

SUMMARY
Native states of proteins are flexible, populating more than just the unique native conformation. The energetics and dynamics resulting from this conformational ensemble are inherently linked to protein function and regulation. Proteolytic susceptibility is one feature determined by this conformational energy landscape. As an attempt to investigate energetics of proteins on a proteomic scale, we challenged the E. coli proteome with extensive proteolysis and determined which proteins, if any, have optimized their energy landscape for resistance to proteolysis. To our surprise, multiple soluble proteins survived the challenge. Maltose binding protein, a survivor from thermolysin digestion, was characterized by in vitro biophysical studies to identify the physical origin of proteolytic resistance. This experimental characterization shows that kinetic stability is responsible for the unusual resistance in maltose binding protein. The biochemical functions of the identified survivors suggest that many of these proteins may have evolved extreme proteolytic resistance because of their critical roles under stressed conditions. Our results suggest that under functional selection proteins can evolve extreme proteolysis resistance by modulating their conformational energy landscapes without the need to invent new folds, and that proteins can be profiled on a proteomic scale according to their energetic properties by using proteolysis as a structural probe.
Keywords: Protein folding, energy landscape, proteolysis, proteomics, proteolytic susceptibility
INTRODUCTION
Proteins do not adopt unique, static structures; they access many different conformations within the native state ensemble.1-3 This ensemble includes small fluctuations around the native conformation, partially unfolded forms, and even the globally unfolded form. The population of each conformation is determined by its stability according to a Boltzmann distribution. These populations, combined with the dynamics of interconversion among conformations, define the conformational energy landscape of a protein. This energy landscape is encoded within the amino acid sequence, and underlies biological properties such as catalysis, signal transduction, and protein turnover.4-7 Thus, the entire energy landscape is subject to the same types of evolutionary pressures as is the native structure.
In spite of the great interest in energy landscapes, experimental determinations of energetic information on protein conformations have been slow, requiring purification of individual proteins and investigation with traditional biophysical instrumentation. These limitations have impeded acquiring a system-wide perspective on protein energetics. What is the distribution of kinetic and thermodynamic stabilities of proteins within a proteome? Is there a biological reason for the difference in conformational energy landscapes between proteins? Are the conformational energy landscapes of orthologous proteins conserved along with their structures and functions? These questions demand a new approach to studying protein energetics.
Here we report an investigation of proteolytic susceptibility as an attempt for energetic profiling of proteins on a proteomic scale. A protein’s susceptibility to proteolytic digestion is a functional attribute linked to its energy landscape.8-10 In order to be cleaved, the substrate polypeptide chain must be extended to fit into the substrate-binding sites of a protease,11 which make compactly folded proteins poor substrates for proteolysis. Proteolysis of compactly folded proteins requires access to high-energy cleavable states, where cleavage sites are exposed to proteases through local or global unfolding (Fig 1A).8,12-14 Proteolytic susceptibility of folded proteins, therefore, is determined by the thermodynamic or kinetic accessibility of these cleavable states. The nominal energy landscape diagrams in Fig 1B depict how proteolytic susceptibility is dictated by energy landscapes. Each diagram shows the energy levels of folded, intermediate, and globally-unfolded states. When a protein has an unstructured region in its native conformations, the protein can be cleaved by a protease without unfolding (1 in Fig 1B). Proteins 2 and 3 show different global stability but the same susceptibility, while Proteins 3 and 4 have the same global stability but different susceptibility. Protein 5 has a cleavable state relatively low in energy but a high kinetic barrier to access the cleavable state, which confers the protein’s proteolytic resistance. A protein’s proteolytic susceptibility is, therefore, determined by its conformational energy landscape and not by its global stability.8,15
Figure 1. Proteolysis of proteins under native conditions.
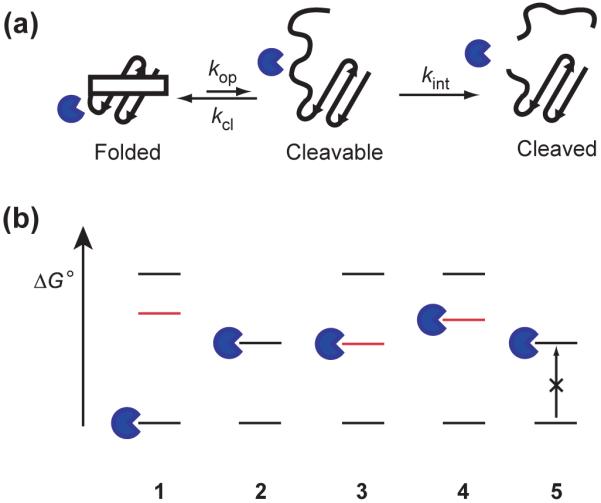
(a) Schematic representation of the mechanism of proteolytic cleavage of a protein in its native state. Proteins without flexible loops or unstructured regions in the folded conformation are protected from proteolysis. These proteins are cleaved only by accessing cleavable states. kop, kcl, and kint are the kinetic constants for opening, closing, and intrinsic proteolysis steps, respectively. (b) Nominal energy landscape of proteins to explain proteolytic susceptibility. Lowest lines in the energy diagram indicate native forms, and the highest lines indicate fully unfolded forms. Lines in red indicate the cleavable states between folded and globally-unfolded states. Proteins with flexible loops or unstructured regions (1) are digested even in their native conformation. Otherwise, proteins need to unfold fully (2), or transform to a cleavable form (3 and 4), to be digested. Kinetic barriers can also result in resistance to proteolysis by making it difficult to access the cleavable state which is low in energy (5).
Recent studies on the protein α-lytic protease provide an unusual example of a conformational energy landscape resistant to proteolysis.16 This protein, itself a protease in a harsh extracellular environment, ensures proteolytic resistance with an unusually high kinetic barrier to unfolding (local or global). How unusual are proteins whose energy landscapes encode resistance to proteolysis? We need to determine proteolytic susceptibility of proteins on a proteomic scale to answer this question. To profile proteins according to their proteolytic susceptibility in a high-throughput fashion, we devised a survival assay, where proteins in a cell lysate are subjected to extensive proteolysis; the survivors are then identified using genomic data. We chose the proteome of E. coli for our first investigation.
RESULTS
Extensive proteolysis of an E. coli lysate
Proteolytic digestion was carried out on an E. coli K12 lysate prepared from an overnight culture. In the first assay, the lysate was digested with 0.40 mg/mL trypsin (approximately 5~10% of the total protein in the reaction) at 25 °C for four days, and the reaction was monitored by SDS-PAGE (Fig. 2A). A significant number of proteins were digested within the first 30 minutes; some proteins, however, survived proteolysis and were visible throughout the four-day experiment.
Figure 2. Digestion of E. coli lysate with trypsin.

(a) SDS-PAGE gel of samples taken at the designated time points from the proteolysis reaction of E. coli lysate by 0.40 mg/mL trypsin. (b) SDS-PAGE gel of samples taken at the designated time points from the proteolysis reaction of E. coli lysate by 0.40 mg/mL thermolysin.
