

Abstract
The release of phosphate (Pi) is an important element in actomyosin function and has been shown to be accelerated by the binding of myosin to actin. To provide information about the structural elements important for Pi release, possible escape pathways from various isolated myosin II structures have been determined by molecular dynamics simulations designed for studying such slow processes. The residues forming the pathways were identified and their role evaluated by mutant simulations. Pi release is slow in the pre-powerstroke structure, an important element in preventing the powerstroke prior to actin binding, and is much more rapid for Pi modeled into the post-rigor and rigor-like structures. The backdoor route suggested by Yount et al. is dominant in the pre-powerstroke and post-rigor states, while a different path is most important in the rigor-like state. This finding suggests a novel mechanism for the actin-activated acceleration of Pi release.
INTRODUCTION
Myosins form a superfamily whose members use ATP to control their interactions with actin filaments, thereby creating force and directed movements [1, 2]. All members of the family, like other molecular motors, such as the kinesins, utilize the differential binding of ATP and its hydrolysis products to induce the conformational changes involved in the motion [3]. A schematic description of the global structural changes of myosin and its interaction with actin was given by Lymn and Taylor in 1971 [4] before any high resolution X- ray structure of myosin or actin were available (see Figure 1A). In such a scheme, the myosin cycle can be regarded as consisting of two parts: the states corresponding to free myosin (i.e., myosin not bound to actin) and the states of the actomyosin complex. Binding of ATP to the strongly bound actomyosin complex (the rigor state) promotes the conformational transition to the post-rigor state, which causes myosin to dissociate from actin. ATP hydrolysis follows coupled to a major change in the orientation of the lever arm, the so-called “recovery stroke”. The latter leads to the pre-powerstroke state and the motor domain weakly rebinds to actin. Release of Pi and the “powerstroke”, the force generating step of the cycle, follow. Dissociation of ADP leads the system back to the rigor state.
FIG. 1.

The Lymn-Taylor cycle. Figure (A) shows the functional details of the cycle; Figure (B) shows the position of the switches for each state. (I) Rigor state - both switch I and switch II are open; no ligand. (II) Post-rigor state - switch I closed, switch II open; ATP. (III) Post-recovery state - switch I closed, switch II closed; ATP. (IV) Pre-powerstroke state - both switches are closed; ADP.Pi. The post-recovery state is assumed to have the same lever arm position as in pre-powerstroke, but ATP has not been hydrolyzed yet; it is in equilibrium with the post-rigor state (no structure is available but see Ref. [45]).
Crystal structures have provided detailed information on the myosin conformations in the parts of the Lymn-Taylor cycle not involving actin; i.e., the post-rigor and the pre-powerstroke states (see Figure 1). As yet, no high resolution structures for the states of myosin bound to actin exist, although there is evidence from cryoelectron microscopy [5] that one of the structures recently obtained for myosin V with no nucleotide bound corresponds to the strongly bound actomyosin complex [6] (the so-called rigor state). This structure along with the nucleotide-free structure of Dictyostelium myosin II [7], which shows partial closure of the actin-binding cleft and a very similar twisting of the β-sheet, are referred to as the rigor-like state. The available X-ray structures show that the conformations of the myosin motor domain can be described in terms of the relative positions of pseudo-rigid subdomains with connecting elements; see the description in Refs. [8, 9]. The important subdomains are referred to as U50, L50, N terminal, and converter (with the attached lever arm); see Figure S1. The major difference in the subdomain positions between the post-rigor and the pre-powerstroke conformations is the orientation of the lever arm, which is “down” in the former and “up” in the latter. Rigor-like structures have a down position of the lever arm, and differ from the post-rigor state in having a “closed” cleft between the U50 and L50 subdomains; the cleft is fully open in the post-rigor state and is partially open in the pre-powerstroke state. Based primarily on cryoelectron microscopy reconstructions [10, 11], closing of the cleft is believed to be essential for strong binding to actin.
Important differences among the various structures exist for loops in the neighborhood of the ligand binding site. They are switch I, which is part of the U50 subdomain, the P-loop, which is part of the N terminal subdomain, and switch II, which links U50 to L50 near the γ-phosphate pocket. Switch I, switch II, and the P-loop form a Walker-type structural motif [12], which is generally found in the neighborhood of the nucleotide binding site in proteins in which ATP is required for conformational change [13, 14]; they are collectively referred to as the nucleotide binding elements. In the literature [7] the positions of switch I is referred to as closed (C) if Ser 237 interacts with the Mg2+ ion and otherwise open (O); switch II is defined as closed (C) if Gly 457 forms a main-chain hydrogen bond to the γ-phosphate of ATP and otherwise is open (O); the Dictyostelium sequence numbering is used. Thus, the (switch I/switch II) elements can be categorized in a qualitative sense as (O/O) for the rigor-like state, (C/O) for the post-rigor state and (C/C) for the pre-powerstroke state. No (O/C) structure is known, though its existence has been postulated [7, 15]. The position of the P-loop has been categorized as “up” in the rigor state and “down” in the post-rigor and pre-powerstroke states [16], though considerably less attention has been given to it.
For myosin V, kinetic studies indicate that there exists a pre-powerstroke state bound to actin from which Pi is released [17]. The molecular mechanism of Pi release that is presumed to be activated by actin and coupled to the powerstroke is not known [16].
Because of the importance of the Pi release step in the actomyosin cycle, we study here possible pathways for the escape of Pi in a number of myosin II structures. Since the time scale of the actual Pi release is on the order of milliseconds or longer [18], we use a variant of the multicopy simulation method [19], termed multicopy enhanced sampling (MCES), to accelerate the phenomenon so that it can be studied by molecular dynamics simulations. In the MCES approach a number of copies of the ligand (30 in the present case) are simulated at high temperature while the protein is kept at 300 K. The method has been employed in studies of ligand escape from myoglobin after photolysis [19, 20] and of the exit pathway for retinoic acid from the retinoic acid receptor [21]. A number of alternative techniques exist for studying the escape of a small ligand from the interior of the protein. Examples are the “random-expulsion molecular dynamics” method [22], the “implicit ligand sampling” [23] and metadynamics [24]. Interestingly, the latter two [23, 25] and MCES [19] have been applied to study the exit pathways of CO from myoglobin and give similar results. If quantitative information on the free-energy profile is desired the “implicit ligand sampling” and metadynamics are useful approaches. However, the MCES method appears best suited for searching for likely pathways, the purpose of the present paper.
We have applied the MCES approach to a series of structures available for myosin II from the slime mold (Dictyostelium discoideum; we refer to it as Dicty, in accord with standard usage). The structures are listed in Table I with their designation as parts of the Lymn-Taylor cycle and the positions of the elements that play a role in the subsequent analysis. The structures studied fall into two major classes: those with switch II open for which Pi release is fast, and those with switch II closed with significantly slower release of Pi. The pre-powerstroke structure of myosin II falls in the latter class, the rigor-like and the post-rigor structures in the former. The simulated release shows that multiple pathways can be used by the phosphate to escape the protein but only one is dominant in each structure. The calculations suggest that Pi release from myosin involves different molecular mechanisms in the presence or absence of actin. Detailed analysis of the Pi release trajectories from the pre-powerstroke structure is used to obtain a microscopic interpretation of the high activation barrier observed in the absence of actin. The residues that contribute to the release barrier are determined by alanine scanning mutagenesis in silico. The results obtained for the rigor-like structure suggest a possible molecular mechanism for the actin-activated acceleration of Pi release.
