetic and equilibrium studies of Ah receptor-ligand binding: Use of [125I]2-iodo-7,8-dibromodibenzo-p-dioxin. Mol Pharmacol. 1988;34:229–237. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brauze D. Comparison of the induction of a 4S β-naphthoflavone-binding protein, cytochrome P450 1A1 and NAD(P)H:quinone oxidoreductase in β-naphthoflavone-treated rats. Toxicol Lett. 2004;152:111–116. doi: 10.1016/j.toxlet.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Brown MM, Schneider UA, Petrulis JR, Bunce NJ. Additive binding of polychlorinated biphenyls and 2,3,7,8-tetrachlorodibenzo-p-dioxin to the murine hepatic Ah receptor. Toxicol Appl Pharmacol. 1994;129:243–251. doi: 10.1006/taap.1994.1249. [DOI] [PubMed] [Google Scholar]
- Bunce NJ, Landers JP, Safe SH. Kinetic models for association of 2,3,7,8-tetrachlorodibenzo-p-dioxin with the Ah receptor. Arch Biochem Biophys. 1988;267:384–397. doi: 10.1016/0003-9861(88)90044-6. [DOI] [PubMed] [Google Scholar]
- Butler RB, Kelley ML, Powell WH, Hahn ME, Van Beneden RJ. An aryl hydrocarbon receptor homologue from the soft-shell clam, Mya arenaria: Evidence that invertebrate AHR homologues lack TCDD and BNF binding. Gene. 2001;278:223–234. doi: 10.1016/s0378-1119(01)00724-7. [DOI] [PubMed] [Google Scholar]
- Chen YH, Riby J, Srivastava P, Bartholomew J, Denison M, Bjeldanes L. Regulation of CYP1A1 by indolo[3,2-b]carbazole in murine hepatoma cells. J Biol Chem. 1995;270:22548–22555. doi: 10.1074/jbc.270.38.22548. [DOI] [PubMed] [Google Scholar]
- Delescluse C, Ledirac N, de Sousa G, Pralavorio M, Lesca P, Rahmani R. Cytotoxic effects and induction of cytochromes P450 1A1/2 by insecticides, in hepatic or epidermal cells: Binding capability to the Ah receptor. Toxicol Lett. 1998;96-97:33–39. doi: 10.1016/s0378-4274(98)00047-2. [DOI] [PubMed] [Google Scholar]
- Denison MS. Heterogeneity of rat hepatic Ah receptor: Identification of two receptor forms which differ in their biochemical properties. J Biochem Toxicol. 1992;7:249–256. doi: 10.1002/jbt.2570070408. [DOI] [PubMed] [Google Scholar]
- Denison MS, Elferink CF, Phelan D. The Ah receptor signal transduction pathway. In: Denison MS, Helferich WG, editors. Toxicant-Receptor Interactions in the Modulation of Signal Transduction and Gene Expression. Taylor and Francis; Philadelphia: 1998a. pp. 3–33. [Google Scholar]
- Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- Denison MS, Pandini A, Nagy SR, Baldwin EP, Bonati L. Ligand binding and activation of the Ah receptor. Chem Biol Interact. 2002a;141:3–24. doi: 10.1016/s0009-2797(02)00063-7. [DOI] [PubMed] [Google Scholar]
- Denison MS, Phelan D, Winter GM, Ziccardi MH. Carbaryl, a carbamate insecticide, is a ligand for the hepatic Ah (dioxin) receptor. Toxicol Appl Pharmacol. 1998b;152:406–414. doi: 10.1006/taap.1998.9999. [DOI] [PubMed] [Google Scholar]
- Denison MS, Phelps CL, DeHoog J, Kim HJ, Bank PA, Yao EF. Banbury Report 35: Biological Basis for Risk Assessment of Dioxins and Related Compounds. Cold Spring Harbor Laboratory Press; New York: 1991. pp. 337–350. [Google Scholar]
- Denison MS, Rogers JM, Rushing SR, Jones CL, Tetangco SC, Heath-Pagliuso S. Analysis of the aryl hydrocarbon receptor (AhR) signal transduction pathway. In: Maines MD, Costa LG, Reed DJ, Sassa S, Sipes G, editors. Current Protocols in Toxicology. Vol. 4. John Wiley & Sons, Inc.; New York: 2002b. pp. 4.8.1–4.8.45. [DOI] [PubMed] [Google Scholar]
- Denison MS, Seidel SD, Rogers WJ, Ziccardi M, Winter GM, Heath-Pagliuso S. Natural and synthetic ligands for the Ah receptor. In: Puga A, Wallace KB, editors. Molecular Biology of the Toxic Response. Taylor & Francis; Philadelphia: 1999. pp. 393–410. [Google Scholar]
- Denison MS, Vella LM. The hepatic Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin: Species differences in subunit dissociation. Arch Biochem Biophys. 1990;277:382–388. doi: 10.1016/0003-9861(90)90594-o. [DOI] [PubMed] [Google Scholar]
- Denison MS, Vella LM, Okey AB. Structure and function of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Species difference in molecular properties of the receptors from mouse and rat hepatic cytosols. J Biol Chem. 1986b;261:3987–3995. [PubMed] [Google Scholar]
- Denison M, Wilkinson CF, Okey AB. Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin: Comparative studies in mammalian and nonmammalian species. Chemosphere. 1986a;15:1665–1672. [Google Scholar]
- Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, Ikawa S, Fujii-Kuriyama Y. Dioxin binding activities of polymorphic forms of mouse and human arylhydrocarbon receptors. J Biol Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- Farrell K, Safe S. Absence of positive co-operativity in the binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin to its cytosolic receptor protein. Biochem J. 1987;244:539–546. doi: 10.1042/bj2440539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell K, Safe L, Safe S. Synthesis and aryl hydrocarbon receptor binding properties of radiolabeled polychlorinated dibenzofuran congeners. Arch Biochem Biophys. 1987;259:185–195. doi: 10.1016/0003-9861(87)90485-1. [DOI] [PubMed] [Google Scholar]
- Fontaine F, Delescluse C, de Sousa G, Lesca P, Rahmani R. Cytochrome 1A1 induction by primaquine in human hepatocytes and HepG2 cells: Absence of binding to the aryl hydrocarbon receptor. Biochem Pharmacol. 1999;57:255–262. doi: 10.1016/s0006-2952(98)00304-9. [DOI] [PubMed] [Google Scholar]
- Gobas FA, Lahittete JM, Garofalo G, Shiu WY, Mackay D. A novel method for measuring membrane-water partition coefficients of hydrophobic organic chemicals: Comparison with 1-octanol-water partitioning. J Pharm Sci. 1988;77:265–272. doi: 10.1002/jps.2600770317. [DOI] [PubMed] [Google Scholar]
- Gradelet S, Astorg P, Pineau T, Canivenc MC, Siess MH, Leclerc J, Lesca P. Ah receptor-dependent CYP1A induction by two carotenoids, canthaxanthin and beta-apo-8′-carotenal, with no affinity for the TCDD binding site. Biochem Pharmacol. 1997;54:307–315. doi: 10.1016/s0006-2952(97)00176-7. [DOI] [PubMed] [Google Scholar]
- Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- Henry EC, Gasiewicz TA. Transformation of the aryl hydrocarbon receptor to a DNA-binding form is accompanied by release of the 90 kDa heat-shock protein and increased affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem J. 1993;294:95–101. doi: 10.1042/bj2940095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester JE, Gasiewicz TA. Characterization of the in vitro stability of the rat hepatic receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Arch Biochem Biophys. 1987;252:606–265. doi: 10.1016/0003-9861(87)90067-1. [DOI] [PubMed] [Google Scholar]
- Lashley MR, Niedzinski EJ, Rogers JM, Denison MS, Nantz MH. Synthesis and estrogen receptor affinity of a 4-hydroxytamoxifen-labeled ligand for diagnostic imaging. Bioorg Med Chem. 2002;10:4075–4082. doi: 10.1016/s0968-0896(02)00329-2. [DOI] [PubMed] [Google Scholar]
- Ledirac N, Delescluse C, de Sousa G, Pralavorio M, Lesca P, Amichot M, Berge JB, Rahmani R. Carbaryl induces CYP1A1 gene expression in HepG2 and HaCaT cells but is not a ligand of the human hepatic Ah receptor. Toxicol Appl Pharmacol. 1997;144:177–182. doi: 10.1006/taap.1997.8120. [DOI] [PubMed] [Google Scholar]
- Lesca P, Peryt B, Larrieu G, Alvinerie M, Galtier P, Daujat M, Maurel P, Hoogenboom L. Evidence for the ligand-independent activation of the AH receptor. Biochem Biophys Res Commun. 1995;209:474–482. doi: 10.1006/bbrc.1995.1526. [DOI] [PubMed] [Google Scholar]
- Meylan WM, Howard PH. Atom/fragment contribution method for estimating octanol-water partition coefficients. J Pharm Sci. 1995;84:83–92. doi: 10.1002/jps.2600840120. [DOI] [PubMed] [Google Scholar]
- Moorthy B. Persistent expression of 3-methylcholanthrene-inducible cytochromes P4501A in rat hepatic and extrahepatic tissues. J Pharmacol Exp Ther. 2000;294:313–322. [PubMed] [Google Scholar]
- Notides AC, Weichman BM, Lerner N, de Boer W. The role of ligand-binding as a determinant of the structure and activation of the estrogen receptor. Adv Exp Med Biol. 1979;117:297–307. doi: 10.1007/978-1-4757-6589-2_16. [DOI] [PubMed] [Google Scholar]
- Ogle TF. Characterization of progesterone binding to nuclear receptors in rat placenta. J Steroid Biochem. 1986;24:945–951. doi: 10.1016/0022-4731(86)90345-6. [DOI] [PubMed] [Google Scholar]
- Pandini A, Denison MS, Song Y, Soshilov AA, Bonati L. Structural and functional characterization of the AhR ligand binding domain by homology modeling and mutational analysis. Biochemistry. 2007;46:696–708. doi: 10.1021/bi061460t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrulis JR, Perdew GH. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem Biol Interact. 2002;141:25–40. doi: 10.1016/s0009-2797(02)00064-9. [DOI] [PubMed] [Google Scholar]