Trypsin cleaves specifically after lysine and arginine residues, and some proteins with proteolytically-sensitive conformations may survive the assay due to this specificity. We therefore repeated the same survival assay using thermolysin (0.40 mg/mL at 25 °C) which cleaves before hydrophobic and aromatic residues (Ile, Leu, Val, Ala, Met, Phe).17 Again, many proteins survived this four-day incubation (Fig. 2B). Even more survivors were observed with thermolysin than with trypsin. The differences are not surprising since the proteases have different catalytic activities and specificities. The results, however, clearly indicate that there are also survivors to a protease with broader substrate specificity than trypsin.
In order to account for any loss in protease activity due to autodigestion during the course of the assay, protease activity was monitored over the four-day incubation. No apparent decrease in protease activity was observed under our reaction conditions where 10 mM CaCl2 was included (Data not shown), suggesting that survival is indeed a consequence of a protein’s resistance to proteolysis within the experimental time scale.
Identification of survivors
We used 2-dimensional (2-D) gel electrophoresis to identify survivors. An E. coli K12 lysate was incubated with 0.40 mg/mL trypsin or thermolysin. The reactions were quenched after 1 day and 4 days. Comparison of 2-D gels of 1-day digestion and 4-day digestion allowed us to monitor any apparent decrease in intensity between 1 day and 4 days. We selected 30 spots from 2-D gels of 4-day trypsin digestion and 40 spots from 2-D gels of 4-day thermolysin digestion (Fig 3 and Table 1). To minimize the redundancy of the identified proteins, only one spot was chosen when a series of spots exist in a horizontal arrangement, which frequently indicate variants of one protein with different charges due to modifications during sample preparation. Spots showing any noticeable decrease in intensity from 1-day to 4-day digestion were indicated in Table 1. Proteins corresponding to these spots are likely to have the minimal resistance required to survive the current challenge. The same amount of untreated lysate was also run on a 2-D gel to estimate roughly how many proteins in E. coli proteome are sampled with the current approach. About 500 spots were observed from the cell lysate with the staining method used in this study.
Figure 3. Identification of survivors by 2-D electrophoresis.

E.coli soluble fraction digested with 0.40 mg/mL trypsin (A) or 0.40 mg/mL thermolysin (B) for 4 days were analyzed by 2-D electrophoresis. The numbers on the gels indicate the spots analyzed by in-gel digestion and mass spectrometry. The identity of the protein in each spot is listed in Table 1. Black arrows indicate spots corresponding proteases.
Table 1. Identification of proteins from 2-Dimensional electrophoresis gels.
| A. Identified proteins from spots on the 2-D gel of E. coli lysate digested with 0.40 mg/mL trypsin for 4 days. | |||||
|---|---|---|---|---|---|
| Spot number | Protein ID | Gene name | Length | M.W. (kDa) | pI |
| 1 | P21179 | katE | 753 | 84.2 | 5.54 |
| 2 | P06715 | Gor | 450 | 48.8 | 5.64 |
| 3 | P76108 | ydcS | 359 | 40.0 | 6.27 |
| 4 | P06977 | gapA | 330 | 35.4 | 6.58 |
| 5 | P25887 | yghA | 294 | 31.4 | 6.32 |
| 6 | P60651 | speB | 306 | 33.6 | 5.14 |
| 7 | P24223 | pdxJ | 242 | 26.3 | 5.61 |
| 8* | P00882 | deoC | 259 | 27.7 | 5.50 |
| 9 | P04790 | tpiA | 255 | 27.0 | 5.64 |
| 10 | P02925 | rbsB | 271 | 28.5 | 5.99 |
| 11 | P09551 | argT | 238 | 25.8 | 5.22 |
| 12 | P12758 | udp | 252 | 27.0 | 5.81 |
| 13 | P12758 | udp | 252 | 27.0 | 5.81 |
| 14 | P75743 | ybgI | 247 | 26.9 | 5.07 |
| 15 | P21367 | ycaC | 208 | 23.1 | 5.20 |
| P32661 | rpe | 225 | 24.6 | 5.13 | |
| 16 | P12758 | udp | 252 | 27.0 | 5.81 |
| 17* | P12758 | udp | 252 | 27.0 | 5.81 |
| 18 | P09743 | deoD | 238 | 25.8 | 5.42 |
| 19 | P10344 | glnH | 226 | 25.0 | 6.87 |
| 20 | P00448 | sodA | 205 | 23.0 | 6.44 |
| 21 | P00448 | sodA | 205 | 23.0 | 6.44 |
| 22 | P10344 | glnH | 226 | 25.0 | 6.87 |
| 23 | P09157 | sodB | 192 | 21.1 | 5.58 |
| 24 | P09157 | sodB | 192 | 21.1 | 5.58 |
| 25* | P17288 | ppa | 175 | 19.6 | 5.03 |
| 26† | P04790 | tpiA | 255 | 27.0 | 5.64 |
| 27† | P12758 | udp | 252 | 27.0 | 5.81 |
| 28* | P11056 | bfr | 158 | 18.5 | 4.69 |
| 29† | P27430 | dps | 166 | 18.6 | 5.72 |
| 30 | P23827 | eco | 142 | 16.1 | 5.94 |
| B. Identified proteins from spots on the 2-D gel of E. coli lysate digested with 0.40 mg/mL thermolysin for 4 days. | |||||
|---|---|---|---|---|---|
| Spot number | Protein ID | Gene name | Length | M.W. (kDa) | pI |
| 1 | P21179 | katE | 753 | 84.2 | 5.54 |
| 2* | P07024 | ushA | 525 | 58.2 | 5.40 |
| 3* | P13482 | treA | 535 | 60.5 | 5.36 |
| 4 | P14178 | pykF | 470 | 50.7 | 5.77 |
| 5* | P23843 | oppA | 517 | 58.4 | 5.85 |
| 6 | P00391 | ipdA | 473 | 50.6 | 5.79 |
| 7 | P06715 | gor | 450 | 48.8 | 5.64 |
| 8* | P22259 | pckA | 540 | 59.6 | 5.46 |
| 9* | P19926 | agp | 391 | 43.6 | 5.38 |
| 10 | P37095 | pepB | 427 | 46.2 | 5.60 |
| 11* | P75691 | yahK | 349 | 38.0 | 5.80 |
| 12 | P00509 | aspC | 396 | 43.6 | 5.54 |
| 13 | P11665 | Pgk | 386 | 41.0 | 5.08 |
| 14 | P02928 | malE | 370 | 40.7 | 5.22 |
| 15 | P31133 | potF | 344 | 38.3 | 5.53 |
| 16 | P76108 | ydcS | 359 | 40.0 | 6.27 |
| 17* | P06977 | gapA | 330 | 35.4 | 6.58 |
| 18 | P13652 | cdd | 294 | 31.5 | 5.42 |
| 19† | P14178 | pykF | 470 | 50.7 | 5.77 |
| 20† | P14178 | pykF | 470 | 50.7 | 5.77 |
| 21 | P02925 | rbsB | 271 | 28.5 | 5.99 |
| 22 | P00882 | deoC | 259 | 27.7 | 5.50 |
| 23 | P09551 | argT | 238 | 25.8 | 5.22 |
| 24 | P30859 | artI | 224 | 25.0 | 5.32 |
| 25 | P04790 | tpiA | 255 | 27.0 | 5.64 |
| 26 | P32697 | aphA | 212 | 23.5 | 5.94 |
| 27 | P10344 | glnH | 226 | 25.0 | 6.87 |
| P30860 | artJ | 224 | 24.9 | 5.97 | |
| 28 | P10344 | glnH | 226 | 25.0 | 6.87 |
| 29 | P00448 | sodA | 205 | 23.0 | 6.44 |
| 30 | P12758 | udp | 252 | 27.0 | 5.81 |
| 31 | P21367 | ycaC | 208 | 23.1 | 5.20 |
| 32 | P32661 | rpe | 225 | 24.6 | 5.13 |
| 33 | P17288 | ppa | 175 | 19.6 | 5.03 |
| 34 | P09157 | sodB | 192 | 21.1 | 5.58 |
| 35† | P07651 | deoB | 407 | 44.4 | 5.11 |
| 36*† | P12758 | Udp | 252 | 27.0 | 5.81 |
| 37† | P12758 | Udp | 252 | 27.0 | 5.81 |
| 38 | P11056 | Bfr | 158 | 18.5 | 4.69 |
| 39 | P27430 | Dps | 166 | 18.6 | 5.72 |
| 40† | P05313 | aceA | 434 | 47.5 | 5.16 |
The spot intensity has been decreased noticeably compared with the same spot on the 2-D gel of the lysate digested for 1 day under the same condition.
