

Abstract
Endolymphatic hydrops (ELH), hearing loss and neuronal degeneration occur together in a variety of clinically significant disorders, including Meniere’s disease (MD). However, the sequence of these pathological changes and their relationship to each other are not well understood. In this regard, an animal model that spontaneously develops these features postnatally would be useful for research purposes. A search for such a model led us to the PhexHyp-Duk mouse, a mutant allele of the Phex gene causing X-linked hypophosphatemic rickets. The hemizygous male (PhexHyp-Duk/Y) was previously reported to exhibit various abnormalities during adulthood, including thickening of bone, ELH and hearing loss. The reported inner-ear phenotype was suggestive of progressive pathology and spontaneous development of ELH postnatally, but not conclusive. The main focuses of this report are to further characterize the inner ear phenotype in PhexHyp-Duk/Y mice and to test the hypotheses that (a) the PhexHyp-Duk/Y mouse develops ELH and hearing loss postnatally, and (b) the development of ELH in the PhexHyp-Duk/Y mouse is associated with obstruction of the endolymphatic duct (ED) due to thickening of the surrounding bone. Auditory brainstem response (ABR) recordings at various times points and histological analysis of representative temporal bones reveal that PhexHyp-Duk/Y mice typically develop adult onset, asymmetric, progressive hearing loss closely followed by the onset of ELH. ABR and histological data show that functional degeneration precedes structural degeneration. The major degenerative correlate of hearing loss and ELH in the mutants is the primary loss of spiral ganglion cells. Further, PhexHyp-Duk/Y mice develop ELH without evidence of ED obstruction, supporting the idea that ELH can be induced by a mechanism other than the blockade of longitudinal flow of endolymphatic fluid, and occlusion of ED is not a prerequisite for the development of ELH in patients.
Keywords: Endolymphatic hydrops, Meniere’s disease, Phex mutant
1. Introduction
Endolymphatic hydrops was first found to be associated with Meniere’s disease (MD) by Hallpike and Cairns (1938) and Yamakawa (1938). The significance of ELH in the development of symptoms in patients with MD has been recognized by many investigators, and the histological finding of Reissner’s membrane distension is taken to be the hallmark of ELH (Arenberg et al., 1970; Belal and Ylikoski, 1980; Fraysse et al., 1980; Paparella, 1984; Schuknecht, 1976). However, ELH occurs in conditions other than Meniere’s disease, including delayed endolymphatic hydrops following trauma (Dodson et al., 2007), relapsing polychondritis (Murata et al., 2006), and some cases of acoustic neuroma (Mahmud et al., 2003). Unfortunately, current therapies directed at alleviating symptoms via presumed amelioration of ELH using diuretics and shunt surgery have been largely unsuccessful in preventing the progression of hearing deterioration in patients with ELH-related disorders. This is due, in part, to a lack of understanding of the relationship among ELH, hearing loss and neural degeneration. Animal models are necessary since appropriate human specimens are difficult to obtain.
Kimura and Schucknecht developed in 1965 a guinea pig model of ELH (Kimura and Schuknecht, 1965). ELH is achieved in this model by surgical obliteration of the endolymphatic duct (ED), and distention of Reissner’s membrane ensues days to weeks after surgery. The guinea pig model has proven to be useful in the study of ELH, particularly in relation to the human MD condition (Aran et al., 1984; Hott et al., 2003; Nadol, 1990). However, this model suffers from some important limitations. First, it requires surgery to produce the phenotype. Second, this model does not reliably produce vestibular symptoms (Horner, 1995). Lastly, and most importantly, induction of ELH by surgical obliteration of the ED is a mechanism that deviates from the spontaneous development of ELH that usually occurs in MD. An animal model that spontaneously develops ELH and hearing loss some time after birth would be a good supplement to the classic guinea pig model.
Mouse mutants have served as useful models for a wide variety of inner ear disorders. A survey of literature for a potential mouse model for hearing loss and ELH drew our attention to a mutant called PhexHyp-Duk (Lorenz-Depiereux et al., 2004). The PhexHyp-Duk mouse harbors a loss-of-function mutation in the Phex gene, and hemizygous males (PhexHyp-Duk/Y) exhibit various phenotypic features including shortened hind legs and tail, hypophosphatemia, hypocalcemia and rickets-like bone disease. In the BALB/cAnBomUrd genetic background, the PhexHyp-Duk/Y mouse develops an ear phenotype that includes variable hearing loss and compromised balance function. A cochlear duct section from a 5-month-old PhexHyp-Duk/Y mouse (only one time point was reported) showed ELH and thickening of the bone surrounding the cochlea with the presence of a precipitate in the scala tympani (Lorenz-Depiereux et al., 2004). This report led us to hypothesize that (a) PhexHyp-Duk/Y mice develop ELH and hearing loss postnatally, and (b) development of ELH in PhexHyp-Duk/Y mice is associated with obstruction of the endolymphatic duct (ED) due to thickening of the surrounding bone. To test these hypotheses, we undertook a detailed characterization of the inner ear phenotype in PhexHyp-Duk/Y mice, and the results are presented here.
2. Methods
2.1. Mice
The PhexHyp-Duk allele, a spontaneous mutation in the BALB/cAnBomUrd background (now to be shortened to BALB/cUrd), was obtained from The Jackson Laboratory (TJL). PhexHyp-Duk mutation maintained in the C57BL/6J (B6) background was also available/obtained from TJL. To obtain Phex males and control littermates, BALB/cUrd (or B6) PhexHyp-Duk carrier females (+/PhexHyp-Duk) were bred with BALB/cUrd (or B6) Phex wildtype (+/Y) mice. The Animal Care and Use Committee of CWRU approved the care and use of the mice in this study.
Male mice carrying the PhexHyp-Duk allele in the BALB/cUrd background typically showed at least one sign of vestibular dysfunction, including head bobbing, unstable gait and/or circling behavior. Although carrier females also displayed periodic circling behavior, the overall phenotype, including small body size and slight abnormal shape of the skull, was more consistent in PhexHyp-Duk/Y males and was reliably confirmed by the Phex genotyping scheme described later. We, therefore, used PhexHyp-Duk/Y mice and compared the results to +/Y sibling mice for all analyses.
