

Abstract
Genetic modifiers can be detected in mice by looking for strain background differences in inheritance or phenotype of a mutation. They can be mapped by analyses of appropriate linkage crosses and congenic lines, and modifier genes of large effect can be identified by positional-candidate gene testing. Inbred strains of mice vary widely in onset and severity of age-related hearing loss (AHL), an important consideration when assessing hearing in mutant mice. At least 8 mapped loci and a mitochondrial variant (mt-Tr) are known to contribute to AHL in mouse strains; one locus (ahl) has been identified as a variant of the cadherin 23 gene (Cdh23753A/G). This variant also was shown to modify hearing loss associated with the Atp2b2dfw-2J and Mass1frings mutations. The hearing modifier (Moth1) of tubby (Tubtub) mutant mice was shown to be a strain variant of the Mtap1a gene. Human hearing modifiers include DFNM1, which suppresses recessive deafness DFNB26, and a nuclear gene that modulates the severity of hearing loss associated with a mitochondrial mutation. Recently, a variant of the human ATP2B2 gene was shown to exacerbate hearing loss in individuals homozygous for a CDH23 mutation, similar to the Atp2b2dfw-2J–Cdh23753A/G interaction affecting hearing in mice. Because modifier genes and digenic inheritance are not always distinguishable, we also include in this review several examples of digenic inheritance of hearing loss that have been reported in both mice and humans.
Keywords: Modifier gene, Genetic background, Inbred mouse strain, Age-related hearing loss, Digenic inheritance, AHL, QTL
1. Phenotypic diversity, strain background, and modifier genes
Genetic modifiers are heritable factors capable of modifying the phenotype of a mutant gene without having an obvious effect on the normal condition. Modifier genes are found in all genetically mixed populations and their collective influence on a mutant phenotype is referred to as the genetic background effect or, in the case of inbred strains of mice, the strain background effect. Previously published review papers are replete with examples of the effects of modifier genes on the phenotypic diversity of a wide variety of genetic disorders in humans and mice (Barthold, 2004; Erickson, 1996; Houlston and Tomlinson, 1998; Montagutelli, 2000; Nadeau, 2001, 2003; Romeo and McKusick, 1994; Slavotinek and Biesecker, 2003). Examples of modifier genes are especially prevalent in genetically heterogeneous phenotypes like sensorineural deafness (Slavotinek and Biesecker, 2003), and some reviews have focused specifically on modifiers of hearing (Ahituv and Avraham, 2002; Friedman et al., 2000; Haider et al., 2002; Riazuddin et al., 2002). Here, we present an updated review on strain background effects and genetic modifiers of hearing in mice including some related results from studies of human populations.
Genetic modifiers are one of several factors that can contribute to the phenotypic diversity of a genetic disorder. Nonheritable sources of phenotypic variation include environmental effects, somatic cell mutations, and stochastic events. Environmental factors that are known to contribute to hearing loss include viral and bacterial infections, ototoxic drugs, and exposure to loud sounds. Diet, maternal effects, and differences in the in utero environment may also contribute to phenotypic variation. Somatic cell mutations or stochastic events that occur early in development may lead to abnormal cell lineages that could potentially alter phenotypes. Epigenetic effects, including imprinting and chromosome condensation, can affect the phenotype of an individual by altering the expression patterns of particular genes.
Genetic sources of phenotypic variation include multiple different genes contributing to the phenotype (genetic heterogeneity), allelic heterogeneity within each of these loci, and genetic background effects (modifier loci). Hereditary hearing disorders exhibit a high degree of genetic heterogeneity. There are more than 35 different genes and more than 100 mapped loci that have been shown to underlie human hereditary non-syndromic deafness disorders (Hereditary Hearing Loss Home-page, http://webhost.ua.ac.be/hhh/). Likewise, there are more than 200 mouse mutations associated with inner ear dysfunction (Hereditary Hearing Impairment in Mice, http://www.jax.org/hmr/index.html). Allelic heterogeneity, when different mutations of the same gene cause distinctive disease phenotypes, may also be a common source of phenotypic variation (Romeo and McKusick, 1994). A collection of allelic mutations exhibiting variable phenotypes is called an allelic series. Correlation of mutations with phenotypes in such a series can provide valuable functional information about different structural components of a gene. For example, different mutations of the MYO7A gene underlie the distinctive human deafness disorders USH1B, DFNA11, and DFNB2 (Liu et al., 1997; Weil et al., 1997), and different mutations of the orthologous mouse Myo7a gene underlie different deafness phenotypes (Mburu et al., 1997). Allelic heterogeneity versus genetic background effects are often difficult to distinguish in human genetic diseases because of the largely unknown genetic composition of human populations. In mice, where genetic background can be easily controlled, the effects of modifier genes and allelic diversity are easily distinguished.