The size of the peptide estimated from the gel suggests the spot corresponds to a fragment from proteolysis of the protein.
Selected spots were analyzed using peptide-mass mapping by MALDI-TOF to identify proteins (Table 1). A protein is considered as a survivor only when the molecular weight of the protein estimated from the gel matches within 10% with that expected from the sequence. Using this approach, we identified 22 survivors from digestion with trypsin and 34 survivors from digestion with thermolysin (Table 2). Sixteen of the identified trypsin survivors (73%) were also identified as thermolysin survivors (Fig. 4 and Table 2). The existence of so many common survivors suggests that the survival is not due to substrate specificities of the proteases but due to the unusual energy landscapes of the survivors. The apparent pI values of most survivors are consistent with the calculated pI values. Any inconsistent pI values may indicate posttranslational modifications or oxidation during the incubation.
Table 2. Identified survivors.
Information on each protein was collected from Swiss-Prot/TrEMBL (http://us.expasy.org/sprot/). Only periplasmic proteins are indicated so in the localization column
| gene name |
Description | PDB ID | Subunits | Localization |
|---|---|---|---|---|
| Common survivors | ||||
| katE | catalase HPII | 1GGE | 4 | |
| gor | glutathione reductase | 1GET | 2 | |
| ydcS | putative periplasmic binding protein, ydcS | periplasmic† | ||
| gapA | glyceraldehyde-3-phosphate dehydrogenase A | 1GAD | 4 | |
| deoC | deoxyriboaldolase | 1JCL | 2 | |
| tpiA | triosephosphate isomerase | 1TRE | 2 | |
| rbsB | ribose binding periplasmic protein | 2DRI | 1 | periplasmic |
| argT | LAO-binding periplasmic protein | 1 | periplasmic | |
| udp | uridine phosphorylase | 1K3F | 6 | |
| ycaC | ycaC | 1YAC | 8 | |
| rpe | ribulose phosphate 3-epimerase | |||
| glnH | glutamine-binding periplasmic protein | 1WDN | 1 | periplasmic |
| soda | superoxide dismutase (Mn) | 1D5N | 2 | |
| sodB | superoxide dismutase (Fe) | 1ISA | 2 | |
| ppa | inorganic phosphatase | 1JFD | 6 | |
| bfr | bacterioferritin | 1BFR | 24 | |
| Identified as survivors only in trypsin digestion | ||||
| yghA | hypothetical oxidoreductase, yghA | |||
| speB | agmatinase | |||
| pdxJ | PNP synthase | 1M5W | 8 | |
| ybgI | hypothetical UPF0135 protein ybgI | 1NMO | 6* | |
| deoD | purine nucleoside phosphorylase | 1A69 | 6 | |
| eco | ecotin | 1ECY | 2 | periplasmic |
| Identified as survivors only in thermolysin digestion | ||||
| ushA | UDP-sugar hydrolase (5′-nucloetidase) | 1HP1 | 1 | periplasmic |
| treA | trehalase | 1 | periplasmic | |
| pykF | pyruvate kinase I | 1PKY | 4 | |
| oppA | periplasmic oligopeptide-binding protein | 1 | periplasmic | |
| ipdA | dihydrolipoyl dehydrogenase | 2 | ||
| pckA | phosphoenolpyruvate carboxykinase | 1AYL | 1 | |
| agp | glucose-1-phosphatase | 1NT4 | 2 | periplasmic |
| pepB | peptidase B | 6 | ||
| yahK | yahK (alcohol dehydrogenase-like) | 1UUF | 2* | |
| aspC | aspartate aminotransferase | 1AAW | 2 | |
| pgk | phosphoglycerate kinase | 1 | ||
| malE | maltose binding protein | 1ANF | 1 | periplasmic |
| potF | putrescine-binding protein | 1A99 | 1 | periplasmic |
| cdd | cytidine deaminase | 1CTT | 2 | |
| artI | arginine-binding protein 1 | 1 | periplasmic† | |
| aphA | class B acid phosphatase | 1N8N | 4 | periplasmic† |
| artJ | arginine-binding protein 2 | 1 | periplasmic† | |
| dps | DNA protection during starvation protein | 1DPS | 12 | |
The quaternary structure of this protein is based on the structure determined by X-ray crystallography and is not confirmed under physiological conditions.
The localization information on this protein is inferred from sequence analyses and is not determined experimentally.
Figure 4. Common survivors from assays with trypsin and thermolysin.
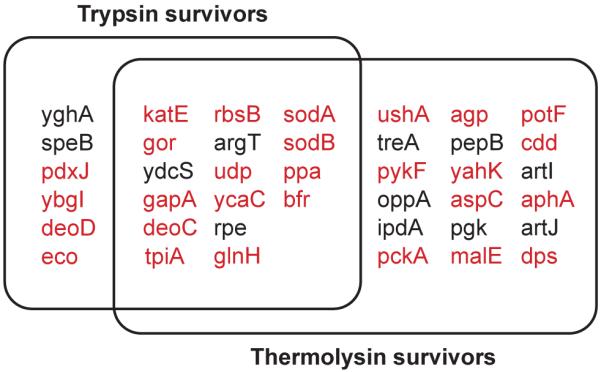
The proteins in the overlapping region survived 4-day digestion with 0.40 mg/mL trypsin and 0.40 mg/mL thermolysin. For convenience, gene names are used for corresponding proteins. The proteins shown in red have structural coordinates deposited in the protein data bank (http://www.rcsb.org/pdb/).
Sequence analysis of survivors
To identify any general rules that encode survival to such extensive proteolysis, we looked for common features within the amino acid compositions and the three dimensional structures of the survivors. An analysis of amino acid composition shows that the survivors from trypsin digestion contain plenty of lysine and arginine residues, potential trypsin cleavage sites. Lysine/arginine residues comprise (10.4 ± 2.6)% of the total number of residues of each survivor. For comparison, we determined the average lysine/arginine content of all open reading frames in the E. coli genome to be (10.3 ± 3.4)%. Resistance to digestion is not due to a lack of potential cleavage sites.