2.2. Auditory-evoked brainstem response (ABR)
Auditory brainstem response (ABR) testing was conducted as previously described (Zheng et al., 2006). Briefly, mice aged P21 and older were anesthetized with an intraperitoneal injection of ketamine, xylazine and acepromazine at doses of 40 mg/kg, 5 mg/kg, and 1 mg/kg, respectively. Some mice aged P21–P25 were anesthetized by an IP injection of Avertin (tri-bromo-ethanol, concentration 0.31 mg g−1 body weight) as we found that this increased the anesthetic survival of this younger subset.
Body temperature was maintained at 37–38 °C by placing the mice on a homeothermic heating pad (Harvard apparatus, Holliston, Massachusetts) within a soundproof chamber during testing. ABR testing was carried out using a SmartEP system from Intelligent Hearing Systems (Miami, FL). Platinum subdermal needle electrodes were inserted at the vertex and ventrolaterally to the right ear and to the left ear. For an initial survey of hearing function in the PhexHyp-Duk allele in the BALB/cUrd background from P21 to P300, we used a click stimulus to cover a broad range of frequencies. Click stimuli of 100 ms duration were presented for at least 500 sweeps to both the left and right ears (one at a time) through high frequency transducers (a closed system). ABR thresholds were obtained from both ears for each animal by reducing the stimulus intensity from 100 dB SPL in 10 dB steps and finally repeated in 5 dB steps until the lowest intensity that could evoke a reproducible ABR pattern was detected.
We examined B6-PhexH-D/Y mice at various time points after birth with a special focus on the young-adult stage (P21–P130 days old), a period during which inner ear phenotype was clearly observed in the BALB/cUrd background. For an initial survey of the hearing function in the PhexHyp-Duk allele in the BALB/cUrd background, we tested hearing function in mutant males and sibling controls at approximately P21 (at weaning), P28 (~1 month old), P61 (~2 months old), P93 (~3 months old) and P131 (~4 months old). At each time point, many mutants (n ≥ 5) and several control siblings (n ≥ 3) were tested.
2.3. Anatomical analysis
For morphological studies, the inner ears of PhexHyp-Duk/Y mice in the BALB/cUrd background were examined at postnatal days 0, 5, 10, 15, 21, 25, 40, 60, 90 and 300. Five or more PhexHyp-Duk/Y and 2 control (+/Y) mice at each time point were processed for histological analysis; all mice P21 or older were tested for ABR threshold before they were processed for histological analysis. Many mutants (P21 n = 12, P25 n = 10, P30 n = 14, P40 n = 12, P60 n = 11, P90 n = 7) and several control siblings (n > 5) per time point were tested. In all cases, inner ear tissues were fixed by perilymphatic perfusion with phosphate-buffered 2.5% glutaraldehyde through the round window after removing the stapes and puncturing the round window membrane. The dissected temporal bone was then immersed in fixative for 24 h at 4 °C and rinsed in 0.1 M sodium phosphate buffer (pH 7.4). For light microscopic study, the temporal bones were subsequently decalcified in 0.35 M EDTA and embedded in glycol methacrylate. Tissue sections were then cut to a thickness of 2–5 μm and stained with toluidine blue. The inner ear sections were examined for abnormalities involving the cochlear duct, stria vascularis, and spiral ganglion. Distention of Reissner’s membrane in the cochlear duct was taken to be the criterion for ELH in the affected ear (Kimura and Schuknecht, 1965).
Following ABR recordings of B6-PhexH-D/Y mice at specific time points, the inner ears were processed for histological analysis as described previously. As the study was underway, we noticed that B6-PhexH-D/Y mice had normal hearing or only mild hearing loss in one ear, and the structures in the cochlear duct of both ears appeared normal. The presence of fluid (otitis media) in the middle ear could cause the mild-hearing loss. We examined the middle ear of the B6-PhexH-D/Y mice and control siblings for otitis media. To avoid bias, the researcher who carried out the middle ear observation/ear dissection was blind to the genotype and ABR results. The results were complied later, and an example of that is shown in Table 2.
Table 2.
Hearing loss and middle ear pathology in B6-PhexHyp-Duk/Y mice
| Mouse ID | Age (P) | Genotype | LE (dB SPL) | RE (dB SPL) | LE | RE |
|---|---|---|---|---|---|---|
| 701 | 61 | H-D/Y | 15 dB | 5 dB | Clear | Clear |
| 702 | 61 | +/Y | 10 dB | 5 dB | Clear | Clear |
| 685 | 93 | H-D/Y | 20 dB | 25 dB | OM | OM |
| 684 | 93 | +/Y | 10 dB | 5 dB | Clear | Clear |
| 686 | 131 | H-D/Y | 25 dB | 5 dB | OM | Clear |
| 687 | 131 | H-D/Y | 25 dB | 10 dB | OM | Clear |
| 688 | 131 | +/Y | 10 dB | 5 dB | Clear | Clear |
P = days postnatal; OM = otitis media.
2.4. Genotyping
We compared the hemizygous male (PhexHyp-Duk/Y) to the wild-type male (+/Y) mice in all the analyses described here. Our breeding scheme was as follows: PhexHyp-Duk/+ females crossed to +/Y males. Offspring from this breeding scheme could include normal (+/Y) and mutant (PhexHyp-Duk/Y) males and wild-type (+/+) and heterozygous (+/Phex) females. PhexHyp-Duk/Y mice were identified from control littermates using a protocol similar to that described previously (Lorenz-Depiereux et al., 2004) (Fig. 1A and B). Briefly, genomic PCR amplification of the Phex gene was used to screen for the presence or absence of exon 14. DNA was isolated from tail biopsies using a Qiagen DNeasy tissue kit (Valencia, CA). Primers were designed to amplify exon 10 of the Phex gene as a positive control for PCR (forward primer KA552 5′-TTGCCAAC AGTTTTCCAAAGG-3′; reverse primer KA553 5′-AAGCTCCCTACATCCCATCC-3′) and to mutant deleted exon 14 to identify mice carrying the PhexHyp-Duk allele (forward primer KA554 5′-ATAGCGTCTCTT CTGGTTGC-3′; reverse primer KA555 5′-GCTGGC TACCCTGAGTTGAG-3′). Touchdown-PCR amplification was performed using Platinum Taq Polymerase (Invitrogen). The cycling program began with an initial denaturation at 94 °C for 2 min, then 94 °C for 30 s, 62 °C for 30 s and 72 °C for 30 s, followed by 8 cycles at decreasing annealing temperatures of 1 °C, and then by an additional 26 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s, and a final extension for 2 min at 72 °C. Expected product sizes for 552/553 were 293 bp and for 554/555 were 308 bp. Both negative controls (containing no template DNA) and positive controls (containing DNA from normal siblings) were conducted for each round of genomic PCR. Products were resolved on a 2% agarose gel stained with 5% ethidium bromide and were observed under UV light.
Fig. 1.