TABLE 1.
Structures of Dicty myosin II used in the study. The rigor-like [7] and post-rigor [8] structures were downloaded from the Protein Data Bank; the pre-powerstroke structure was provided by Jon Kull (personal communication).
| State | PDB code | Ligand(s) | switch I | switch II | P-loop | β-sheet | U50/L50 cleft |
|---|---|---|---|---|---|---|---|
| rigor-like | 1Q5G | - | open | open | up | twisted | closed |
| post-rigor | 1MMD | ADP, Mg2+, BeF3 | closed | open | down | flattened | open |
| pre-powerstroke | M754 | ADP, Mg2+, BeF3 | closed | closed | down | flattened | partially closed |
The simulation results are presented in the next section. The interpretation of the simulations and a discussion on their biological significance follow. The preparation of the structures along with the description of the methods is given in the last section.
RESULTS
MCES simulations results
Pi release was studied in the myosin structures listed with the positions of switch I, switch II, and the P-loop in Table I; i.e., the pre-powerstroke, the post-rigor, and the rigor-like states of Dicty myosin II. For each protein structure, 50 explicit-water MCES simulations were carried out as described in the “Experimental Procedures” with the following temperatures for the ligand (Pi): 300, 450, 600, 900, 1500, 2000, and 3000 K. These simulations correspond to 350 MCES runs per protein structure. Each run was 100 ps long and included 30 phosphate copies. Release probabilities were computed at each temperature as the number of Pi replicas released divided by 1500 (i.e., 30 replicas times 50 runs). Pi release was based on a distance criterion using a cutoff of 30 Å from its initial position; the cutoff was chosen large enough to avoid miscounting of replicas trapped in the interior of the protein and far away from the initial binding pocket. The release probabilities for the various myosin structures are shown in Figure 2 as a function of temperature. The data indicate that there is a high activation barrier for Pi release in the pre-powerstroke structure and that the barrier is much lower in the rigor-like and post-rigor structures; i.e., Pi replicas start to escape the protein at 2000 K in the pre-powerstroke state, while significant release is observed at 900 K from the other myosin conformations. Moreover, the three structures show different release trends as a function of temperature suggesting that the nature of the barrier(s) is different in the various structures (see below).
FIG. 2.

Phosphate release probabilities at various temperature values for the post-rigor, rigor-like, and pre-powerstroke states of Dicty myosin II. Release probabilities were evaluated by computing the fraction of Pi replicas released in fifty explicit-water MCES runs. Pi-release was judged on a distance criterion by using a cutoff of 30 Å from the initial position of the ligand. Uncertainties were estimated from the standard deviation of the probability distributions obtained at various temperatures of the ligand.
Visual inspection of the “successful” trajectories (i.e., those where Pi actually left the protein) indicates that several pathways exist for the ligand to escape in each protein structure. The observed paths can be grouped into six routes which include the previously suggested “backdoor” and “frontdoor” routes [26], as well as four additional paths which are referred to as “backdoor II”, “side”, “top” and “rear”; a graphical representation of the various pathways is given in Figure 3. The paths differ in that they connect the Pi binding pocket to the solvent by making use of distinct protein tubes and thus involve different amino acids. The backdoor route (back I) goes through switch I and switch II in the upward direction and the phosphate is released through the U50/L50 cleft; the frontdoor route (front) goes rightward from the Pi binding pocket through the P-loop and switch I and the phosphate is released by the ATP binding pocket; the backdoor II path (back II) goes through the P-loop and switch II and releases the phosphate in the proximity to helix SH2; the “side” path involves all three nucleotide binding elements and is somewhat located between the two backdoor routes; finally, the “top” and the “rear” routes share the first portion of the path, which is almost orthogonal to backdoor I and proceeds between the relay helix and the U50/L50 linker, to finally access the solvent by making use of different tubes on the top and the rear of the protein, respectively. Interestingly, all escape paths are visited in the three myosin states at some temperature, but with very different probabilities.
FIG. 3.

Structural representation of the calculated Pi-release pathways (left) along with their relative probabilities (right) in the three Dicty myosin II structures. Six distinct exit routes were identified: the backdoor I (red), backdoor II (blue), side (cyan), frontdoor (violet), rear (green), and top (yellow) paths. The simulations show that all escape paths can be visited in the three myosin states though with different probabilities. The most populated path per structure is given in larger spheres. On the right-hand side, release probabilities per path are shown as a function of the temperature of the ligand. The dominant path for Pi release is shown to be backdoor I for both the pre-powerstroke and post-rigor structures, and backdoor II in rigor-like.
A “sentinel” residue analysis (see “Experimental Procedures”) was used to determine the populations of the individual paths in each structure. Figure 3 shows the projections of the overall release probabilities onto the different paths, so as to highlight the individual contributions. The results indicate that despite the existence of multiple pathways in all states, one path in each structure is largely dominant; i.e., backdoor I is dominant in the pre-powerstroke and post-rigor states, while backdoor II is most probable in the rigor-like state. The biological relevance of this finding and its possible implications are described in the “Discussion” section.
Pi release in the pre-powerstroke state
The release of the phosphate from the pre-powerstroke structure was analyzed in most detail because it is the only state considered where Pi (and Mg.ADP) are actually present. The primary aim of the analysis was to identify residues blocking or hindering the escape of the phosphate along the backdoor I path, which is dominant in the pre-powerstroke state (see above). For this purpose, the contact analysis described in the “Experimental Procedures” was performed on the Pi replicas that do not escape in the MCES runs carried out at the different temperatures of the ligand. The comparison of the results obtained for increasing temperature values makes it possible to find the barriers along the escape path and to identify the residues involved. Statistics collected in the pre-release temperature range highlight the amino acids that contribute to retaining the phosphate in the interior of the protein, whereas statistics in the post-release range provide information on the ones making up the exit tube; the post-release range is defined as the interval of ligand temperature for which significant Pi release (> 10%) is observed. The results are reported in Table II. The data show that in the pre-release temperature range (temperature values between 300 and 1500 K) the phosphate is confined in a small cavity formed by a dozen residues belonging solely to the three nucleotide binding elements (i.e., the P-loop, switch I and switch II). These residues, which include the salt-bridging R238 and E459, are interconnected by a complex network of H-bonding interactions (see Figure S2) which confer a high mechanical strength to the Pi binding pocket. Actually, the binding pocket is found to be so tight that only small deformations occur in the pre-release temperature range; only two new residues are found by increasing the temperature of the ligand up to 900 K (see Table II on top, residue numbers in bold); also, all residues identified by the contact analysis in that temperature range belong to the nucleotide binding elements. Thus, the Pi-binding pocket acts as a real “cage” for the phosphate. In the post-release range (ligand temperatures equal to or higher than 2000 K), the cage breaks in the direction of backdoor I and thirteen additional residues are identified along the exit tube (see underlined residue numbers in Table II); they are not part of any nucleotide-binding element. Among them, seven amino acids are found at a ligand temperature of 2000 K, and six at 3000 K. It suggests that there are two layers of blocking residues along the exit path. Overall, the residues identified by the contact analysis as having a role in the release of Pi from the pre-powerstroke structure are listed in Table S1. The picture which emerges is that of a myosin structure with a very tight Pi binding pocket followed by two layers of residues that further hinder the release of the phosphate along the backdoor path. The rotameric state of the identified residues were monitored by computing the time series of the χ1 dihedral angle on the MCES runs at temperatures compatible with Pi release (T ≥ 2000 K). The rotameric distributions show that S181, R232, S237, R238, and E459 in the close proximity to the cage, and F461, E467, Q468, and I471 in the outer layers populate a second rotameric state (see Figure S3). These conformational rearrangements occur only at high temperature and are expected to have a role in the release process. Visual inspection of the “successful” trajectories makes it clear that simulated Pi release from the pre-powerstroke state is a consequence of local conformational transitions (see Video S1).