α-lytic protease, a bacterial enzyme known for its kinetic stability and protease resistance, has 16% glycines, while its proteolysis-sensitive homolog chymotrypsin has only 9% glycines. This high glycine content was proposed to be a structural factor that enables tight and cooperative packing within the core of this protein.18 We determined the average glycine content of all identified survivors to be (7.6 ± 1.6)%, which is clearly much lower than that for α-lytic protease and more in line with our determination for the average glycine content of all open reading frames in E. coli genome, (7.1 ± 2.5)%. Therefore, the high glycine content is not likely to be a common reason for proteolytic resistance.
Proteolysis kinetics of maltose binding protein
To confirm the validity of the proteomic survival assay, we cloned, expressed, and purified maltose binding protein (MBP), a survivor identified from thermolysin digestion, but not from trypsin digestion. Digestion of purified MBP by 0.40 mg/mL thermolysin was so slow that the reaction was monitored for 20 days. The kinetic constant for the proteolysis of MBP by 0.40 mg/mL thermolysin was determined to be 1.2 × 10−6 s−1, corresponding to a half life of 6.6 days, which confirmed the result of the survival assay on a proteomic scale. MBP was also digested with 0.40 mg/mL trypsin with a greater rate constant, 4.4 × 10−5 s−1 (half life = 4.3 hours), which is also consistent with the result that MBP was not found as a survivor from the tryptic digestion of E. coli lysate. Unfolded MBP was observed to be quite susceptible to thermolysin,19 indicating the structure of this protein protects it from being digested by proteases.
To understand the physical origin of MBP’s resistance to proteolysis by thermolysin, we determined proteolysis kinetics of the protein at different thermolysin and trypsin concentration (Fig. 5). When proteolysis of a protein occurs by the kinetic mechanism shown in Fig. 1A, the overall proteolysis rate constant (kp) is expressed as:
| (1) |
where kop and kcl are the rate constants for the forward and the backward reaction from the folded state to the cleavable state, and kint is the intrinsic proteolysis rate for an unstructured peptide. When kint is estimated as the product of kcat/Km and protease concentration ([E]),8 Eq. 1 can be rewritten as:
| (2) |
where Kop (= kop/kcl) is the equilibrium constant between the folded and the cleavable states in Fig. 1A.8 By determining kp at different concentration of a protease, we can determine kop and Kop of the opening step leading to the cleavable conformation. When kcl >> kint, however, Eq. 1 is simplified as
| (3) |
by which we can only determine Kop.
Figure 5. Proteolysis mechanism of maltose binding protein by thermolysin and trypsin.

Maltose binding protein (MBP) was incubated at 25°C with 0.40 mg/mL thermolysin (a) or 0.40 mg/mL trypsin (b) in 20 mM Tris–HCl buffer (pH 8.0) containing 50 mM NaCl and 10 mM CaCl2. The kp values for thermolysin digestion were fit to Eq. 2 to determine kop and Kop(kcat/KM). From the kp values for trypsin digestion only Kop(kcat/KM) value was determined by a linear regression using Eq. 3.
The plot of the proteolysis rates of MBP determined at different thermolysin concentrations shows an asymptotic behavior (Fig. 5A), suggesting that the kinetics to access the cleavable state in MBP (kop in Fig. 1A) determines the overall proteolysis rate at high concentration of protease. However, the rate of proteolysis of MBP by trypsin is linearly dependent on the protease concentration without any indication of the asymptotic pattern shown in proteolysis by thermolysin (Fig 5B), indicating that kcl >> kint under the given assay condition. By fitting kp of proteolysis by thermolysin to Eq. 2, kop and Kop were determined to be 1.8 × 10−6 s−1 and 4.9 × 10−7. The kp values of proteolysis by trypsin was fit to Eq. 3, and Kop was determined to be 8.3 × 10−6. To determine Kop, kcat/KM values measured with peptide substrates were used: ABZ-Ala-Gly-Leu-Ala-pNA for thermolysin (7.3 × 105 M−1s−1) and insulin β-chain for trypsin (3.0 × 105 M−1s−1).19,20
The kop value for proteolysis by thermolysin (1.8 × 10−6 s−1 is in a quite good agreement with the global unfolding rate constant for MBP determined by urea denaturation (1.3 × 10−6 s−1). This result strongly suggests that proteolysis of MBP by thermolysin is limited by the same kinetic barrier limiting global unfolding. The energies of the cleavable states for thermolysin and trypsin digestion are calculated to be 8.6 kcal/mol and 6.9 kcal/mol, respectively, with the determined Kop values. We also determined the global stability of MBP (ΔGunf°) to be 14.6 ± 0.7 kcal/mol by monitoring unfolding of the protein in urea by circular dichroism. The smaller energies of the cleavable states than ΔGunf° suggest the proteolysis of MBP occurs through intermediate states, not through globally-unfolded state.
Discussion
Structures of survivors
Seventeen out of the 22 trypsin survivors (77%) and 24 out of 34 thermolysin survivors (71%) have had their structures solved by X-ray crystallography (Fig. 4 and Table 2). This unusually high proportion of proteins with known structures suggests that proteolytically resistant proteins are advantageous for X-ray crystallography, perhaps due to ease of crystallization or purification. Inspection of these available protein structures did not reveal any common structural features to explain their proteolytic resistance. The gallery of structures in Fig. 6 does not reveal any characteristic structural elements, such as tight loops, specific arrangement of secondary structures, or common motifs. For instance, only 4 out of 28 proteins with known structures have disulfide bonds: ecotin, UDP-sugar hydrolase, glucose-1-phosphatase, and putrescine-binding protein. Therefore, there does not seem to be any specific structure or fold required for protease resistance, again suggesting a fine-tuning of the energy landscape.
Figure 6. Ribbon representation of the structures of proteins that survived both trypsin and thermolysin.

Structures of assumed biological molecules are shown for multimeric proteins. (a) Catalase HPII (PDB entry: 1GGE). (b) Glutathione reductase (PDB entry: 1GET). (c) Glyceraldehyde-3-phosphate dehydrogenase A (PDB entry: 1GAD). (d) deoxyriboaldolase (PDB entry: 1JCL). (e) triosephosphate isomerase (PDB entry: 1TRE). (f) Ribose binding periplasmic protein (PDB entry: 2DRI). (g) Uridine phosphorylase (PDB entry: 1K3F). (h) ycaC (1YAC). (i) Glutamine-binding periplasmic protein (PDB entry: 1WDN). (j) Manganese superoxide dismutase (PDB entry: 1D5N). (k) Iron superoxide dismutase (PDB entry: 1ISA). (l) Inorganic pyrophosphatase (PDB entry: 1JFD). (m) Bacterioferritin (PDB entry: 1BFR). Ribbon diagrams were made with the program MOLSCRIPT.41
The wide array of protein folds observed among the survivors (Fig. 6) is perhaps not surprising. Themophilic proteins encode significantly different thermodynamic properties from their mesophilic homologues, even though they have the same three-dimensional folds.21,22 Apparently, just like the thermophilic proteins, under functional selection the survivors have evolved such extreme protease resistance by modulating their conformational energy landscapes without the need to invent new structures or folds.