Genotyping scheme to identify PHEXHyp-Duk/Y and +/Y littermates. (A) Schematic (not drawn to scale) showing the deleted exons in the mutant allele and the position of PCR primers used for genotyping. Primers to exon 10 (KA 552/553) serve as positive controls and primers to exon 14 (KA 554/555) are used to screen for the PHEXHyp-Duk mutation. (B) and (C) Gel pictures of Phex (B) and Sry genotyping (C) schemes used to genotype a litter of P5 mice numbered 1–5. PCR results in panel B show that mouse #4 is a PHEXHyp-Duk/Y male and mice 1, 2, 3 and 5 are either female (+/Phex or +/+) or male (+/Y); screening for Sry gene confirmed that mouse #4 was male and identified 1 and 5 as +/Y males and 2 and 3 as females. M = ΦX174/HaeIII molecular weight marker; N = no template control lane; * = non-specific product.
For positive identification of males in young litters (<P15), we used a PCR approach to detect the male specific gene, Sry, on the Y chromosome. The PCR protocol used to detect Sry was described previously (Kunieda et al., 1992) (Fig. 1C). Briefly, a two-step “nesting” PCR ampli.- cation was performed, and the first round consisted of 10 cycles of 94 °C for 30 s, 54 °C for 30 s, and 72 °C for 12 s using primers KA608 (5′-TCTTAAACTCTGAAGAAGA GA-3′) and KA609 (5′-GTCTTGCCTGTATGTGAT GG-3′). Five microlitre of first round PCR was added to 45 μl PCR mix containing primers KA610 (5′-GTGAGA GGCACAAGTTGGC-3′) and KA611 (5′-CTCTGTGT AGGATCTTCAATC-3′), and underwent 30 cycles of 94 °C for 30 s, 52 °C for 1 min, and 72 °C for 12 s. The Y chromosome specific product expected as a result of the second amplification step was 147 bp. Products were resolved on a 3% agarose gel stained with 5% ethidium bromide and observed under UV light.
3. Results
3.1. Onset and deterioration of inner ear function
Signs of vestibular dysfunction in PhexHyp-Duk/Y mice were typically observed around P15. This time varied between affected mice from the same litter, although the balance dysfunction became obvious beyond P30 in most cases. Signs of vestibular dysfunction in PhexHyp-Duk/Y mice at 2–3 weeks suggested hearing function might be affected at a young age.
To determine the onset and progression of hearing loss in PhexHyp-Duk/Y mice, click-evoked ABR was measured starting at P21, the age of mature hearing sensitivity (Fig. 2A). Representative results from different mutants aged P21–P90 are shown in Fig. 2B. At P21, most mutants tested showed close to normal hearing thresholds in one ear, but showed mild-to-moderate hearing loss in the other; only 2 mutants out of the 12 observed showed significant bilateral hearing loss by P21. At P25, 7 of the 10 mutants tested showed detectable bilateral hearing loss exceeding the threshold of the control siblings by 30 dB SPL. This hearing loss varied between the left and right ears; 1 animal had near normal thresholds and 2 had unilateral hearing loss. Beyond P25, the ABR data showed that, with age, the majority of mutants developed bilateral and asymmetric hearing loss as shown in Fig. 2B. The few remaining mutants had unilateral near-normal thresholds. To determine whether hearing loss was progressive in PhexHyp-Duk/Y mice, we carried out a separate experiment in which mutants and control mice were tested longitudinally from P21 to P90 (Fig. 2C). Mutant H-D/Y #255 showed a progressive trend with mild fluctuation; mutant H-D/Y #256 showed significant ABR threshold increase within a span of 10 days; mutant H-D/Y #257 showed stable but elevated thresholds in the right ear and slightly elevated thresholds and mild fluctuating hearing loss in the left ear during that time course. A second litter was tested longitudinally from P21 to P60 (Fig. 2D). From this litter, mutant H-D/Y #289 showed severe bilateral hearing loss by P21 and quickly escalated to a 100 dB threshold by P25. Mutant H-D/Y #290 showed a fluctuating trend with an asymmetrical progression of hearing loss from P40 to P60. Subsequent experiments on a small but different group of mice focused on frequency specific hearing loss in young mutants (P21–P40). Again, mutants showed bilateral asymmetric hearing loss, which became worse with age. The results suggest that mutants tend to acquire low frequency hearing loss sooner than high frequency hearing loss (Table 1). In sum, PhexHyp-Duk/Y mice spontaneously develop bilateral, asymmetric, and progressive hearing loss, with the onset of inner ear dysfunction in most cases centered between P25 and P30. Results also showed that mutants can display fluctuating hearing loss or hearing loss that is steady over time.
Fig. 2.