TABLE 2.
Residues identified by the contact analysis in the pre-release (top) and post-release (bottom) temperature range for the investigated myosin structures. For increasing temperature of the ligand, newly identified residues are shown in bold. The results of the analysis in the pre-release temperature range highlight residues which contribute to blocking Pi in the interior of the protein, the ones in the post-release temperature range highlight residues making up the exit tube. Underlined numbers in the pre-powerstroke post-release range indicate blocking residues along the “backdoor” exit tube.
| Ligand Temperature [K] | |||||||
|---|---|---|---|---|---|---|---|
| Myosin state | Element | 300 | 450 | 600 | 900 | 1500 | |
| Pre-release range | Pre-powerstroke | P-loop | 181, 185, 186 | 181, 185, 186 | 181, 182, 185, 186 | 181, 182, 185, 186 | 181, 182, 185, 186 |
| switch I | 233, 236, 237, 238 | 233, 236, 237, 238 | 230, 233, 236, 237, 238 | 230, 233, 236, 237, 238 | 230, 232, 233, 236, 237, 238 | ||
| switch II | 455, 456, 457, 459 | 455, 456, 457, 459 | 455, 456, 457, 459 | 455, 456, 457, 459 | 455, 456, 457, 459 | ||
| other | - | - | - | - | 223, 227, 239, 241 | ||
| Post-rigor | P-loop | 181 | 180, 181 | 180, 181 | 180, 181, 185 | ||
| switch I | 236, 237, 238 | 236, 237, 238 | 236, 237, 238 | 230, 236, 237, 238 | |||
| switch II | 456 | 456 | 456 | 456, 459 | |||
| other | - | 239 | 239 | 239, 262, 471 | |||
| Rigor-like | P-loop | 179, 180 | 178, 179, 180, 185 | 178, 179, 180, 181, 185 | 178, 179, 180, 181, 185 | ||
| switch I | 232, 233, 236, 237, 238 | 232, 233, 235, 236, 237, 238 | 232, 233, 235, 236, 237, 238 | 232, 233, 235, 236, 237, 238 | |||
| switch II | 455, 456, 457 | 455, 456, 457, 458 | 455, 456, 457, 458, 459 | 455, 456, 457, 458, 459 | |||
| other | - | 678, 679 | 675, 678, 679 | 655, 675, 678, 679 | |||
| Myosin state | Element | 1500 | 2000 | 3000 | |
|---|---|---|---|---|---|
| Post-release range | Pre-powerstroke | P-loop | 181, 182, 185, 186 | 181, 182,185, 186 | |
| switch I | 230, 232, 233, 236, 237, 238 | 230, 231, 232, 233, 236, 237, 238 | |||
| switch II | 455, 456, 457, 459 | 454, 455, 456, 457, 459 | |||
| other | 219, 223, 227, 228, 239, 241, 264, 265, 267, 461, 468, 471 | 219, 222, 223, 227, 228, 229 | |||
| 587 | 239, 241, 243, 264, 265, 266 | ||||
| 267, 270, 272, 275, 460, 461 | |||||
| 465, 467, 468, 471, 587 | |||||
| Post-rigor | P-loop | 180, 181, 185 | 181, 185 | 181, 185 | |
| switch I | 230, 232, 236, 237, 238 | 230, 232, 236, 237, 238 | 230, 232, 236, 237, 238 | ||
| switch II | 456, 459 | 456, 459 | 456, 459 | ||
| other | 239, 262, 263, 265, 266, 267, 461, 467, 468, 471, 587, 590 | 239, 262, 263, 265, 266, 267 | 239, 262, 265, 266, 423, 467, 468, 470, 471, 587 | ||
| 423, 461, 467, 468, 470, 471 | 467, 468, 470, 471, 587 | ||||
| 587, 590 | |||||
| Rigor-like | P-loop | 178, 179, 180, 181, 185, 186 | 178, 179, 180, 181, 185, 186 | 178, 179, 180, 181, 185, 186 | |
| switch I | 232, 233, 235, 236, 237, 238 | 232, 233, 235, 236, 237, 238 | 232, 233, 235, 236, 237, 238 | ||
| switch II | 454, 455, 456, 457, 458, 459 | 454, 455, 456, 457, 458, 459 | 454, 455, 456, 457, 458, 459 | ||
| other | 189, 219, 223, 227, 239, 241, 655, 675, 678, 679 | 189, 219, 223, 227, 239, 241, 475, 478, 654, 655, 675, 678 | 189, 219, 223, 239, 241, 265 | ||
| 475, 478, 479, 654, 655, 675 | |||||
| 679 | 678, 679 | ||||
The results show a mechanism for the high temperature release of the phosphate from the pre-powerstroke state in the simulations. A corresponding mechanism is suggested to hold at ambient temperature, but results in much longer escape times, as observed experimentally. The release occurs through backdoor I; the other escape routes are so much less probable that they can be neglected (see Figure 3). The pathway is located between the U50 and the L50 subdomains and guides the phosphate to the solvent through the large cleft between them (see Figure 4A). The exit route is composed of two segments, a short “inner tube” which confines the Pi in the interior of the protein, and a longer “outer tube” that involves two successive layers of residues. The release process involves local changes of residues, mainly dihedral angle transitions, in both the inner and the outer tube. Since the residues belonging to the latter were found in the post-release temperature range, the former is predicted to be associated with the highest free-energy barrier. In the inner tube, the most important rearrangement involves the salt-bridging residues R238 and E459, which undergo a large deformation to allow the displacement of the phosphate into the outer tube (see Figure 4B). The rearrangement of the salt bridge is coupled to the rotameric transition of the nearby I471, which makes space to accommodate the large rotation of E459. Once in the outer tube, the phosphate searches for an escape route. At this stage, steric clashes with the first layer of residues located at the beginning of the tube hinder the diffusion of Pi. The phosphate hits the layer and rebounds back to the inner cage several times. Repeated escape attempts promote the rotameric transition of two residues in the first layer (i.e., F461 and Q468) and one residue in the second layer (i.e., E467), plus a series of minor changes of nearby residues. These rearrangements involve residues coupled by H-bonding interactions, such as E264 and R267, which must be at least temporarily broken, or bulky residues partially exposed to the solvent, such as F270, whose movement corresponds to a solvation penalty. In both cases, these rather local changes involve energy barriers which contribute to the overall activation barrier for Pi release (see “Discussion”). After that, the exit tube opens wide to the solvent and the phosphate is free to diffuse away from the protein. The qualitative description of the release mechanism is complemented by more quantitative data coming from mutant simulations (see below).