The contribution of genetic background to phenotypic diversity reflects the additive and interactive (epistasis) effects of multiple genes. Because individual genes do not act alone but rather in concert with many other genes, it is not surprising that modifier genes are a common source of phenotypic variation in genetically heterogeneous populations. Differences in genetic background can alter the inheritance, expressivity, and pleiotropy of mutant phenotypes. Incomplete penetrance and dominance modification can alter the type of inheritance that is observed on different strain backgrounds. Incomplete penetrance occurs when some mice with predisposing genotypes do not manifest the mutant phenotype. Penetrance can be reduced to the point that a mutant phenotype may not be seen at all on certain strain backgrounds. Dominance modification occurs when the expression of a mutant phenotype in heterozygotes varies with strain background such that in one strain the mutant phenotype may appear to be recessive while on another background it may appear dominant. Changes in expressivity may manifest as qualitatively different phenotypes on different genetic backgrounds or as quantitative differences in the severity, time of onset, or progression of the mutant phenotype. Changes in pleiotropy are seen as a gain or loss in the manifestation of additional traits that are distinct from the mutant phenotype. Genetic models invoking variation in expression thresholds of mutant phenotypes have been described that help explain these different modifier effects (Nadeau, 2001).
Products of modifier genes may include proteins that directly interact with the mutant protein (such as receptor-ligand or heteromeric protein subunits), factors that affect the timing or rate of transcription of the mutated gene, factors that affect the degradation of mutant protein or RNA or are involved in apoptotic pathways, and functionally related proteins in alternate pathways that can compensate for (or worsen) the dysfunction caused by the mutation. Modifier genes in mice can be detected by looking for strain background differences in inheritance or phenotype of a mutation. Modifier loci can be mapped by analyses of appropriate linkage crosses and congenic lines, and modifier genes of large effect can be identified by positional-candidate gene testing. In the following section, we provide a detailed strategy for the genetic analysis of modifier genes in mice.
2. Strategy for the analysis of genetic modifiers in inbred mouse strains
The effect of modifying alleles on the mutant allele is masked on a coisogenic background. Hence, detecting a modifier requires crossing the mutant allele with different inbred strains to allow for random segregation of mutant and modifying alleles. There is no clear-cut strategy as to which outcross strain to use, but strains that provide a higher degree of genetic diversity, such as wild-derived strains or strains from a different genealogical subgroup, have been used successfully (Ikeda et al., 1999). Using homozygous mutants to generate the F1 hybrids leads to a genetically uniform population, which can be used to ascertain potentially dominant modifier effects. Initiating a cross with heterozygous mutants generates two genotypes in F1 hybrids, which provides useful information of background effects that are independent of the mutant allele. F1 hybrid carriers may be intercrossed or backcrossed to the mutant parental strain. An intercross provides three genotypes per polymorphic locus and 3n genotypes for n number of loci, and a backcross yields 2n genotypes. Thus, an intercross provides a higher degree of allele combinations and a greater chance to detect different phenotypic, epistatic, and dominant effects. In addition, an intercross generates twice as many informative meioses and provides a higher genetic resolution for additive effects. For dominance effects, however, in which a modifier affects the mutant allele only when in a homozygous state, the backcross provides a larger phenotypic population for linkage detection. Because of the reduction in residual genetic variance, the required significance threshold for a back-cross is lower than that for an intercross (Darvasi, 1998).