Auditory brainstem response (ABR) threshold levels for Phex mice in the BALB/cUrd background. (A) ABR chart for click stimulus from a mutant at P21 (left panel) and a control +/Y littermate (right panel). (B) Sample of ABR results using click stimulus from mutants at various time points. Control littermate ABR results not shown, but consistently gave results similar to above right panel with no more than a 5 dB difference. (C) Progression of hearing loss in one litter that was tested and retested from P21 to P90. *Mice HD/Y #256 and #257 died ~P35 and ~P72, respectively. (D) A second litter was tested from P21 to P60. For panels C and D, X-axis represents days after birth; Y-axis represents sound intensity (dB SPL).
Table 1.
ABR thresholdsa from PhexHyp-Duk/Y and +/Y mice to pure-tone stimuli
| Mouse # | Genotype | Frequency (kHz) | Hearing threshold, dB SPL |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| P21 |
P25 |
P30 |
P40 |
|||||||
| Left | Right | Left | Right | Left | Right | Left | Right | |||
| 383 | PhexH-D/Y | 2 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 4 | >100 | 90 | >100 | >100 | >100 | >100 | >100 | >100 | ||
| 8 | 85 | 80 | >100 | >100 | >100 | >100 | >100 | >100 | ||
| 16 | 75 | 75 | 100 | 85 | >100 | 90 | >100 | 90 | ||
| 32 | 60 | 60 | 80 | 85 | 75 | >100 | 75 | 90 | ||
| 400 | PhexH-D/Y | 2 | 40 | 65 | 35 | 55 | 65 | 40 | 45 | 55 |
| 4 | 30 | 65 | 30 | 55 | 55 | 35 | 35 | 60 | ||
| 8 | 20 | 60 | 20 | 45 | 45 | 35 | 25 | 55 | ||
| 16 | 5 | 30 | 15 | 15 | 40 | 15 | 15 | 35 | ||
| 32 | 25 | 40 | 20 | 30 | 40 | 30 | 25 | 40 | ||
| 406 | PhexH-D/Y | 2 | 75 | 40 | 70 | 40 | 80 | 80 | >100 | 80 |
| 4 | 65 | 35 | 65 | 35 | 65 | 70 | >100 | 75 | ||
| 8 | 50 | 25 | 55 | 25 | 65 | 65 | 80 | 75 | ||
| 16 | 45 | 10 | 45 | 10 | 50 | 50 | 80 | 65 | ||
| 32 | 45 | 35 | 45 | 35 | 35 | 50 | 80 | 75 | ||
| 385 | +/Y | 2 | 45 | 30 | 40 | 30 | 45 | 35 | 45 | 40 |
| 4 | 35 | 25 | 30 | 30 | 30 | 30 | 30 | 30 | ||
| 8 | 25 | 15 | 20 | 15 | 20 | 20 | 20 | 20 | ||
| 16 | 20 | 15 | 15 | 10 | 20 | 10 | 15 | 10 | ||
| 32 | 25 | 25 | 20 | 25 | 25 | 25 | 20 | 30 | ||
| 411 | +/Y | 2 | 40 | 35 | 40 | 30 | 40 | 30 | 40 | 35 |
| 4 | 30 | 30 | 25 | 25 | 25 | 25 | 30 | 25 | ||
| 8 | 20 | 20 | 20 | 20 | 15 | 15 | 20 | 10 | ||
| 16 | 10 | 10 | 20 | 10 | 10 | 10 | 20 | 5 | ||
| 32 | 15 | 20 | 25 | 30 | 20 | 30 | 25 | 25 | ||
Thresholds were determined by testing mice at P21, and retesting them at P25, 30 and 40.
3.2. Onset and sequence of inner ear pathology
No histological abnormalities in the cochlear duct were seen at birth. The morphology of the functional domains in the cochlear duct (Reissner’s membrane, the organ of Corti and the stria vascularis) appeared normal. The ED and crus commune (CC) were comparable to the control ear without evidence of obstruction and, most importantly, there was no sign of ELH at birth (Fig. 3). Typically, cochlear ducts appeared healthy at P5 (data not shown) and P10 (Fig. 4) with no obvious indication of cellular damage or cell death in the stria vascularis, organ of Corti or spiral ganglion.
Fig. 3.