FIG. 4.
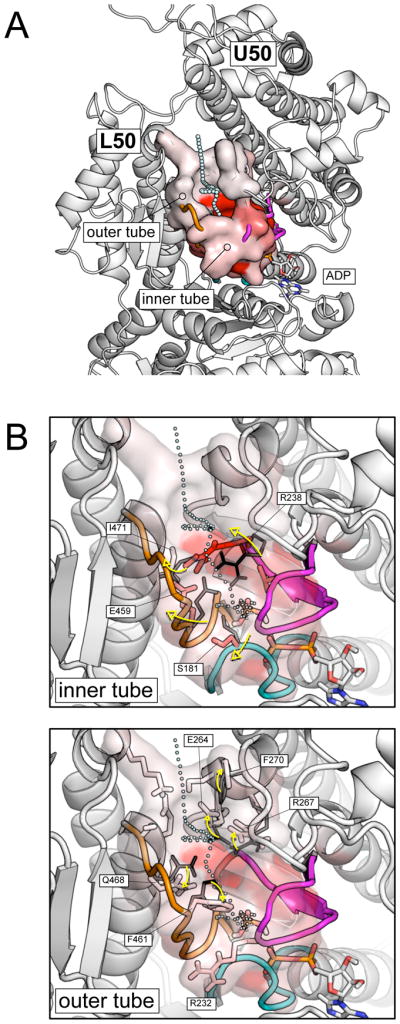
Pi release from the pre-powerstroke state. (A) A pictorial representation of the most populated exit pathway (backdoor I) from the pre-powerstroke myosin in the post-release temperature range (T > 2000 K). The exit path is shown by cyan spheres; the molecular surface of the residues making up the exit tube is color-coded from white to red according to the number of contacts they make with the phosphate along the simulations; i.e., the red color corresponds to a fraction of contacts close to one, the white color to a fraction close to zero. The exit pathway is located between the U50 and L50 subdomains and is composed of a short “inner tube”, which cages the Pi in the interior of the protein, and a longer “outer tube”, which involves two successive layers of residues. (B) Local conformational rearrangements in the inner (top) and the outer tube (bottom) that allows the release of Pi. The P-loop, switch I and switch II are shown in cyan, magenta, and orange, respectively. Residues identified by the rotameric analysis as playing a role in gating Pi are labeled. Black thin and white to red color-coded thick sticks show the equilibrium and the release-prone conformations of the identified residues.
Mutational studies in silico
The actual contribution of the residues identified as having a role in Pi release was determined by mutational studies in silico. Eight alanine myosin mutants in the pre-powerstroke conformation were constructed and tested: S181A, T230A, R232A, R238A, E459A, R267A, F461A, and Q468A. The first five mutations involve residues in the inner tube; the remaining three residues are in the outer tube. Given the limited character of alanine mutagenesis [27], the structures of the mutants were obtained from the wild-type pre-powerstroke conformation by removing the atoms that are no longer present and energy minimizing the resulting systems. All mutants were heated up and equilibrated in the presence of a sphere of explicit waters with the stochastic boundary, as done for the wild type (see “Experimental Procedures”). Twenty MCES release simulations were performed per mutant at a temperature of 1500 K for the ligand. This value is the highest temperature in the pre-release range for the wild type. At this temperature, it is expected that single-point mutations that lower barriers would show higher release probabilities than the wild type. Release probabilities computed for the eight mutants are given in Figure 5 (on top). All of them show increased release, with the salt-bridging residues R238 and E459 being most important and the others having somewhat smaller effects. Alanine site-directed mutagenesis of residues S181 and R232 also increases the overall release probability but favors the release through the sidedoor (see Table III), which is not the dominant path in the pre-powerstroke state. Overall, the in-silico mutational studies indicate that residues R238, E459, F461, and Q468 have a role in Pi release from the pre-powerstroke structure. The first two, which are located in the inner tube and form the salt-bridge between switch I and switch II, make the largest contribution to the release barrier and are crucial for gating. The others, which are located in the outer tube, have significantly lower effects.
FIG. 5.

Release probabilities from pre-powerstroke, post-rigor, and rigor-like structures for the wild-type myosin and a series of single-point alanine mutants. The results were obtained from twenty MCES runs carried out in the pre-release temperature range; i.e., a ligand temperature of 1500 K was employed in the pre-powerstroke state and 600 K in the rigor-like and post-rigor states. Uncertainties were estimated from the standard deviation of the probability distributions. White boxes correspond to the release probability of the wild-type myosin in the post-release temperature range (i.e., a ligand temperature of 2000 K in the pre-powerstroke state and 900 K in the rigor-like and post-rigor states) and are used for comparison. The mutational studies show that: (i) alanine substitutions of the salt-bridging residues R238 and E459 significantly lower the release barrier in the pre-powerstroke state (top); (ii) the same is true for R238 in the post-rigor state (middle); (iii) mutations of residues E180 and R232 into alanines have large effects in the rigor-like but not in the pre-powerstroke state.
TABLE 3.
Release probability per path for the wild-type pre-powerstroke myosin and eight single-point alanine mutants. The results were obtained from a series of MCES simulations in the pre-release temperature range for the wild-type, i.e., at a ligand temperature of 1500 K. The data indicate that residues R238 and E459, which form the salt-bridge crucial for hydrolysis [39], make the largest contribution to the release barrier along the backdoor I path. Alanine substitutions of S181, T230, and R232 slightly increase the release probability but favor side-door release.
| Protein sequence | Exit path (%) | |||||
|---|---|---|---|---|---|---|
| back I | back II | side | front | rear | top | |
| wt | x | x | 1.0 | x | 0.2 | 0.1 |
| S181A | 3.7 | x | 7.5 | 0.2 | 0.5 | 0.2 |
| T230A | x | x | 2.8 | x | x | x |
| R232A | 2.3 | x | 8.8 | x | 0.3 | x |
| R238A | 38.5 | x | 2.8 | x | x | 0.3 |
| E459A | 23.7 | x | 1.3 | x | x | x |
| R267A | 2.7 | x | x | x | x | x |
| F461A | 5.3 | x | 1.3 | x | x | x |
| Q468A | 4.0 | x | 2.5 | x | x | x |
| wt (2000 K) | 11.9 | x | 1.9 | 0.1 | 0.3 | x |
Pi release in the post-rigor and rigor-like states
The above analysis was repeated for the post-rigor and the rigor-like states; for their properties see Table I. These two structures are studied in comparison with the pre-powerstroke state to obtain insights into conformational aspects of myosin which could play a role in gating Pi release. The results of the contact analysis are reported in Table II. This time the pre-release range is narrower and includes ligand temperatures between 300 and 600 K; it indicates a lower barrier for Pi release than that in the pre-powerstroke state. In this range of temperatures, only seven blocking residues were identified in the post-rigor state, whereas 17 residues were found in the rigor-like state. These data along with the result that the main exit pathway differs in these two myosin states suggest that Pi release from post-rigor and rigor-like is different, although the energetics are similar. By monitoring the rotameric state of the blocking residues during Pi release, i.e., analysis of the χ1 dihedral time series, two groups of five and six residues having a role in gating from the post-rigor and rigor-like myosin, respectively, were identified (data not shown). The individual contributions of these residues were accessed by alanine mutagenesis in silico. Release probabilities computed for the post-rigor and rigor-like myosin mutants are reported in Figure 5. In neither case, single-point alanine substitutions led to a significant change in the release path (see Tables S2 and S3).