Mutant progeny from intercrosses (F2 generation) or back-crosses (N2 generation) are examined carefully for variation in phenotype. If the mutant mice exhibit differential—that is modified—phenotypes, they are genotyped at approximately 100 marker loci, which are roughly spaced in 15 cM intervals across the genome. Modifiers of hearing in mice are generally treated as quantitative trait loci (QTLs). For initial linkage scanning, selecting mice with extreme phenotypes maximizes information without sacrificing detection capability (Darvasi, 1997a). Association between marker loci and phenotypes can be detected using linkage analysis programs such MapManager (Manly and Meer, 2001), R/QTL (Broman et al., 2003), or PseudoMarker (Sen and Churchill, 2001). Web sites for these computer programs are listed below.
MapManager—http://www.mapmanager.org/
PseudoMarker—http://www.jax.org/staff/churchill/labsite/software/pseudomarker/index.html
Marker associations with lod scores above 3.3 (for a backcross) or 4.3 (for an intercross), which correspond to a genome-wide significance level of P = 0.05, are considered as significant and warrant further genetic investigation (Lander and Kruglyak, 1995). After a significant linkage is detected, it is important to refine the map position to a high resolution so that few candidate genes need be considered. For further refinement, the segregating mapping cross may be expanded or congenic lines may be produced. A congenic line provides the advantage of isolating the modifier effect, but also presents a risk of losing the modifier effect during backcrossing. The congenic line may also be used for high-resolution genetic mapping to narrow the modifier interval and delimit the number of candidate genes (Darvasi, 1997b).
In some cases, previously constructed and characterized recombinant inbred (RI) strains (Williams et al., 2001) can provide an efficient means for mapping modifiers provided that the modifier effect differs between the progenitor strains of the RI set. Each strain of an RI set is an inbred genetic mosaic derived from different chromosomal contributions of its progenitor strains. A dominant or semidominant mutant deafness allele can be crossed against each strain of an RI set and carrier offspring phenotyped. Phenotypic differences among RI strains that segregate with the mutant allele can readily be mapped because the precise locations of breakpoints already have been determined for each RI strain so there is no need to genotype any of the progeny. RI strains have the added advantage that multiple isogenic mice from each strain can be examined to obtain more reliable phenotypes. RI mapping analysis, however, cannot be applied to modifiers of recessive mutations. Chromosome substitution (CS) strains (Nadeau et al., 2000) also can be used in modifier mapping studies if the modifier effect differs between the host and donor strains of the CS set. Each strain of a CS set contains primarily the genome of the host strain with the contribution of the donor strain limited to a single chromosome. CS strains can be used to rapidly produce congenic strains to evaluate candidate chromosomal regions for modifier effects. Starting with a CS strain speeds congenic construction because only the target chromosome (containing the modifier) needs to be typed for parental contributions at each backcross generation.
Although it is difficult to prove with a single experiment that a particular candidate gene is responsible for a strain-specific modifier effect, an accumulation of supporting evidence can provide a compelling case (Flint et al., 2005). Such evidence may include the following: (1) a highly refined genetic interval that limits the number of candidate genes, (2) an obvious functional relationship of the candidate gene with the trait phenotype, (3) a DNA alteration of the candidate gene that strongly associates with the trait and occurs in an evolutionarily conserved coding or regulatory region, (4) a consistent association of trait phenotypes with biallelic gene variants in multiple inbred strains, (5) phenotype conversion by transgene complementation, and (6) phenotype conversion by targeted gene “knock-in.” Phenotype conversion by transgenic or gene knock-in approaches is considered the most definitive support for gene identity.
3. Age-related hearing loss (AHL) in inbred mouse strains
Loci that contribute to AHL in inbred strains may also act as hearing modifiers of mutant genes. The mapping and identification of genes contributing to AHL in inbred mouse strains may, therefore, indirectly identify modifier genes and thus provide important functional information about the genes they modify. AHL in mice is a complex trait resulting from the interaction of multiple predisposing-genes and environmental factors. Therefore, genetic factors underlying AHL are best treated as quantitative trait loci (QTLs) that affect hearing loss susceptibility and not as mutant genes that directly cause hearing loss. An individual mouse with a predisposing AHL genotype may not exhibit hearing loss; however, collectively, mice with the predisposing genotype will on average exhibit below normal hearing levels. AHL is a very age sensitive trait, so it is important that only mice of the same age classes are compared. One of the best means for assessing hearing in mice is by auditory-evoked brainstem response (ABR) threshold measurements. A survey of more than 80 inbred mouse strains at The Jackson Laboratory has identified 18 strains that exhibit significantly elevated ABR thresholds before the age of 3 months (including 129P1/ReJ, A/J, BUB/BnJ, DBA/2J, and NOD/LtJ) and 16 additional strains (including C57BL/6J and BALB/cJ) that exhibit elevated ABR thresholds at older ages (Zheng et al., 1999) (updated in Hereditary Hearing Impairment in Mice, http://www.jax.org/hmr/index.html). AHL in mice is progressive with high frequency losses occurring earliest, a consequence of cochlear hair cell loss progressing from base to apex (Henry and Chole, 1980; Ohlemiller and Gagnon, 2004; Spongr et al., 1997). Outer hair cells are affected before inner hair cells and ganglion cell loss appears secondary to hair cell degeneration.