Cross sections of the cochlear duct (basal and apical turns) and endolymphatic duct in a control (A and B) and mutant (C and D) mouse at P0. In the control (+/Y) and PhexHyp-Duk/Y (H-D/Y) mouse, Reissner’s membrane (arrows) is in normal position and the endolymphatic duct (ED) is patent. CC, crus commune. All scale bars indicate 100 μm.
Fig. 4.
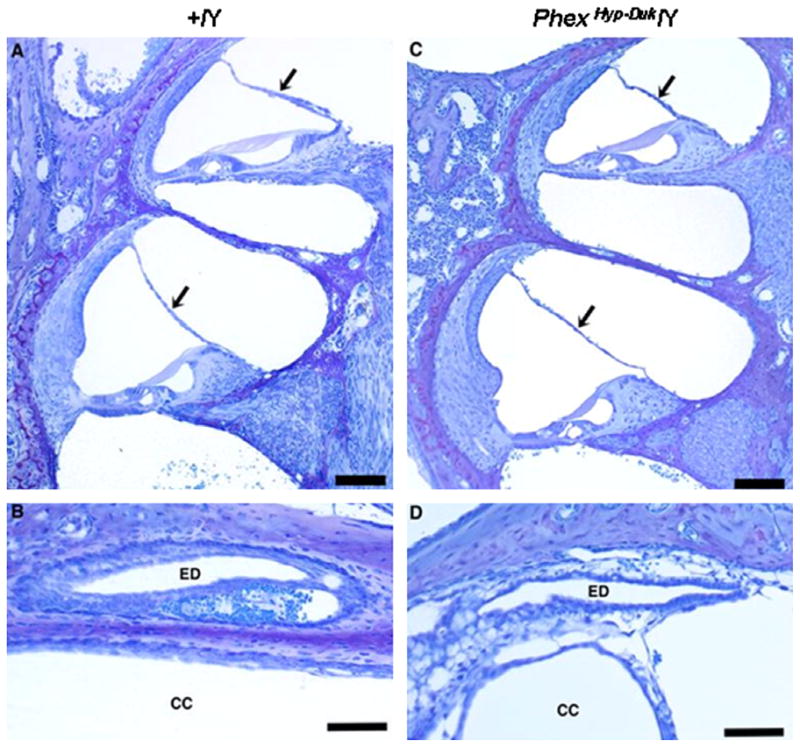
Cross sections of the cochlear duct (basal and apical turns) and endolymphatic duct in a control (A and B) and mutant (C and D) mouse at P10. In the control (+/Y) and PhexHyp-Duk/Y (H-D/Y) mouse, Reissner’s membrane (arrows) is in normal position and the endolymphatic duct (ED) is patent. CC, crus commune. All scale bars indicate 100 μm.
Around weaning age (P21–P25), some of the mutants examined began to show mild apical hydrops. In other cases, hydrops was apparent in the basal as well as apical turns. A prominent case is shown in Fig. 5. However, no significant hair cell loss was observed in any of the cochlear sections examined at P25 (Fig. 6). In all cases, the ED appeared normal along its entire course, and the path of the duct showed no occlusion or dilation. The endolymphatic sac (ES) appeared normal at P25 as well (Fig. 6). Around P25, the otic bony capsule of PhexHyp-Duk/Y was markedly thickened and surrounded by non-mineralized bone (Fig. 7).
Fig. 5.

Cross sections of the cochlear duct (basal and apical turns) and endolymphatic duct in a control (A and B) and mutant (C and D) mouse at P21. The PhexHyp-Duk/Y (H-D/Y) mouse shows well-developed endolymphatic hydrops compared to the control (+/Y); arrows indicate displaced Reissner’s membrane. The endolymphatic ducts (ED) are patent in both the affected and control mice. CC, crus commune. All scale bars indicate 100 μm.
Fig. 6.

Cross sections of cochlear duct (A), endolymphatic duct (B and C) and endolymphatic sac (D) from a PhexHyp-Duk/Y(H-D/Y) mouse at P25. The cochlear duct of the apical turn (A) shows moderate endolymphatic hydrops (the arrow indicates displaced Reissner’s membrane). Scale bar indicates 100 μm. The proximal portion of the patent endolymphatic duct (ED) in the area near the union of the saccular and utricular ducts is seen in (B). Sc, saccular lumen; arrow points in direction of saccular duct leading into the endolymphatic duct (ED); Ut, utricular lumen; V, utriculoendolymphatic valve at origin of utricular duct. Scale bar indicates 150 μm. (C) illustrates the patent endolymphatic duct (ED) approximately midway along its course near the crus commune (CC). Vn, vein that accompanies the duct. Scale bar indicates 100 μm. (D) Cross section of the endolymphatic sac (ES), which is open and appears normal. Scale bar indicates 125 μm.
Fig. 7.