In the post-rigor state, R238 clearly makes the dominant contribution to the escape barrier. Other residues, such as S456, also contribute to it although to a much lower extent. Considering that in the post-rigor state the dominant escape path was found to be backdoor I (see Figure 3), these results indicate that Pi release is essentially the same in the post-rigor and the pre-powerstroke states; see Figure 6A. The only important difference regards the activation barrier of the process, which is much lower in the former. Our in silico mutagenesis analysis, which reported rather large contributions for both R238 and E459 in the pre-powerstroke state, shows that the escape barrier in the post-rigor state is lower because the salt-bridge between R238 and E459 is not formed. Breaking of this salt-bridge opens the gate of the backdoor I exit route.
FIG. 6.

Comparison of Pi release paths in various myosin structures. (A) Pi release from the pre-powerstroke (grey) and the post-rigor (blue) states; the structures were aligned on the N-terminal subdomain. The simulation results indicate that in both cases Pi is released through backdoor I between the U50 and L50 subdomains. The exit path goes between switch I and switch II and mainly involves the salt-bridging residues R238 (part of switch I) and E459 (part of switch II). The result that the release barrier was found to be much higher in the pre-powerstroke state along with the mutagenesis data strongly indicate that R238 and E459 are crucial for Pi release through backdoor I. When switch II is open (i.e., post-rigor), the salt-bridge is not formed and the activation barrier is sensibly lower. Closing of switch II, while going from the post-rigor to the pre-powerstroke state (yellow dashed arrow), restores the salt-bridge and the activation barrier increases. (B) Pi release from the pre-powerstroke (grey) and the rigor-like (red) states; the structures were aligned on the L50 subdomain. In the rigor-like state Pi release proceeds through an alternative path, here named backdoor II. The latter involves residues of switch II and the P-loop and releases the phosphate through a tube located between the N-terminal and L50 subdomains. Backdoor II opening requires the displacement of the P-loop (black dashed arrow), which moves away from the switches. The P-loop movement does not involve the breaking of the salt-bridge between R238 and E459, which is distant from the rigor-like exit path.
In the rigor-like state, the largest contributions come from E180 and R232 (see Figure 5, on bottom). Mutations of these residues into alanines result in a considerable decrease of the escape barrier, so that the Pi release rate is expected to be significantly accelerated in these mutants. The simulation results for the rigor-like state show that: (i) Pi release occurs through backdoor II; (ii) the salt-bridge between residues R238 and E459, which is formed in this state, does not play a gating role; and (iii) residues E180 and R232, which were not detected in the analysis of Pi release from the pre-powerstroke structure, make an important contribution to the release barrier. Thus and by contrast to post-rigor, the rigor-like state appears to behave quite differently from the pre-powerstroke state; see Figure 6B. The simulation data suggest that the conformational change which occurs during the powerstroke, i.e., the actin-activated transition connecting the pre-powerstroke and the rigor-like structures, may sensibly change the Pi release properties of the myosin molecule. The significance of these results is discussed below.
DISCUSSION
Myosin function involves a large conformational change (the so-called powerstroke) when myosin is attached to actin. This internal motion engenders a relative displacement of actin with respect to myosin. In muscle, the concerted powerstrokes of multiple myosins (myosin II) pull actin so as to shorten the sarcomere, while in the case of myosins V and VI, the powerstroke results in the walking motion of individual two-headed myosins on actin. It is generally accepted that in myosin V the powerstroke is triggered by Pi release, after ATP hydrolysis in the pre-powerstroke state [28]; for myosins II, the exact role of Pi release is not fully resolved [29]. Since hydrolysis occurs in free myosin, Pi release must be slow enough to permit the actin binding step to take place before Pi escape. After actin binding, more rapid Pi release should occur to activate the powerstroke, as it does in myosin V. Kinetic measurements have shown that the Pi release rate from free myosin II is in the range 0.01s−1 to 0.06s−1 at room temperature [18] and 120s−1 for free myosin V [17], while in the bound form the rate is accelerated to 35s−1 in myosin II [30] and to >200s−1 for myosin V [17]. Thus, the essential point related to Pi release for a functional actomyosin cycle is that the escape barrier must be relatively high in the free pre-powerstroke state and be lowered significantly by binding to actin. Since there are no pre-powerstroke structures available for actin-bound myosin, the molecular mechanism of this accelerated release is not known. For the other myosin states (e.g. rigor and post-rigor) the Pi release rate does not play a role since free Pi is not present. However, these states can be used to obtain insights into conformational aspects of myosin which could be important in gating.
Given Pi release even in the presence of actin (s to ms) is slow on the simulation time scale (ps to ns), we have used a specialized molecular dynamics technique called multicopy enhanced sampling (MCES) to investigate possible escape pathways in various X-ray structures of myosin II from Dictyostelium discoideum (Dicty): the rigor-like, the post-rigor, and the pre-powerstroke structures; the recently reported myosin II structures from squid and sea scallop [31], which more closely resemble the rigor-like state of myosin V than that of Dicty, have not been considered to avoid secondary effects associated with protein sequence variation. In the MCES method the temperature of the multiple Pi replicas (30 in this case) is varied while that of the protein and water is kept at 300 K. Rate enhancement is required to obtain release events during the simulations, which are much shorter (on the order of 100 ps) than the actual time for release. In the present analysis, Pi release was simulated with an explicit treatment of the solvent by employing a sphere of water molecules large enough to solvate all cavities leading to the protein surface. The simulations were carried out over a wide range of temperatures (from 300 to 3000 K) so as to determine the accessible paths and obtain qualitative information concerning the relative activation energies for Pi release in the various structures of myosin. Hundreds of events were sampled for each protein structure at different temperatures for Pi. The comparison of the temperature required for significant escape of Pi on the simulation time scale shows that the escape barrier is high (≥1500 K) for the pre-powerstroke structure, while it is lower (≥900 K) for the rigor-like and post-rigor structures. Since Pi can only be released after hydrolysis, the results confirm that the barrier is high where it needs to be; i.e., in the pre-powerstroke state of myosin not bound to actin. Using the standard open/closed terminology (see Table I and Ref. [7]) the results indicate that the position of switch II is the essential element determining the escape barrier; the barrier is low when switch II is “open” (rigor-like and post-rigor) and high (pre-powerstroke) when switch II is “closed” independent of the position of switch I. However, we note that this description for the positions of the elements in the ATP binding pocket needs modification for a valid microscopic interpretation of the phenomenon. An important element is the position of the P-loop (see Ref. [16]); i.e., the so-called opening of the switches actually corresponds to an increase in their distance relative to the P-loop and is not simply related to their positions.