Proteolytic resistance does not apparently imply a lack of conformational change or allostery. For instance, the periplasmic binding proteins switch from open forms to closed forms when they bind to their cognate ligands.23,24 Inorganic pyrophosphatase, purine nucleotide phosphorylase, and glyceraldehyde-3-phosphate dehydrogenase are multimeric enzymes that show cooperativity in their catalysis.25-27 Survival of these proteins suggests that these dynamic processes do not necessarily result in proteolytically-susceptible conformations.
The survivors are comprise a group of diverse quaternary structures (Table 2). Because the energetics of oligomeric proteins is dependent on protein concentration, proteolytic susceptibility of these proteins could be dependent on protein concentration. Since the protein concentration in the survival assay is much lower than their concentrations in vivo, survivors in this dilute condition would be still resistant at protein concentrations close to those in vivo. Several of the survivors are also known to bind various cofactors including metals. Th observed resistance observed may reflect a property of holo-enzymes complexed with cofactors. It should be noted, however, that the free cofactor concentrations in the assay must be quite low because the lysate used in this study was carefully dialyzed.
Resistance to proteolysis and biological functions
Energy landscapes encoding such apparent rigidity may be an important functional feature subject to natural selection. Many of the survivors in Table 2 belong to two categories of biochemical functions: a family of periplasmic binding proteins and a group of stress-related proteins. It is important to note that our screen was not comprehensive and is undoubtedly biased by the culture conditions and experimental protocol. Absence of a protein from the list should not imply proteolysis sensitivity.
Nine of the identified survivors are periplasmic binding proteins (Table 2). This appears particularly significant, considering E. coli has only ~40 periplasmic binding proteins.28 Periplasmic proteins of E. coli are likely to be more exposed to exogenous proteases than are cytosolic proteins. The presence of ecotin, one of the survivors and an endogenous protease inhibitor in E. coli periplasm, indicates the necessity of protection against exogenous proteases in the periplasmic space. Our results suggest that proteolytic resistance might be a common property of periplasmic binding proteins in E. coli.
Many of the surviving proteins have biological functions associated with the stationary phase (Table 2), in which E. coli needs to survive starvation and oxidative stress. Dps has a role in protecting DNA against oxidative stress during starvation,29 and Dps is one of the genes induced most strongly by hydrogen peroxide.30 Iron and manganese superoxide dismutases are also important in protecting E. coli against oxidative stress during starvation.31 Bacterioferritin, a Dps homolog, also sequesters excess iron in a non-toxic form in the central cavity inside the spherical 24-mer.32 These biochemical functions strongly suggest that their proteolytic resistance is related to their role in stress response.
Since cytosolic proteins are not likely to be exposed to exogenous proteases, the biological benefit of proteolytic resistance for the cytosolic survivors is less obvious. It might increase the lifetime of these proteins by protecting them from endogenous protease activities;33 or, it might be an indication of conformational rigidity evolved to minimize any unwanted modification, such as deamidation.34 Increasing the lifetime of proteins essential for stationary phase would thereby decrease the need for protein synthesis, which is an expensive process for E. coli in the stationary phase.
In vivo degradation of proteolytically resistant proteins
How are these proteolytically-resistant proteins degraded in vivo? Proteins induced specifically in stationary phase, such as Dps, need to be degraded rapidly when E. coli re-enters growth phase. Recently, Dps was found to have a degradation sequence at its N-terminus for an ATP-dependent protease, ClpXP.35 Interestingly, N-terminal sequencing of Dps from the 2-D gel showed that trypsin cleaved this degradation sequence (Data not shown). These N-terminal residues are also not ordered in the Dps structure solved by X-ray crystallography.36 Therefore, Dps has a highly flexible N-terminus which is cleaved readily by trypsin or recognized by ClpXP. The rest of the protein is, however, resistant to proteolysis, which would ensure in vivo stability of the protein in the absence of proteolysis by ClpXP. This rigid structure tagged with a degradation signal within a flexible terminus seems to be an effective strategy to control the degradation of a protein exclusively by ATP-dependent proteases.
Thermodynamic and kinetic requirements for survival
What does it mean to be a survivor? When the overall proteolysis rate constant is kp, the fraction of survival as a function of time (fs) is:
| (4) |
where N0 and N are the concentrations of intact proteins at t = 0 and after incubating for time t, respectively. Using the protease concentration (12 μM) and kcat/KM for the cleavage of ABZ-Ala-Gly-Leu-Ala-pNA, a generic thermolysin substrate (7.3 × 105 M−1s−1)19, the kint value under the conditions of the survival assay with thermolysin is estimated to be 8.8 s−1. With this estimated kint value, kp can be calculated for any given kop and kcl using Eq. 1. Also, the fraction of survival (fs) can be calculated for given kop and kcl with Eq. 4. Fig. 7 shows the color-coded contour diagram of the fraction of survival at each kop and kcl. The diagram shows a clear transition zone between a phase of survival (purple) and complete digestion (red).
Figure 7. Contour diagram of the fraction of survival.
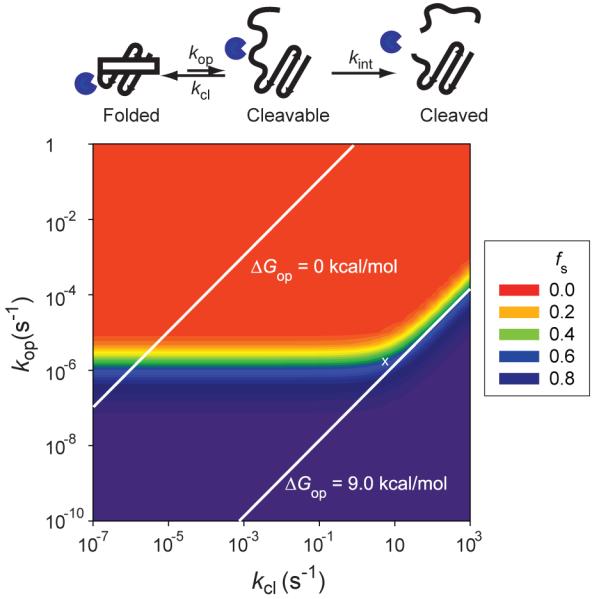
The fraction of survival (fs) at the end of the assay with thermolysin is determined with Eqs. 1 and 4 using kint of 8.8 s−1. kop and kcl are the forward and reverse rate constants for opening to the cleavable state. kcl is the intrinsic rate constant for proteolysis of proteins in the cleavable state. The kop and kcl values giving ΔGop of 0 kcal/mol and 9.0 kcal/mol are indicated with white solid lines. Cleavable states in the red region are accessible under the assay condition and proteins with a cleavable state in the red region cannot pass the survival assay. The white ‘×’ symbol indicates the location of the cleavable state of MBP through which this protein is digested by thermolysin.