Otic capsule bone. Low-power micrographs showing cross sections of otic capsules (A) from a control (+/Y) mouse at P25 and (B) from a PhexHyp-Duk/Y mouse at P30. The mutant shows endochondrial ossification without formation of mineralized bone. The scale bars indicate 300 μm.
By P40, most mutants demonstrated severe ELH as evidenced by distension of Reissner’s membrane. The ED was patent in both the affected and control animals (Fig. 8). The cells of the spiral ganglion and the organ of Corti remained intact despite well documented hearing loss at this age. By P90, the inner ear phenotype of mutants varied but was generally severe and included profound hearing loss. Mutants showed severe hydrops in all parts of the labyrinth, and cochlear duct sections showed displaced Reissner’s membrane (Fig. 9). In Fig. 9A–D, the apical cochlear turn from a mutant animal is compared to an unaffected sibling. In this animal, the ED was not occluded or narrowed and was open throughout its entire course. The endolymphatic sac was open and looked essentially normal. In mutant mice, all turns of the cochlea had hydrops, and in some specimens, Reissner’s membrane appeared to be ruptured in the basal-most part of the first turn. In some cases, the membranous wall of the saccule was ruptured as well, apparently due to hydrops (data not shown). Analysis of additional specimens at P90 showed no obvious strial pathology (Fig. 9E and F) in PhexHyp-Duk/Y. By P90, spiral ganglion degeneration in most turns of the cochlea was apparent. In some instances, the entire apical portion of the spiral ganglion was completely degenerated before degeneration of ganglion cells in the basal turn of the cochlea (Fig. 9C apical turn; Fig. 10C basal turn). Although spiral ganglion cells showed extensive degeneration, the hair cells were largely preserved, suggesting ganglion cell loss may precede loss of hair cells (Figs. 9D and 10). The ABR and histology data from control siblings (+/Y) demonstrates that the inner ear phenotype in the young-adult mutants (P21–P90) is associated with the Phex mutation, and age-related effect does not set in until later. Age-related hearing loss and spiral ganglion cell loss were observed in control siblings at P300, but the phenotype was much more severe in PhexHyp-Duk/Y mice by P300 (Figs. 11–13).
Fig. 8.

Cross sections of the cochlear duct (upper basal turn) and endolymphatic duct in a control (A and B) and PhexHyp-Duk/Y (C and D) mouse at P40. The arrows indicate Reissner’s membrane in control (A) or PhexHyp-Duk/Y mouse (C). The endolymphatic ducts (ED) are patent in both the affected and control animals. CC, crus commune. All scale bars indicate 100 μm.
Fig. 9.

Cochlear cross sections from control (+/Y) and PhexHyp-Duk/Y (H-D/Y) mice at P90. A,B: low and high power views of apical turn from a control animal. Reissner’s membrane (straight arrow) is in normal position, and the spiral ganglion (G) appears normal. The organ of Corti is intact, including the inner hair cell (curved arrow in “B”). C,D: low and high power views of apical turn from a Phex mutant. There is severe endolymphatic hydrops (arrows indicate distended Reissner’s membrane in “C”), and the modiolus (M) is nearly devoid of ganglion cells. The inner hair cell remains intact in the organ of Corti (curved arrow in “D”). E,F: high power views of the stria vascularis (S) and spiral ligament (L) in a control animal (E) and a Phex mutant (F). In both cases, the lateral wall tissues appear essentially normal. “Bn” indicates bone of the otic capsule, which, in the Phex mutant (F), is thickened and of abnormal texture and staining quality. Scale bars indicate 50 microns in “A” and “C” and 20 μm in “B”, “D”, “E” and “F.”
Fig. 10.

Basal turn cochlear cross sections from control (+/Y) and PhexHyp-Duk/Y (H-D/Y) mice at P90. A,B: low and high power views of basal turn from a control animal. Reissner’s membrane (straight arrow) and the spiral ganglion (G) are normal. All cellular components of the organ of Corti (curved arrow in “B”) are intact. C,D: low and high power views of basal turn from a Phex mutant. There is severe endolymphatic hydrops (arrows indicate distended Reissner’s membrane) and relative preservation of spiral ganglion (G) in the basal turn. In the organ of Corti (curved arrow in “D”), the outer hair cells are missing, but the inner hair cell remains intact. Scale bars indicate 50 μm in “A” and “C”, 20 μm in “B” and “D.”
Fig. 11.

Cross sections of cochlear duct (A) and endolymphatic duct (B) from a PhexHyp-Duk/Y (H-D/Y) mouse at P300. Severe endolymphatic hydrops (arrows) and spiral ganglion degeneration are seen in the cochlear duct (A). The endolymphatic duct (arrow) is patent (B). All scale bars indicate 100 μm.
Fig. 13.

Cochlear cross sections from control (+/Y) mice in the BALB/cUrd background at 10 months of age. A,B: low and high power views from an animal in which there was a full complement of ganglion cells and cochlear nerve fibers in the modiolus (M), Reissner’s membrane (straight arrow) was in normal position and the organ of Corti was intact (curved arrows). The higher power view in “B” shows normal-appearing hair cells in the organ of Corti (curved arrow). In a second control specimen (C), there were reduced numbers of ganglion cells in the modiolus (M) of the apical portion of the cochlea. Inner hair cells (curved arrow) of the organ of Corti were intact, and Reissner’s membrane was in normal position (straight arrow). Another section from the same specimen is shown in “D,” in which the apical portion of the modiolus is seen at higher magnification. Arrows indicate areas in which there is thinning of the ganglion cell population. Scale bars indicate 100 microns in “A”, 25 μm in “B” and 50 μm in “C” and “D.”
3.3. Inner ear phenotype of the PhexHyp-Duk/Y mouse in the C57BL/6J background
We examined B6-PhexHyp-Duk/Y mice at various time points after birth with a special focus on P21–P130 days old. Compared to sibling control, the B6-PhexHyp-Duk/Y mice showed small body size and slight abnormal shape of the skull. However, signs of inner ear dysfunction were not apparent: mutants’ startle response was comparable to their littermates, and no signs of head-bobbing or circling were observed. ABR recordings from young B6-PhexHyp-Duk/Y mice (P21–P50) to click stimulus showed no significant difference compared to sibling controls (data not shown). Cochlear duct section from ~1 month old B6-PhexHyp-Duk/Y mice showed no ELH or other degenerative changes in the cochlear sensory epithelia (Fig. 14). However, the otic capsule of B6-PhexHyp-Duk/Y was markedly thickened and surrounded by non-mineralized bone (Fig. 14A), similar to that observed in the PhexHyp-Duk/Y in the BALB/cUrd background. We then recorded ABR from 2-, 3-, and 4-month-old B6-PhexHyp-Duk/Y mice to determine if they develop cochlear phenotype comparable to the mutants in the BALB/cUrd background. The absence of significant hearing loss in these mice was striking. However, in some of the mutants (≥2 months old), one ear had normal threshold but the other showed a slight elevation in threshold. This was 10–15 dB SPL over normal threshold and typically associated with the presence of otitis media in that ear (Table 2). The cochlear section from 4-month-old B6-PhexHyp-Duk/Y mice showed no signs of ELH, spiral ganglion or hair cell degeneration, but the otic capsule of B6-PhexHyp-Duk/Y was markedly thickened and the malformation of the capsule bone was obvious (Fig. 15).
Fig. 14.