The atomistic detail provided by the MCES simulations allows for a structural interpretation of the calculated rate for Pi release in the various myosin structures. The simulations showed that at least six pathways exist for Pi to escape from the protein at the appropriate temperature, with one path dominant in each structure. In this context, the “backdoor” route suggested by Yount et al. [26, 32] was found to be dominant in the pre-powerstroke and post-rigor states, while another escape route, here named backdoor II, was found to be dominant in the rigor-like state. The latter pathway appears to be similar to the one suggested by Cope et al. [33], though no direct evidence for its existence was given. The two backdoor paths are very different: backdoor I path is located between switch I and switch II and guides the phosphate into the large cleft between the U50 and L50 subdomains (see Video S2); backdoor II path goes between the P-loop and switch II and releases Pi in proximity to helix SH2 through a short tube between the N-terminal and the L50 subdomain (see Video S3 and Figure 6). These results indicate that Pi release, if it could occur, would follow essentially the same path in the post-rigor and the pre-powerstroke states, although the energetic barriers are different; a distinct pathway exists in the rigor-like state.
The existence of backdoor II suggests that it should be considered, as well as backdoor I, as a low-barrier escape route in the actin-activated acceleration of Pi release. A state prior to the powerstroke when myosin is bound to actin has been identified by kinetic studies [34] and is referred to as the Sleep-Hutton state [28]. However, there are no high resolution data concerning its structure nor have cryoelectron microscopy measurements identified this state. Based on early structures of myosin II corresponding to what are now called the post-rigor and the pre-powerstroke states [35, 36], Houdusse and Sweeney [9] (also reviewed in Ref. [28]) speculated that actin might induce myosin to transiently adopt a post-rigor like conformation, so as to release Pi prior to the formation of the strongly bound actomyosin complex. This hypothesis followed the observation of Yount et al. [26] that a backdoor exit route for Pi (here identified as backdoor I) was visible in the crystal structure of the post-rigor state, where switch II is open, but not in the structure for the pre-powerstroke state, where switch II is closed. It was suggested that the re-opening of switch II after hydrolysis, as well as the U50/L50 cleft, was the essential element controlling the release rate from myosin. Such a model was ruled out by later mant-nucleotide fluorescence experiments on myosin V which showed that the Sleep-Hutton state is not equivalent to the post-rigor state [37]. The fluorescence data was interpreted as requiring a structural transition with a rearrangement of switch I for Pi release; the data did not provide information on the position of switch II. Thus, the repositioning of switch I was suggested to be essential to create an exit route for Pi release [37]. This conclusion was in line with the model proposed by Reubold et al. from the analysis of the crystal structure of nucleotide-free myosin II [7], which showed the first myosin II conformation in which both switch I and switch II have moved away from the nucleotide-binding pocket; i.e., they are both open in the standard terminology. Based on this structure, Reubold et al. speculated that the weak association of myosin to actin results in the opening of switch I. The latter was presumed to break the salt-bridge between R238 and E459 and it was suggested that Pi release is accelerated through a “trapdoor” mechanism, which promotes Pi release through a path alternative to the backdoor route of Yount et al. [26]. However, no details were given concerning the nature of the path. A similar model was proposed by Holmes et al. based on electron cryo-microscopy reconstructions of decorated actin [15]. In both papers, the Sleep-Hutton state would have switch I open and switch II closed, with no role assigned to the latter for Pi release; also the position of the P-loop was not discussed.
The simulation results suggest a possible alternative mechanism for accelerated Pi release from actomyosin. They show that Pi release is fast (i.e., it has a low energy barrier) when both switches are open with the release proceeding through the backdoor II path, which was found to be dominant in the rigor-like state. We suggest that increased binding to actin results in the progressive displacement of the P-loop away from the phosphate binding pocket. Such an actin-activated “opening” of the P-loop corresponds to an increase of the distance between the P-loop and both switches and is equivalent to the simultaneous opening of the latter. The P-loop movement results in the opening of backdoor II (see Figure 6) which is the critical event in the release of Pi from the rigor-like state. One important question related to the above concerns the molecular mechanism by which actin binding triggers the opening of the P-loop. A recent normal mode analysis of the transition between the rigor-like and post-rigor states of myosin V has shown that cleft closure is mechanically coupled to the twisting of the β-sheet through the large swing of the U50 subdomain, which opens and shuts the cleft [38]. Furthermore, the analysis made clear that there is a strong coupling between the relative positions of the P-loop and switch I and the twist of the β-sheet, so that an increased twisting of the latter corresponds to an increased distance between the former two, and vice versa [38]. These results suggest that the twisting of the central β-sheet to a more stable conformation is the essential element in the actin-activated “opening” of the P-loop. During the transition from a weakly-bound to a more strongly-bound (Sleep-Hutton) actomyosin complex, the closure of the cleft that is required for strong binding to actin [5, 15] would induce a more pronounced twisting of the β-sheet, which in turn results in the displacement of the P-loop from the switches and opening of backdoor II (see Figure 7). An important element of the mechanism is that this can occur with a pre-powerstroke state position for the converter and lever arm. The actin-activated acceleration of Pi release would thus proceed without involving the re-opening of the U50/L50 cleft nor the breaking of the salt-bridge between residues R238 and E459. The mutational studies carried out in silico for the myosin rigor-like structure indicated that alanine substitutions of residues E180 and R232 result in a considerable decrease of the escape barrier. Moreover, these residues were shown to contribute to the barrier for Pi release only when it proceeds through backdoor II. Thus, mutation of E180 and R232 to alanines is expected to sensibly accelerate Pi release in the presence of actin (if backdoor II is used) and not to have sizeable effects in the absence of actin (if backdoor I is used). If these mutations did not have other effects on activity, they would provide a test of the proposed mechanism of Pi release in actomyosin.
FIG. 7.

The molecular mechanism proposed for the actin-activated Pi release from actomyosin. The pre-powerstroke (grey) and the rigor-like (red) structures of myosin II have been aligned by fitting the Cα atoms of the L50 subdomain. The nucleotide-binding elements (i.e., the P-loop, switch I and switch II) are shown in pale colors for the pre-powerstroke structure, while brighter colors are used for the rigor-like structure. During the transition from a weakly-bound to a more strongly-bound actomyosin complex, the closure of the U50/L50 cleft, which is required for strong binding to actin [5], induces a more pronounced twisting of the β-sheet; the latter results from the coordinated twisting of the U50 and the N-terminal subdomains about the axis τ̂ (shown in green). The β-sheet twisting promotes the “opening” of the P-loop, i.e., a large displacement of the P-loop from both switches, which opens the backdoor II path (shown as red dots). In such a model, the actin-activated acceleration of Pi release involves a backdoor path alternative to the one proposed by Yount et al. [26] and proceeds without involving the re-opening of the cleft nor the breaking of the salt-bridge between R238 (part of switch I) and E459 (part of switch II).