The transition zone shows a kink in the region where kcl ~ kint (8.8 s−1). When kcl << kint, the survival is independent of k, which is equivalent to the EX1 regime in hydrogen exchange.8 In this kinetic regime, overall proteolysis is determined only by kop. Under the given assay condition 50% of a protein remains intact if the protein has kop ~ 2 × 10−6 s−1 (t1/2 ~ 4 days) regardless of kcl. When kcl >> kint, the transition zone in Fig. 7 shows another kinetic regime where the fraction of survival depends on both of kop and kcl, which is equivalent to the EX2 regime in hydrogen exchange.8 Therefore, in this kinetic regime the survival depends on the free energy, and not the rate, of the opening step (ΔGop). Under the given assay condition 50% of a protein remains intact if the ΔGop for the lowest cleavable state is 9.0 kcal/mol. The kop and kcl giving ΔGop values of 9.0 kcal/mol are indicated with a white line in Fig. 7. Cleavable states lying below this line are not accessible thermodynamically under the given assay condition. Also, the kop and kcl giving ΔGop values of 0 kcal/mol is indicated in Fig. 7. This line shows a region where kinetic stability (extremely small kop) can protect a protein in spite of thermodynamic instability (a triangular purple region above the white line of ΔGop = 0 kcal/mol). Indeed this region has already been proven by the example of α-lytic protease.16
Globally unfolded states of a protein are also cleavable states. To be a survivor, the globally unfolded states should not be accessible kinetically or thermodynamically under the assay condition. In other words, it is a necessity that the globally unfolded states of survivors should be within the purple region in Fig. 7.
Kinetic barrier protecting maltose binding protein from proteolysis by thermolysin
The location in Fig. 7 of a cleavable state can be determined experimentally by measuring the rate of proteolysis, kp. The kop and Kop values for the cleavable state of MBP by thermolysin were determined to be 1.8 × 10−6 s−1 and 4.9 × 10−7 from kp measured at different protease concentration (Fig. 5A). From these values, kcl is also calculated as 3.6 s−1. The cleavable state of this protein is at the edge of the survival zone in Fig. 7 (marked with a white ‘X’). The kcl value (3.6 s−1) is close to, but still smaller than, kint (8.8 s−1), which locates the proteolysis kinetics of this protein is at the boundary of the EX1 regime. The energy of the cleavable state (8.6 kcal/mol) is smaller than ΔGop for 50% survival of proteins in EX2 regime (9.0 kcal/mol). Therefore, the energy of the cleavable state would not be high enough to protect the protein, if proteolysis of MBP by thermolysin were in the EX2 regime.
The energy diagram of MBP proteolysis by thermolysin is depicted in Fig. 8 using the determined kinetic constants. The energy of the cleavable state is much lower than the global stability of MBP (14.6 ± 0.7 kcal/mol). However, kop (1.8 × 10−6 s−1) is quite close to the global unfolding rate constant for MBP determined by urea denaturation (1.3 × 10−6 s−1), which suggests the kinetic barrier to the cleavable state is the same kinetic barrier determining the global unfolding rate. It is also likely that this cleavable state is one of the kinetic intermediates on the unfolding pathway. This kinetic intermediate does not accumulate during unfolding, because the state exists after the rate-determining step. Considering the reversibility of protein folding, this intermediate could be on the folding trajectory of MBP.
Figure 8. Reaction energy diagrams of MBP proteolysis by thermolysin and trypsin.

The energy diagrams were depicted based on the kinetic data from proteolysis of MBP and the kcat/KM values determined with peptide substrates. The energy of globally unfolded state of MBP (14.6 kcal/mol) is indicated by a dashed line. The access to the cleavable state is rate-determining in proteolysis of MBP by thermolysin, while the intrinsic proteolysis step is rate-determining in proteolysis by trypsin. A question mark indicates the absolute height of the energy barrier is not known from the available data.
This analysis of proteolysis kinetics suggests how MBP achieves proteolytic resistance to thermolysin. First, local fluctuations under native states are minimal. MBP does not expose cleavable sequences for thermolysin digestion without crossing the major kinetic barrier for global unfolding. Next, this kinetic barrier is considerably high. This slow unfolding controls overall proteolysis rate when the protein is surrounded with high concentration of proteases. This strategy used by MBP for proteolytic resistance against proteolysis is well consistent with the case in α-lytic protease that employs the same strategy to the more extreme degree.16 Kinetic stability has been proposed as a result of the evolution- creating the protective role of a high unfolding barrier.37-39 The slow unfolding and proteolytic resistance of MBP is supports this protective role of kinetic stability.
The proteolysis of MBP by trypsin is distinct from that with thermolysin digestion (Fig 5B). Even at 0.40 mg/mL trypsin, the kinetic constant does not show any sign of saturation. Therefore, only Kop could be determined (8.3 × 10−6). The energy diagram of proteolysis of MBP by trypsin is also depicted in Fig. 8. The kint value was calculated to be 5.1 s-1 with the concentration of trypsin (0.40 mg/mL; 17 μM) and the kcat/KM values measured with insulin β-chain for trypsin (3.0 × 105 M−1s−1).20 From the comparison of proteolysis kinetics of MBP by trypsin and thermolysin, it is clear that the cleavable states for the two proteases are distinct and the cleavable state for trypsin digestion is apparently not susceptible to thermolysin. The cleavable state for trypsin digestion is lower by 1.7 kcal/mol than the cleavable state for thermolysin digestion (6.9 kcal/mol versus 8.6 kcal/mol). The kinetic barrier to the cleavable state is also much lower for trypsin digestion, which does not show the saturation behavior; the intrinsic proteolysis step is still rate-limiting even with 0.40 mg/mL of trypsin. Therefore, the kop value, which cannot be determined with the data in Fig. 5B, is much greater than the proteolysis rate with 0.40 mg/mL trypsin (4.4 × 10−5 s−1) and kcl is much greater than kint (5.1 s−1). Overall, proteolysis of MBP by 0.40 mg/mL trypsin (4.4 × 10−5 s−1) is faster than the opening to the cleavable state for thermolysin digestion (1.8 × 10−6 s−1); the rate-limiting step of trypsin digestion is lower by 1.9 kcal/mol than that of thermolysin digestion. The cleavable state for trypsin digestion seems to be accessible by local fluctuation — localized unfolding without global conformational change. Proteolysis through local fluctuation is consistent with the observations that this conformation does not expose any sequences cleavable by thermolysin and the transition from the cleavable to the folded conformation seems quite fast, compared with the intrinsic proteolysis (Fig. 8).
Energetics-based protein profiling
Proteomic studies have characteristically focused on the functions, interactions, and regulation of proteins on a genome-wide scale. We have developed a novel approach of applying proteomic methods to studying energetic properties of proteins. In the studies reported here, we used proteolysis as a structural probe to identify proteins with energy landscapes resistant to proteolysis. The identified rigid proteins may have biotechnological applications. For instance, proteolytic resistance in proteins can ensure a longer lifetime in harsh environments. The proteins identified through this survival assay may be suitable for such engineering applications as is, or as templates for protein engineering. Modification of this method should enable us to analyze proteomes according to other interesting energetic properties, such as thermal stability, kinetic stability, and resistance to chemical denaturants. Energetics-based protein profiling on a proteomic scale will allow us to understand better how conformational energy landscapes are encoded by sequences and structures, and how these energetic properties are related to biological functions. This understanding also will provide important basic knowledge for designing functional proteins with proper dynamics and energetics for their biochemical functions.