Cochlear duct from B6-PhexHyp-Duk/Y mice at P28. (A) Low-power overview of temporal bone cross section from a 28-day-old animal showing thickened bone of abnormal texture. Scale bar indicates 300 μm. (B) Higher-power view showing cochlear duct of normal appearance; there is no evidence of endolymphatic hydrops. Scale bar indicates 100 μm.
Fig. 15.

Cochlear duct from B6-PhexHyp-Duk/Y mice at P131. (A) Overview of cochlear duct showing Reissner’s membrane (straight arrow) and organ of Corti (curved arrow) in a control mouse. The bone (Bn) surrounding the cochlea appears normal. (B) Cochlear duct showing normal Reissner’s membrane (straight arrow) and organ of Corti (curved arrow) in a B6-PhexHyp-Duk/Y mouse. The otic capsule bone (Bn) is thickened and has abnormal texture. The scale bars indicate 100 μm in both “A” and “B.”
4. Discussion
Our search for an animal model that spontaneously develops ELH, hearing loss and degeneration postnatally led us to investigate the PhexHyp-Duk mice in the BALB/cUrd and later in the B6 background. Collectively, data from both studies show that (a) PhexHyp-Duk mice typically develop adult onset, asymmetric, progressive hearing loss closely followed by the onset of ELH (as observed by distention of the Reissner’s membrane), (b) functional degeneration precedes structural degeneration, according to ABR and histological data, (c) the major degenerative correlate of hearing loss and ELH in the mutants is the primary loss of spiral ganglion cells, (d) neuronal loss and hair cell loss are downstream events and not the cause of initial hearing loss, (e) PhexHyp-Duk mice develop ELH without evidence of ED obstruction, and occlusion of ED is not a prerequisite for the development of ELH in patients, (f) the expression of the cochlear duct pathology could vary depending on the genetic background and (g) hearing loss could be mildly exacerbated due to susceptibility to otitis media.
The Phex gene is one of the genes implicated in the pathogenesis of human X-linked hypophosphatemic (XLH) rickets. Fishman et al. concluded that hearing impairment is not a feature of XLH in childhood; however, 3 of 10 adult patients with XLH show sensorineural hearing loss, suggesting that hearing loss in adults is linked to XLH (Fishman et al., 2004). Though no inner ear abnormalities other than hearing loss were reported in these patients, future studies involving vestibular testing are needed, as are genetic tests for mutation of the human Phex gene.
Phex mouse mutants lack the ability to mineralize newly formed bone matrix. The non-mineralized bone is spongy and its composition is altered. It is presumed that the thickened otic capsule in PhexHyp-Duk mice represents a defective bone matrix, and whether this is responsible for some of the observed findings or whether Phex has a more direct role in cochlear fluid homeostasis are unclear. Temporal and spatial localization of the Phex gene expression within the cochlear duct may shed some light on this issue.
Lack of observable cochlear duct pathology and significant hearing loss in B6-PhexH-D mouse clearly demonstrates that the bone abnormality alone cannot trigger ELH and associated hearing loss. Rather, other unknown mechanism(s) may be involved in the cochlea, which is suppressed or modified in B6 background. Indeed, mild hearing loss (as measured by elevation in threshold by 10–15 dB SPL) was observed in one ear in some of B6-PhexH-D/Y mice; the contra-lateral ear showed normal thresholds, though thickening of the otic capsule was patent in both ears. Should defective middle ear bones contribute to hearing loss, then both ears in B6-PhexHyp-Duk/Y mice would have registered some degree of hearing loss. Otitis media was observed in 4/5 mutant ears that showed elevated threshold, indicating that defective bone conductance is not a significant issue in the Phex mouse, but otitis media could account for the asymmetric hearing loss observed in Phex mutants.
The onset of hearing loss for most PhexHyp-Duk/Y mice tested occurs between P21-30. Physiological and morphological examination of PhexHyp-Duk/Y mice from birth to late adulthood showed that, in most cases, deterioration of auditory function begins as early as 3–4 weeks after birth with relatively better preservation of hearing in one ear compared to the other. Among all the mice included in this study, none was totally deaf at a young age, which demonstrates that PhexHyp-Duk mice are not born deaf but develop hearing loss postnatally over a period of time. Longitudinal ABR recordings from 8 PhexHyp-Duk/Y mice along with controls clearly show a progressive pattern of hearing deterioration with some element of fluctuation. Results in Table 1 suggest that mutants tend to acquire low frequency hearing loss earlier than high frequency hearing loss, but more mutants need to be examined to confirm this trend.
Histological analysis clearly shows that cochlear pathology is the dominant factor associated with hearing loss, and it is likely that otitis media minimally contributes to asymmetric hearing loss in PhexHyp-Duk/Y mice. One interesting observation is the relative preservation of hair cells in the apical turn of the cochlea at a time when most of the spiral ganglion cells had degenerated (Figs. 9C, 10A and B). This finding has been described in human temporal bone specimens of patients with MD (Nadol, 1990; Nadol et al., 1987, 1995). This feature is unlike what is observed in other mouse models of sensorineural deafness, where loss of spiral ganglion cells is secondary to hair cell loss (Alagramam et al., 2000). Further, in PhexHyp-Duk/Y mice, degeneration of neuronal cells in the cochlea shows an apex to base progression. However, the fact that the first signs of neuronal degeneration are observed some time after P40 clearly shows that functional degeneration is well underway before structural degeneration.
It should be noted that aging mice show a tendency to lose apical spiral ganglion cells before inner hair cells. In these cases, primary neuronal loss is linked to changes in supporting cells and Reissner’s membrane. Though hydrops has not been reported in aging mouse models, it has been proposed that the changes in supporting cells and Reissner’s membrane may be due to disorders of inner ear fluid homeostasis (Keithley et al., 2004; Ohlemiller and Gagnon, 2004; Willott and Erway, 1998). Due to the presence of hydrops in the young PhexHyp-Duk/Y mutants (P21-90), one might expect a defect in cochlear fluid balance associated with strial pathology. Interestingly, no obvious strial pathology was observed in the PhexHyp-Duk/Y mutant (Fig. 9E and F), suggesting that the proximate cause of initial hearing loss does not involve the stria. However, appearance of normal stria does not prove that the endocochlear potential (EP) is normal; measurement of EP at various time points would be necessary to assess the status of stria in the ear.
The PhexHyp-Duk/Y mouse, however, is not the only hydropic mouse noted in recent years. Several others have been described. The Slc26A4 (former PDS gene) knockout mouse develops ELH as early as embryonic day 15, and the mutant mouse is profoundly deaf (Everett et al., 2001). This mutation is associated with embryonic cochlear malformations not seen in most ELH conditions in patients, including MD, making this model less attractive for use in ELH modeling. Another hydropic mouse that develops ELH in utero is the Foxi1 null mouse. Foxi1 acts as an upstream gene that regulates pendrin expression, hence the null mouse exhibits phenotypic features that are similar to the Scl26A4 knockout mouse (Hulander et al., 2003). Finally, a recently described Brn-4-deficient mouse also develops ELH in utero (Xia et al., 2002). The Brn-4 gene belongs to transcription factors of the Pou3f4 family that regulate the transcription of connexin 26, connexin 31, Na/K ATPase and Na–K–Cl cotransporter (Phippard et al., 1999). This prenatal development of ELH deviates from postnatal development of ELH observed in patients, making this model less attractive.
The focus of this report is not whether Phex is a candidate gene for MD or other conditions leading to ELH. Rather, the point is that PhexHyp-Duk mice are valuable models to use in the study of the pathophysiology linked to ELH since the ear phenotype observed in this mutant is reminiscent of the auditory dysfunction characteristic of patients with ELH condition. The potential advantages of the Phex model in the study of ELH are: (a) compared to the guinea pig model, surgery is not required, making it amenable to use by more researchers, (b) the phenotype is reproducibly seen in mutant males in the BALB/curd background (there is a degree of uncertainty with the surgical model and a small percentage of animals die after surgery), (c) identification of genetic factors associated with susceptibility to ELH condition would be easier with a mouse model than with other animal models and (d) since the development of hearing loss and severe signs of neuronal degeneration occur post weaning, this model offers a new vehicle to test putative otoprotective agents.
There are some limitations to the Phex mouse model. First, episodic vertigo is an important clinical feature in patients with ELH condition. It may be difficult to document episodic vertigo in this model. Whether the observed vestibular dysfunction is episodic or not will require more systematic, long-term studies including quantitative assessment of the vestibular evoked potential (Alagramam et al., 2005). Second, the extent of Reissner’s membrane distention is presently used as a measure of the severity of ELH. Although this is a traditional approach, it may not accurately reflect the onset and/or the severity of the disorder in the endolymph fluid homeostatis. Measurement of the EP along with the physical marker (membrane distention) may be a better way to assess severity of ELH in the Phex mutants. Third, mice have a 2-year life span; patients identified with ELH condition are typically 30-years or older. It is hard to equate late onset hearing loss linked to ELH condition in patients to an animal model that lives for about 2-years. It should be noted that these limitations are shared by other animal models as well. Fourth, susceptibility of the Phex mutant to otitis media appears to increase the threshold by 10–15 dB in the affected ear. This, however, is not a significant problem for three reasons. Data from the B6 mice clearly separates the level of hearing loss associated with OM compared to the more severe hearing loss observed with PhexHyp-Duk mice in BALB/cUrd background. The hearing loss caused by cochlear pathology is much more severe than 10–15 dB loss observed in B6-PhexHyp-Duk mice. ABR data from mutant ears without OM can be used to correlate hearing loss with ELH and degeneration in this model.
In summary, we have established the profile of the inner ear phenotype in the PhexHyp-Duk mutant at various time points from birth to adulthood (Fig. 16), and we compare the phenotype in two genetic backgrounds. The spontaneous development of ELH, hearing loss and sensory cell loss are attractive features of the PhexHyp-Duk mouse model as they seem to mirror many of the pathologic features associated with ELH-related disorders both clinically and experimentally. The PhexHyp-Duk mouse is a potentially useful model to explore the relationship among ELH, hearing loss and neuronal degeneration in the cochlea.
Fig. 16.