The present study shows the molecular origin of the barrier in the pre-powerstroke state, which blocks Pi release and prevents myosin from futile ATP hydrolysis; i.e., a powerstroke occurring in the absence of actin. The main exit route (backdoor I) is composed of a short inner tube formed by a dozen residues belonging to the P-loop, switch I and switch II and a longer outer tube leading to the U50/L50 cleft. Interestingly, all of them are conserved in the alignment of 82 myosin sequences [33]. The identified residues undergo local conformational transitions to allow Pi release. These structural changes, which occur in a highly dense medium and involve the breaking of H-bonding interactions, result in the high pre-powerstroke activation barrier. In silico alanine mutagenesis of the residues involved showed that R238 and E459 in the inner tube make the largest contribution to the overall release barrier, though multiple barriers were found along the main escape pathway (backdoor I). The calculations for the pre-powerstroke structure in the absence of actin indicate that both switch I and switch II contribute to the Pi exit barrier as long as the salt-bridge between R238 (part of switch I) and E459 (part of switch II) is formed. Opening of switch II breaks the salt-bridge and results in a significantly lower escape barrier, as was found in the post-rigor state. One possibility suggested by Gyimesi et al. [29] is that, in the absence of actin, the rate determining step in Pi release is the slow conformational transition from the myosin pre-powerstroke to the post-rigor state, from which release is fast. Given the sizable difference in the Pi release rate from free myosin II and myosin V (see above) and the sequence conservation of the residues found to form the exit path in the pre-powerstroke state, our present results indirectly support this model. However, it should be noted that because the pre-powerstroke structure for myosin V is not available, we cannot exclude that the release occurs directly from the pre-powerstroke state. Although R238 and particularly E459 have a functional role in hydrolysis [39], study of Ala mutants in the presence of high concentrations of Pi and ADP could determine whether they have a role in Pi release.
In summary, the MCES calculations of Pi release pathways in the available myosin II crystal structures have provided a molecular basis for the slow release of Pi in the pre-powerstroke structure and suggested a possible mechanism for release of Pi in the actomyosin complex. We hope that these results will stimulate experiments to test the proposals in this paper.
EXPERIMENTAL PROCEDURES
Crystal structures and their preparation
The crystal structures for the rigor-like and post rigor simulations were taken from the Protein Data Bank (PDB), while that for the pre-powerstroke state was provided by Jon Kull (personal communication) (see Table I); the latter is very similar to 1VOM [40] but provides coordinates for the converter subdomain. Details of the preparation of the structures for the simulations are given in the “Supplemental Information”. All calculations were performed with the CHARMM program [41, 42] (version c35).
Multicopy Enhanced Sampling (MCES) Method
The multicopy enhanced sampling molecular dynamics (MCES) is a useful approach for obtaining information about possible ligand escape pathways for cases where the actual rate of the process requires a time much longer than that accessible to molecular dynamics simulations [19, 21]. In this method, the system is divided into two parts: the ligand(s) and the receptor. The ligand of interest (here H2PO4−) is usually a small part of the system, whereas the receptor (protein and other ligands) are much larger. The ligand is replicated so that multiple copies of the ligand are introduced, while only a single copy of the protein is present; both systems are treated in full atomic detail. Replicas do not interact with each other and the interaction energy between the receptor and each ligand copy is scaled down by a factor equal to the total number of the ligand copies. Thus, all copies of the ligand are seen by the protein as one average ligand. The use of multiple copies of the ligand does not significantly increase the computation time, but it dramatically improves the sampling statistics for the ligand escape pathways. In the present study the ligand is H2PO4−, the receptor is myosin plus Mg2+ and ADP; i.e. we are not studying ADP and Mg2+ release. The simulations are carried out with an explicit treatment of the solvent (see Figure S1) to make the protein dynamics more realistic, while the interactions between the phosphate and the water molecules are set to zero to accelerate the diffusion rate of the ligand outside the protein. To overcome any barrier(s) and simultaneously obtain a qualitative estimate of their relative magnitudes, a series of MCES simulations with different ligand temperatures were carried out while keeping the temperature of the protein at 300K. This was achieved by using separate Nosé-Hoover thermostats for the ligand copies and the protein [43, 44]. For details of the MCES simulations, see “Supplemental Information”.
Sentinel residue analysis
The population of the individual Pi release pathways per myosin structure and ligand temperature were determined by a “sentinel” residue analysis. In this analysis, a few optimally located residues, called sentinels, are used to monitor the release events and cluster them into a discrete number of pathways. In practice, the last contact between the phosphate and one of the sentinel residues along the MCES trajectory are used to define the release event. For details, see “Supplemental Information”.
Contact analysis
The aim of the analysis is to identify residues that have a high probability of being in contact with Pi replicas during the MCES simulations. These residues are likely to block or hinder the release of Pi and are of interest to obtain an atomistic interpretation of the activation barrier involved. The analysis is based on the calculation of distances between the Pi replicas and the myosin residues around the binding pocket to determine if the latter are in contact with the former during the time evolution of the system. For details, see “Supplemental Information”.
Supplementary Material
Acknowledgments
We are very grateful to A. Houdusse for discussions about myosin and, more particularly, many helpful suggestions from her based on a careful reading of the manuscript. We thank L. Sweeney for discussions on myosin kinetics. The myosin crystal structure of the pre-powerstroke Dicty myosin II with a resolved converter was kindly provided by J. Kull. We thank A. Blondell for advice on the implementation of the MCES methodology. This work was granted access to the HPC resources of [CCRT/CINES/IDRIS] under the allocation 2009-[x2009075114] made by GENCI (Grand Equipement National de Calcul Intensif ). The analysis was performed on a Beowulf cluster running Linux in Strasbourg and we thank M. Spichty and F. Rao for setting up and maintaining the cluster. The research done at Harvard was supported in part by a grant from the National Institute of Health (USA) and that in Strasbourg by the Centre National de la Recherche Scientifique (France). M.C. and Y.A. were supported by a grant from the Human Frontier Science Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schliwa M. Molecular Motors. Wiley-VCH; 2003. [Google Scholar]
- 2.Geeves M, Holmes K. Structural mechanism of muscle contraction. Ann Rev Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- 3.Karplus M, Gao Y. Biomolecular motors: the F1-ATPase paradigm. Curr Opin Struct Biol. 2004;14:250–259. doi: 10.1016/j.sbi.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Lymn R, Taylor E. Mechanism of adenosine triphosphate hydrolysis by acto-myosin. Biochemistry. 1971;10:4617–4624. doi: 10.1021/bi00801a004. [DOI] [PubMed] [Google Scholar]