Materials and Methods
Preparation of soluble fraction of E. coli
E. coli K12 was grown overnight in 50 mL Luria Bertani (LB) medium and harvested. The cell pellet was resuspended in 50 mL of 20 mM Tris–HCl (pH 8.0), containing 10 mM EDTA (pH 8.0) and 250 mM NaCl, then pelleted again by centrifugation. The washed cell pellet was resuspended in 10 mL of 20 mM Tris–HCl (pH 8.0), containing 1 mM EDTA (pH 8.0) and 50 mM NaCl. The cells were lysed by lysozyme treatment and sonication, and centrifuged to remove cell debris and the membrane fraction. To prevent nucleic acids from protecting proteins against proteolysis by forming complexes, the resulting supernatant was incubated with 0.1 mg/mL DNase I and 0.1 mg/mL RNase A. MgCl2 and CaCl2 were added to 2.5 mM and 1.0 mM, respectively, for this digestion reaction. To minimize the interference from small metabolites and digested nucleic acids, the lysate was dialyzed first against 20 mM Tris–HCl (pH 8.0) containing 250 mM NaCl and then against 20 mM Tris–HCl (pH 8.0). The lysate was sterilized by passing through a 0.20-μm syringe filter and stored at −20 °C until used.
Proteolysis of E. coli proteome
Proteolysis of E. coli lysate was performed in 20 mM Tris-HCl (pH 8.0) containing 10 mM CaCl2 and 50 mM NaCl. The reaction was initiated by adding trypsin (Sigma, St. Louis, MO) or thermolysin (Sigma, St. Louis, MO) to the final concentration of 0.40 mg/mL and incubated at 25 °C for four days. Samples were taken at a designated time points to monitor the progress of proteolytic digestion by SDS-PAGE. The activity of trypsin in the proteolysis reaction was determined by monitoring the cleavage of Nα-p-tosyl-L-arginine methyl ester (TAME) (Sigma, St. Louis, MO) spectrophotometrically at 247 nm. The activity of thermolysin in the reaction was determined by monitoring the cleavage of o-Aminobenzoyl-Ala-Gly-Leu-Ala-p-nitrobenzylamide (ABZ-Ala-Gly-Leu-Ala-pNA; MD Biosciences, St.Paul, MN) with a fluorometer.
2-D Electrophoresis and protein identification
To identify the survivors, E. coli lysate was incubated with 0.40 mg/mL trypsin or 0.40 mg/mL thermolysin in 20 mM Tris-HCl (pH 8.0) containing 10 mM CaCl2 and 50 mM NaCl at 25 °C for 4 days. 10 μL of 0.50 M EDTA (pH 8.0) (for thermolysin) or 0.10 M Phenylmethylsulphonylfluoride (for trypsin) was added to 240 μL of reactions to quench further proteolysis. 2-D gel electrophoresis was performed as described by the manufacturer.40 Proteins in samples were precipitated by acetone. Pellets were dissolved in the rehydration buffer (8 M Urea, 20 mM DTT, 2% CHAPS, 2.0% IPG buffer, 0.002% Bromophenol blue). The proteins in the rehydration buffer were separated with 13-cm Immobiline Drystrips, pH 3-10 NL (Amersham, Piscataway, NJ) in the first dimension and with continuous 15% SDS gels in the second dimension. Gels were stained by Colloidal Blue staining reagent (INVITROGEN, Carlsbad, CA). Spots on the gels were cut and digested with Montage In-Gel DigestZP Kit (MILLIPORE, Billerica, MA). About 1 μL of the tryptic peptide mixture from each gel spot was combined with an equal volume of matrix solution and allowed to dry on a MALDI target. The matrix solution used was a 10 mg/mL solution of alpha-cyano-4-hydroxycinnamic acid in 0.1% TFA/50% acetonitrile. Mass spectra were acquired on a Bruker (Billerica, MA) Reflex III mass spectrometer. Proteins corresponding to each spot were identified by a web-based software, MS-FIT (http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm).
Determination of proteolysis kinetics of maltose binding protein
The coding regions for maltose binding protein (malE) was amplified by polymerase chain reaction, cloned, and expressed under the control of the T7 promoter. Maltose binding protein was purified with ion exchange and gel filtration chromatography. The purity of each protein was verified using SDS-PAGE and mass spectroscopy.
Proteolysis kinetics was determined based on the method reported elsewhere.8 0.50 mg/mL maltose binding protein was incubated at 25 °C with 0.40 mg/mL thermolysin or 0.40 mg/mL trypsin in 20 mM Tris-HCl buffer (pH 8.0), containing 50 mM NaCl and 10 mM CaCl2. For thermolysin digestion, 15 μL of the reaction was removed at each time point and quenched by adding 5 μL of 50 mM EDTA (pH 8.0). For trypsin digestion, 18 μL of the reaction was removed and quenched by adding 2 μL of 0.1 M PMSF in ethanol. 20 μl of SDS sample buffer was then added to each quenched reaction and boiled. 10 μl of the mixture was used for SDS PAGE. Gels were stained with Sypro Red fluorescent dye (Molecular Probes, Eugene, OR) and scanned with Typhoon imaging system (GE Healthcare, Piscataway, NJ). Proteolysis kinetic constants (kp) were determined by monitoring the change in intensity of intact protein bands. Determined kinetic constants were fit to Eq. 2 to determine kop and Kop.