Approximate timeline of events in the inner ear of PhexHyp-Duk/Y. Dotted line indicates onset of phenotype, mild hearing loss (HL), signs of endolymphatic hydrops (ELH), spiral ganglion cell (SGC) degeneration, or hair cell degeneration, with some variability among animals in onset, appearance and severity. Solid line that follows dotted lines indicates that the phenotype is established with age.
Fig. 12.

ABR thresholds from older control siblings in the BALB/cUrd background. ABR chart for click and pure tone stimulus from 10-monthold +/Y mice 757 and 758. LE and RE on the X-axis represent left ear and right ear; dB SPL Y-axis represents sound intensity.
Acknowledgments
We would like to thank Dr. Brian McDermott and Chris Heddon for critical reading of this manuscript. This work was supported by grants to Drs. Alagramam and Megerian from Rainbow Board of Trustees, Rainbow Babies and Children’s Hospitals, University Hospitals Case Medical Center.
Abbreviations
- MD
meniere’s disease
- ELH
endolymphatic hydrops
- ED
endolymphatic duct
- ES
endolymphatic sac
- ABR
auditory-evoked brainstem response
- CC
crus commune
- HD
PhexHyp-Duk
Contributor Information
Cliff A. Megerian, Email: cliff.megerian@uhhospitals.org.
Maroun T. Semaan, Email: maroun.semaan@uhhospitals.org.
Saba Aftab, Email: saba.aftab@uhhospitals.org.
Lauren B. Kisley, Email: lauren.kisley@case.edu.
Qing Yin Zheng, Email: qyz@case.edu.
Karen S. Pawlowski, Email: Pawlowski@UTSouthwestern.edu.
Charles G. Wright, Email: cgwrigh@utdallas.edu.
Kumar N. Alagramam, Email: kna3@cwru.edu, kna3@case.edu.
References
- Alagramam KN, Stahl JS, Jones SM, Pawlowski KS, Wright CG. Characterization of vestibular dysfunction in the mouse model for Usher syndrome 1F. J Assoc Res Otolaryngol. 2005;6:106–118. doi: 10.1007/s10162-004-5032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagramam KN, Zahorsky-Reeves J, Wright CG, Pawlowski KS, Erway LC, Stubbs L, Woychik RP. Neuroepithelial defects of the inner ear in a new allele of the mouse mutation Ames waltzer. Hear Res. 2000;148:181–191. doi: 10.1016/s0378-5955(00)00152-0. [DOI] [PubMed] [Google Scholar]
- Aran JM, Rarey KE, Hawkins JE., Jr Functional and morphological changes in experimental endolymphatic hydrops. Acta Otolaryngol. 1984;97:547–557. doi: 10.3109/00016488409132933. [DOI] [PubMed] [Google Scholar]
- Arenberg IK, Marovitz WF, Shambaugh GE., Jr The role of the endolymphatic sac in the pathogenesis of endolymphatic hydrops in man. Acta Otolaryngol. 1970;275 (Suppl):1–49. [PubMed] [Google Scholar]
- Belal A, Jr, Ylikoski J. Pathologic significance of Meniere’s symptom complex. A histopathologic and electron microscopic study. Am J Otolaryngol. 1980;1:275–284. doi: 10.1016/s0196-0709(80)80030-5. [DOI] [PubMed] [Google Scholar]
- Dodson KM, Kamei T, Sismanis A, Nance WE. Familial unilateral deafness and delayed endolymphatic hydrops. Am J Med Genet A. 2007;143 (14):1661–1665. doi: 10.1002/ajmg.a.31741. [DOI] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- Fishman G, Miller-Hansen D, Jacobsen C, Singhal VK, Alon US. Hearing impairment in familial X-linked hypophosphatemic rickets. Eur J Pediatr. 2004;163:622–623. doi: 10.1007/s00431-004-1504-z. [DOI] [PubMed] [Google Scholar]
- Fraysse BG, Alonso A, House WF. Meniere’s disease and endolymphatic hydrops: clinical-histopathological correlations. Ann Otol Rhinol Laryngol. 1980;89 (Suppl):2–22. doi: 10.1177/00034894800896s201. [DOI] [PubMed] [Google Scholar]
- Hallpike CS, Cairns H. Observations on the pathology of Meniere’s syndrome. J Laryngol Otol. 1938;53:625–655. doi: 10.1177/003591573803101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner KC. Auditory and vestibular function in experimental hydrops. Otolaryngol Head Neck Surg. 1995;112:84–89. doi: 10.1016/S0194-59989570306-3. [DOI] [PubMed] [Google Scholar]
- Hott ME, Graham M, Bonassar LJ, Megerian CA. Correlation between hearing loss and scala media area in guinea pigs with long-standing endolymphatic hydrops. Otol Neurotol. 2003;24:64–72. doi: 10.1097/00129492-200301000-00014. [DOI] [PubMed] [Google Scholar]
- Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerback S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130:2013–2025. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- Keithley EM, Canto C, Zheng QY, Fischel-Ghodsian N, Johnson KR. Age-related hearing loss and the ahl locus in mice. Hear Res. 2004;188:21–28. doi: 10.1016/S0378-5955(03)00365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura RS, Schuknecht HF. Membranous hydrops in the inner ear of the guinea pig after obliteration of the endolymphatic sac. Pract Otolaryngol. 1965;27:343–354. [Google Scholar]
- Kunieda T, Xian M, Kobayashi E, Imamichi T, Moriwaki K, Toyoda Y. Sexing of mouse preimplantation embryos by detection of Y chromosome-specific sequences using polymerase chain reaction. Biol Reprod. 1992;46:692–697. doi: 10.1095/biolreprod46.4.692. [DOI] [PubMed] [Google Scholar]
- Lorenz-Depiereux B, Guido VE, Johnson KR, Zheng QY, Gagnon LH, Bauschatz JD, Davisson MT, Washburn LL, Donahue LR, Strom TM, Eicher EM. New intragenic deletions in the Phex gene clarify X-linked hypophosphatemia-related abnormalities in mice. Mamm Genome. 2004;15:151–161. doi: 10.1007/s00335-003-2310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud MR, Khan AM, Nadol JB., Jr Histopathology of the inner ear in unoperated acoustic neuroma. Ann Otol Rhinol Laryngol. 2003;112 (11):979–986. doi: 10.1177/000348940311201111. [DOI] [PubMed] [Google Scholar]
- Murata J, Horii A, Tamura M, Mitani K, Mizuki M, Kubu T. Endolymphatic hydrops as a cause of audiovestibular manifestations in relapsing polychondritis. Acta Otolaryngol. 2006;126:548–552. doi: 10.1080/00016480500437369. [DOI] [PubMed] [Google Scholar]
- Nadol JB., Jr Degeneration of cochlear neurons as seen in the spiral ganglion of man. Hear Res. 1990;49:141–154. doi: 10.1016/0378-5955(90)90101-t. [DOI] [PubMed] [Google Scholar]
- Nadol JB, Jr, Thornton AR. Ultrastructural findings in a case of Meniere’s disease. Ann Otol Rhinol Laryngol. 1987;96:449–454. doi: 10.1177/000348948709600420. [DOI] [PubMed] [Google Scholar]
- Nadol JB, Jr, Adams JC, Kim JR. Degenerative changes in the organ of Corti and lateral cochlear wall in experimental endolymphatic hydrops and human Meniere’s disease. Acta Otolaryngol. 1995;519 (Suppl):47–59. doi: 10.3109/00016489509121870. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Gagnon PM. Apical-to-basal gradients in agerelated cochlear degeneration and their relationship to “primary” loss of cochlear neurons. J Comp Neurol. 2004;479:103–116. doi: 10.1002/cne.20326. [DOI] [PubMed] [Google Scholar]
- Paparella MM. Pathogenesis of Meniere’s disease and Meniere’s syndrome. Acta Otolaryngol. 1984;406 (Suppl):10–25. doi: 10.3109/00016488309122996. [DOI] [PubMed] [Google Scholar]
- Phippard D, Lu L, Lee D, Saunders JC, Crenshaw EB., 3rd Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. J Neurosci. 1999;19:5980–5989. doi: 10.1523/JNEUROSCI.19-14-05980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuknecht HF. Pathophysiology of endolymphatic hydrops. Arch Otorhinolaryngol. 1976;212:253–262. doi: 10.1007/BF00453673. [DOI] [PubMed] [Google Scholar]
- Willott JF, Erway LC. Genetics of age-related hearing loss in mice. IV Cochlear pathology and hearing loss in 25 BXD recombinant inbred mouse strains. Hear Res. 1998;119:27–36. doi: 10.1016/s0378-5955(98)00029-x. [DOI] [PubMed] [Google Scholar]
- Xia AP, Kikuchi T, Minowa O, Katori Y, Oshima T, Noda T, Ikeda K. Late-onset hearing loss in a mouse model of DFN3 non-syndromic deafness: morphologic and immunohistochemical analyses. Hear Res. 2002;166:150–158. doi: 10.1016/s0378-5955(02)00309-x. [DOI] [PubMed] [Google Scholar]
- Yamakawa K. Uber die pathologische Veranderung bei einem Meniere-Kranken. J Otorhinolaryngol Soc Jpn. 1938;44:2310–2312. [Google Scholar]
- Zheng QY, Yu H, Washington JL, 3rd, Kisley LB, Kikkawa YS, Pawlowski KS, Wright CG, Alagramam KN. A new spontaneous mutation in the mouse protocadherin 15 gene. Hear Res. 2006;219:110–120. doi: 10.1016/j.heares.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]