- 5.Holmes K, Schroder R, Sweeney H, Houdusse A. The structure of the rigor complex and its implications for the power stroke. Philos Trans R Soc Lond B Biol Sci. 2004;359:1819–1828. doi: 10.1098/rstb.2004.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coureux P, Wells A, Ménétrey J, Yengo C, Morris C, Sweeney H, Houdusse A. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425:419–423. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- 7.Reubold T, Eschenburg S, Becker A, Kull F, Manstein D. A structural model for actin-induced nucleotide release in myosin. Nature Struct Biol. 2003;10:826–830. doi: 10.1038/nsb987. [DOI] [PubMed] [Google Scholar]
- 8.Fisher A, Smith C, Thoden J, Smith R, Sutoh K, Holden H, Rayment I. X-ray Structures of the Myosin Motor Domain of Dictyostelium discoideum Complexed with MgADP.BeFx and MgADP.AlF−4. Biochemistry. 1995;34:8960–8972. doi: 10.1021/bi00028a004. [DOI] [PubMed] [Google Scholar]
- 9.Houdusse A, Sweeney H. Myosin motors: missing structures and hidden springs. Curr Opin Struct Biol. 2001;11:182–194. doi: 10.1016/s0959-440x(00)00188-3. [DOI] [PubMed] [Google Scholar]
- 10.Rayment I, Holden H, Whittaker M, Yohn C, Lorenz M, Holmes K, Milligan R. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 11.Holmes K. Abstracts. J Muscle Res Cell Motil. 2004;25:243–281. [Google Scholar]
- 12.Walker J, Saraste M, Runswick M, Gay N. Distantly related sequences in the alpha-subunits and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vale R. Switches, Latches, and Amplifiers: Common Themes of G Proteins and Molecular Motors. J Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith C, Rayment I. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys J. 1996;70:1590–1602. doi: 10.1016/S0006-3495(96)79745-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes K, Angert I, Kull F, Jahn W, Schroder R. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 2003;425:423–427. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- 16.Geeves M, Holmes K. The molecular mechanism of muscle contraction. Advances in Protein Chemistry. 2005;71:161–193. doi: 10.1016/S0065-3233(04)71005-0. [DOI] [PubMed] [Google Scholar]
- 17.De La Cruz E, Wells A, Rosenfeld S, Ostap E, Sweeney H. The kinetic mechanism of myosin V. Proc Natl Acad Sci USA. 1999;96:13726–13731. doi: 10.1073/pnas.96.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakelin S, Conibear P, Woolley R, Floyd D, Bagshaw C, Kovacs M, Malnasi-Csizmadia A. Engineering Dictyostelium discoideum myosin II for the introduction of site-specific fluorescence probes. J Muscle Res Cell Motil. 2002;23:673–683. doi: 10.1023/a:1024411208497. [DOI] [PubMed] [Google Scholar]
- 19.Elber R, Karplus M. Enhanced sampling in molecular dynamics: use of the time-dependent Hartree approximation for a simulation of carbon monoxide diffusion through myoglobin. J Am Chem Soc. 1990;112:9161–9175. [Google Scholar]
- 20.Gibson Q, Regan R, Elber R, Olson J, Carver T. Distal pocket residues affect picosecond ligand recombination in myoglobin. J Biol Chem. 1992;267:22022–22034. [PubMed] [Google Scholar]
- 21.Blondel A, Renaud J, Fischer S, Moras D, Karplus M. Retinoic Acid Receptor: A Simulation Analysis of Retinoic Acid Binding and the Resulting Conformational Changes. J Mol Biol. 1999;291:101–115. doi: 10.1006/jmbi.1999.2879. [DOI] [PubMed] [Google Scholar]
- 22.Lüdemann S, Lounnas V, Wade R. How do Substrates Enter and Products Exit the Buried Active Site of Cytochrome P450cam? 1. Random Expulsion Molecular Dynamics Investigation of Ligand Access Channels and Mechanisms. Journal of Molecular Biology. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 23.Cohen J, Arkhipov A, Braun R, Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophysical journal. 2006;91:1844–1857. doi: 10.1529/biophysj.106.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laio A, Parrinello M. Escaping free-energy minima. Proceedings of the National Academy of Sciences. 2002;99:12562. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ceccarelli M, Anedda R, Casu M, Ruggerone P. CO escape from myoglobin with metadynamics simulations. Proteins. 2008;71:1231. doi: 10.1002/prot.21817. [DOI] [PubMed] [Google Scholar]
- 26.Yount R, Lawson D, Rayment I. Is myosin a back door enzyme? Biophys J. 1995;68:44–49. [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham B, Wells J. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 28.Sweeney H, Houdusse A. The motor mechanism of myosin V: insights for muscle contraction. Philos Trans R Soc Lond B Biol Sci. 2004;359:1829–1842. doi: 10.1098/rstb.2004.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gyimesi M, Kintses B, Bodor A, Perczel A, Fischer S, Bagshaw C, Malnasi-Csizmadia A. The mechanism of the reverse recovery step, phosphate release, and actin activation of dictyostelium myosin II. J Biol Chem. 2008;283:8153. doi: 10.1074/jbc.M708863200. [DOI] [PubMed] [Google Scholar]
- 30.White H, Belknap B, Webb M. Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate. Biochemistry. 1997;36:828–11. doi: 10.1021/bi970540h. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Gourinath S, Kovacs M, Nyitray L, Reutzel R, Himmel D, O’Neall-Hennessey E, Reshetnikova L, Szent-Gyorgyi A, Brown J, et al. Rigor-like Structures from Muscle Myosins Reveal Key Mechanical Elements in the Transduction Pathways of This Allosteric Motor. Structure. 2007;15:553–564. doi: 10.1016/j.str.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Lawson J, Pate E, Rayment I, Yount R. Molecular Dynamics Analysis of Structural Factors Influencing Back Door Pi Release in Myosin. Biophys J. 2004;86:3794–3803. doi: 10.1529/biophysj.103.037390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cope M, Whisstock J, Rayment I, Kendrick-Jones J. Conservation within the myosin motor domain: implications for structure and function. Structure. 1996;4:969–987. doi: 10.1016/s0969-2126(96)00103-7. [DOI] [PubMed] [Google Scholar]
- 34.Sleep J, Hutton R. Exchange between inorganic phosphate and adenosine 5′-triphosphate in the medium by actomyosin subfragment 1. Biochemistry. 1980;19:1276–1283. doi: 10.1021/bi00548a002. [DOI] [PubMed] [Google Scholar]
- 35.Rayment I, Rypniewski W, Schmidt-Base K, Smith R, Tomchick D, Benning M, Winkelmann D, Wesenberg G, Holden H. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 36.Fisher A, Smith C, Thoden J, Smith R, Sutoh K, Holden H, Rayment I. X-ray structures of the myosin motor domain of dictyostelium discoideum complexed with MgADP.BeFx and MgADP.AlF4+ Biochemistry. 1995;34:8960–8972. doi: 10.1021/bi00028a004. [DOI] [PubMed] [Google Scholar]
- 37.Rosenfeld S, Lee Sweeney H. A model of myosin V processivity. J Biol Chem. 2004;279:40100–40111. doi: 10.1074/jbc.M402583200. [DOI] [PubMed] [Google Scholar]
- 38.Cecchini M, Houdusse A, Karplus M. Allosteric Communication in Myosin V: From Small Conformational Changes to Large Directed Movements. PLoS Comput Biol. 2008;4:e1000129. doi: 10.1371/journal.pcbi.1000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onishi H, Kojima S, Katoh K, Fujiwara K, Martinez H, Morales M. Functional transitions in myosin: Formation of a critical salt-bridge and transmission of effect to the sensitive tryptophan. Proc Natl Acad Sci USA. 1998;95:6653–6658. doi: 10.1073/pnas.95.12.6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith C, Rayment I. X-ray structure of the magnesium (II). ADP. vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 Å resolution. Biochemistry. 1996;35:5404–5417. doi: 10.1021/bi952633+. [DOI] [PubMed] [Google Scholar]
- 41.Brooks B, Bruccoleri R, Olafson B, States D, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. Journal of computational chemistry. 1983;4:187–217. [Google Scholar]
- 42.Brooks B, Brooks C, III, Mackerell A, Jr, Nilsson L, Petrella R, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30 doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81:511–519. [Google Scholar]
- 44.Hoover W. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 45.Fischer S, Windshügel B, Horak D, Holmes K, Smith J. Structural mechanism of the recovery stroke in the Myosin molecular motor. Proc Natl Acad Sci USA. 2005;102:6873–6878. doi: 10.1073/pnas.0408784102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.