Acknowledgements
We thank Kael F. Fischer for discussions and suggestions in development of the project; James A. Blair and Arnab Chowdry for their technical assistance; Alan Sachs, Eric N Nicholson, Srebrenka Robic, Erik J. Miller, Elizabeth A. Shank, David E. Wildes, Jason F. Cellitti, and Tracy A. Young for thoughtful comments on this manuscript. This work was supported by Grant GM50945 (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: Native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNase H. Nat. Struct. Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 4.Kern D, Volkman BF, Luginbuhl P, Nohaile MJ, Kustu S, Wemmer DE. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature. 1999;402:894–898. doi: 10.1038/47273. [DOI] [PubMed] [Google Scholar]
- 5.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 6.Parsell DA, Sauer RT. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J. Biol. Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 7.Matouschek A. Protein unfolding - an important process in vivo? Curr. Opin. Struct. Biol. 2003;13:98–109. doi: 10.1016/s0959-440x(03)00010-1. [DOI] [PubMed] [Google Scholar]
- 8.Park C, Marqusee S. Probing the high energy states in proteins by proteolysis. J. Mol. Biol. 2004;343:1467–1476. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 9.Imoto T, Yamada H, Ueda T. Unfolding rates of globular proteins determined by kinetics of proteolysis. J. Mol. Biol. 1986;190:647–649. doi: 10.1016/0022-2836(86)90250-0. [DOI] [PubMed] [Google Scholar]
- 10.Fontana A, Polverino de Laureto P, De Filippis V, Scaramella E, Zambonin M. Probing the partly folded states of proteins by limited proteolysis. Fold. Des. 1997;2:R17–26. doi: 10.1016/S1359-0278(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 11.Tyndall JD, Nall T, Fairlie DP. Proteases universally recognize beta strands in their active sites. Chem. Rev. 2005;105:973–999. doi: 10.1021/cr040669e. [DOI] [PubMed] [Google Scholar]
- 12.Linderstrøm-Lang K. Peptide bonds in globular proteins. Nature. 1938;142:996. [Google Scholar]
- 13.Linderstrøm-Lang K. Structure and enzymatic break-down of proteins. Cold Spring Harbor Symp. Quant. Biol. 1950;14:117–126. doi: 10.1101/sqb.1950.014.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Anfinsen CB, Scheraga HA. Experimental and theoretical aspects of protein folding. Adv. Protein Chem. 1975;29:205–300. doi: 10.1016/s0065-3233(08)60413-1. [DOI] [PubMed] [Google Scholar]
- 15.Wang L, Kallenbach NR. Proteolysis as a measure of the free energy difference between cytochrome c and its derivatives. Protein Sci. 1998;7:2460–2664. doi: 10.1002/pro.5560071124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaswal SS, Sohl JL, Davis JH, Agard DA. Energetic landscape of α-lytic protease optimizes longevity through kinetic stability. Nature. 2002;415:343–346. doi: 10.1038/415343a. [DOI] [PubMed] [Google Scholar]
- 17.Keil B. Specificity of Proteolysis. Springer-Verlag; New York: 1992. [Google Scholar]
- 18.Cunningham EL, Jaswal SS, Sohl JL, Agard DA. Kinetic stability as a mechanism for protease longevity. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11008–11014. doi: 10.1073/pnas.96.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park C, Marqusee S. Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods. 2005;2:207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 20.Corey D, Craik C. An investigation into the minimum requirements for peptide hydrolysis by mutation of the catalytic triad of trypsin. J. Am. Chem. Soc. 1992;114:1784–1790. [Google Scholar]
- 21.Hollien J, Marqusee S. A thermodynamic comparison of mesophilic and thermophilic ribonucleases H. Biochemistry. 1999;38:3831–3836. doi: 10.1021/bi982684h. [DOI] [PubMed] [Google Scholar]
- 22.Szilágyi A, Závodszky P. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: results of a comprehensive survey. Struct. Fold. Des. 2000;8:493–504. doi: 10.1016/s0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 23.Sun YJ, Rose J, Wang BC, Hsiao CD. The structure of glutamine-binding protein complexed with glutamine at 1.94 Å resolution: Comparisons with other amino acid binding proteins. J. Mol. Biol. 1998;278:219–229. doi: 10.1006/jmbi.1998.1675. [DOI] [PubMed] [Google Scholar]
- 24.Quiocho FA, Spurlino JC, Rodseth LE. Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor. Structure. 1997;5:997–1015. doi: 10.1016/s0969-2126(97)00253-0. [DOI] [PubMed] [Google Scholar]
- 25.Avaeva S, Kurilova S, Nazarova T, Rodina E, Vorobyeva N, Sklyankina V, Grigorjeva O, Harutyunyan E, Oganessyan V, Wilson K, Dauter Z, Huber R, Mather T. Crystal structure of Escherichia coli inorganic pyrophosphatase complexed with SO42−. Ligand-induced molecular asymmetry. FEBS Lett. 1997;410:502–508. doi: 10.1016/s0014-5793(97)00650-9. [DOI] [PubMed] [Google Scholar]
- 26.Koellner G, Luic M, Shugar D, Saenger W, Bzowska A. Crystal structure of the ternary complex of E. coli purine nucleoside phosphorylase with formycin B, a structural analogue of the substrate inosine, and phosphate (Sulphate) at 2.1 Å resolution. J. Mol. Biol. 1998;280:153–166. doi: 10.1006/jmbi.1998.1799. [DOI] [PubMed] [Google Scholar]
- 27.Duée E, Olivier-Deyris L, Fanchon E, Corbier C, Branlant G, Dideberg O. Comparison of the structures of wild-type and a N313T mutant of Escherichia coli glyceraldehyde 3-phosphate dehydrogenases: implication for NAD binding and cooperativity. J. Mol. Biol. 1996;257:814–38. doi: 10.1006/jmbi.1996.0204. [DOI] [PubMed] [Google Scholar]
- 28.Oliver DB. Periplasm. In: Neidhardt FC, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. 2 edit. Vol. 1. ASM Press; Washington, D.C.: 1996. pp. 88–103. 2 vols. [Google Scholar]
- 29.Martinez A, Kolter R. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 1997;179:5188–5194. doi: 10.1128/jb.179.16.5188-5194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng M, Wang X, Templeton LJ, Smulski DR, LaRossa RA, Storz G. DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 2001;183:4562–4570. doi: 10.1128/JB.183.15.4562-4570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dukan S, Nystrom T. Oxidative stress defense and deterioration of growth-arrested Escherichia coli cells. J. Biol. Chem. 1999;274:26027–26032. doi: 10.1074/jbc.274.37.26027. [DOI] [PubMed] [Google Scholar]
- 32.Andrews SC, Le Brun NE, Barynin V, Thomson AJ, Moore GR, Guest JR, Harrison PM. Site-directed replacement of the coaxial heme ligands of bacterioferritin generates heme-free variants. J. Biol. Chem. 1995;270:23268–23274. doi: 10.1074/jbc.270.40.23268. [DOI] [PubMed] [Google Scholar]
- 33.Gottesman S. Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- 34.Robinson NE, Robinson AB. Molecular clocks. Proc. Natl. Acad. Sci. U.S.A. 2001;98:944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell. 2003;11:671–83. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 36.Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 1998;5:294–303. doi: 10.1038/nsb0498-294. [DOI] [PubMed] [Google Scholar]
- 37.Plaza del Pino IM, Ibarra-Molero B, Sanchez-Ruiz JM. Lower kinetic limit to protein thermal stability: A proposal regarding protein stability in vivo and its relation with misfolding diseases. Proteins. 2000;40:58–70. doi: 10.1002/(sici)1097-0134(20000701)40:1<58::aid-prot80>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 38.Scalley-Kim M, Baker D. Characterization of the folding energy landscapes of computer generated proteins suggests high folding free energy barriers and cooperativity may be consequences of natural selection. J. Mol. Biol. 2004;338:573–583. doi: 10.1016/j.jmb.2004.02.055. [DOI] [PubMed] [Google Scholar]
- 39.Godoy-Ruiz R, Ariza F, Rodriguez-Larrea D, Perez-Jimenez R, Ibarra-Molero B, Sanchez-Ruiz JM. Natural selection for kinetic stability is a likely origin of correlations between mutational effects on protein energetics and frequencies of amino acid occurrences in sequence alignments. J. Mol. Biol. 2006;362:966–978. doi: 10.1016/j.jmb.2006.07.065. [DOI] [PubMed] [Google Scholar]
- 40.Berkelman T, Stenstedt T. 2-D Electrophoresis Using Immobilized pH Gradients: Principles and Methods, Amersham Bioscience. Piscataway; NJ: 1998. [Google Scholar]
- 41.Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]