- Poland A, Glover E. Comparison of 2,3,7,8-tetrachlorodibenzo-p-dioxin, a potent inducer of aryl hydrocarbon hydroxylase, with 3-methylcholanthrene. Mol Pharmacol. 1974;10:349–359. [PubMed] [Google Scholar]
- Poland A, Glover E. 2,3,7,8,-Tetrachlorodibenzo-p-dioxin: Segregation of toxicity with the Ah locus. Mol Pharmacol. 1980;17:86–94. [PubMed] [Google Scholar]
- Poland A, Knutson JC. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- Pollenz RS. The aryl hydrocarbon receptor but not Arnt protein is rapidly depleted in hepatic and non-hepatic cultured cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol. 1996;49:391–398. [PubMed] [Google Scholar]
- Pollenz RS. The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem Biol Interact. 2002;141:41–61. doi: 10.1016/s0009-2797(02)00065-0. [DOI] [PubMed] [Google Scholar]
- Pollenz RS, Buggy C. Ligand-dependent and -independent degradation of human aryl hydrocarbon receptor (hAHR) in cell culture models. Chem Biol Interact. 2006;164:49–59. doi: 10.1016/j.cbi.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Pollenz RS, Dougherty EJ. Redefining the role of XAP2 and CHIP in the degradation of endogenous AHR in cell culture models. J Biol Chem. 2005;280:3334–33356. doi: 10.1074/jbc.M506619200. [DOI] [PubMed] [Google Scholar]
- Procopio M, Lahm A, Tramontano A, Bonati L, Pitea D. A model for recognition of polychlorinated dibenzo-p-dioxins by the aryl hydrocarbon receptor. Eur J Biochem. 2002;269:13–18. doi: 10.1046/j.0014-2956.2002.02619.x. [DOI] [PubMed] [Google Scholar]
- Ramadoss P, Perdew GH. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol Pharmacol. 2004;66:129–136. doi: 10.1124/mol.66.1.129. [DOI] [PubMed] [Google Scholar]
- Ramadoss P, Perdew GH. The transactivation domain of the Ah receptor is a key determinant of cellular localization and ligand-independent nucleocytoplasmic shuttling properties. Biochemistry. 2005;44:11148–11159. doi: 10.1021/bi050948b. [DOI] [PubMed] [Google Scholar]
- Riddick DS, Huang Y, Harper PA, Okey AB. 2,3,7,8-Tetrachlorodibenzo-p-dioxin versus 3-methylcholanthrene: Comparative studies of Ah receptor binding, transformation, and induction of CYP1A1. J Biol Chem. 1994;269:12118–12128. [PubMed] [Google Scholar]
- Safe S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs) Crit Rev Toxicol. 1990;21:51–88. doi: 10.3109/10408449009089873. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- Song Z, Pollenz RS. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation and gene regulation. Mol Pharmacol. 2002;62:806–816. doi: 10.1124/mol.62.4.806. [DOI] [PubMed] [Google Scholar]
- Taylor P, Insel PA. Molecular basis of pharmacologic selectivity. In: Pratt WB, Taylor P, editors. Principles of Drug Action: The Basis of Pharmacology. Churchill Livingstone Inc.; New York: 1990. pp. 1–56. [Google Scholar]
- Wassenberg DM, Di Giulio RT. Synergistic embryotoxicity of polycyclic aromatic hydrocarbon aryl hydrocarbon receptor agonists with cytochrome P4501A inhibitors in Fundulus heteroclitus. Environ Health Perspect. 2004;112:1658–1664. doi: 10.1289/ehp.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenberg DM, Nerlinger AL, Battle LP, Di Giulio RT. Effects of the polycyclic aromatic hydrocarbon heterocycles, carbazole and dibenzothiophene, on in vivo and in vitro CYP1A activity and polycyclic aromatic hydrocarbon-derived embryonic deformities. Environ Toxicol Chem. 2005;24:2526–2532. doi: 10.1897/04-440r1.1. [DOI] [PubMed] [Google Scholar]
- Wei YD, Helleberg H, Rannug U, Rannug A. Rapid and transient induction of CYP1A1 gene expression in human cells by the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole. Chem Biol Interact. 1998;110:39–55. doi: 10.1016/s0009-2797(97)00111-7. [DOI] [PubMed] [Google Scholar]
- Weichman BM, Notides AC. Estradiol-binding kinetics of the activated and nonactivated estrogen receptor. J Biol Chem. 1977;252:8856–8862. [PubMed] [Google Scholar]
- Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- Wooge CH, Nilsson GM, Heierson A, McDonnell DP, Katzenellenbogen BS. Structural requirements for high affinity ligand binding by estrogen receptors: A comparative analysis of truncated and full length estrogen receptors expressed in bacteria, yeast, and mammalian cells. Mol Endocrinol. 1992;6:861–869. doi: 10.1210/mend.6.6.1495491. [DOI] [PubMed] [Google Scholar]