AHL in the inbred mouse strains so far tested appears to be inherited in a recessive manner: F1 hybrids of AHL-positive strains with inbred strains that do not exhibit AHL (such as CAST/Ei or CBA/CaJ) retain normal hearing beyond 12 months of age. Because of the recessive nature of AHL, most linkage studies have been performed by backcrossing the F1 strain hybrids to the AHL-positive strains. The recessive inheritance and progressive nature of AHL in mouse strains are illustrated in Fig. 1, which show the ABR threshold frequency distributions of parental strains, F1 hybrids, and N2 generation progeny from a (NOD/LtJ × CAST/Ei) × NOD/LtJ backcross. The strongly bimodal pattern of this distribution indicates the segregation of one or a few loci with large effects, whereas a more normally distributed pattern with a single mode would indicate a large number of loci with small effects.
Fig. 1.

Bimodal frequency distribution of backcross mice with different hearing thresholds. Frequency distributions of 16 kHz ABR thresholds are shown for the parental strains (NOD, CAST), the F1 hybrids, and the N2 generation progeny from a (NOD/LtJ × CAST/Ei) × NOD/LtJ backcross. The recessive nature of AHL is shown by the similar thresholds of CAST and F1 hybrid mice as contrasted with the thresholds of NOD mice. The frequency distribution of thresholds in the N2 mice is strongly bimodal, indicating the segregation of one or a few loci with large effects. The progressive nature of the hearing loss can be seen by comparing the threshold distributions of N2 mice tested at 3 and 6 months of age.
3.1. The ahl locus
To map AHL loci, ABR thresholds (or other measures of hearing) of progeny from linkage crosses are examined for their associations with segregating marker loci distributed throughout the genome. By analysis of a (C57BL/6J × CAST/Ei) × C57BL/6J backcross, we originally mapped a locus (symbol ahl) on Chromosome (Chr) 10 that is a major contributor to AHL in C57BL/6J mice (Johnson et al., 1997). We subsequently showed by analyses of additional backcrosses involving different inbred strains and the wild-derived CAST/Ei strain that the same ahl locus contributes to AHL in at least 10 inbred strains (Johnson et al., 2000). Evidence from other investigators indicates that ahl also influences the sensitivity of mice to noise-induced hearing loss (Davis et al., 2001; Erway et al., 1996; Harding et al., 2005). We constructed a congenic strain (designated B6.CAST-+ahl) with a CAST-derived wild type allele of the ahl locus (which confers AHL resistance) transferred onto an otherwise C57BL/6J strain background (Johnson et al., 1997). This congenic strain has proven to be valuable in subsequent studies by us and other investigators to determine the specific effect of the ahl locus on age-related and noise-induced hearing loss (Harding et al., 2005; Keithley et al., 2004; Vazquez et al., 2004).
The ahl locus was mapped near two other loci affecting hearing in mice: modifier of deaf waddler (mdfw) and waltzer (v), which suggested the possibility of allelism. The mdfw locus modifies hearing in mice that are heterozygous for the Atp2b2dfw-2J mutation. A correspondence of ahl and mdfw hearing loss phenotypes was observed among 12 inbred strains (Zheng and Johnson, 2001), and provided evidence that these two independently discovered loci represent the same gene. Waltzer was shown to be a mutation in the cadherin 23 (Cdh23) gene (Di Palma et al., 2001), and high-resolution genetic and physical mapping failed to separate Cdh23 from the mdfw candidate region (Bryda et al., 2001), further supporting the notion that mdfw and, by inference, ahl are alleles of Cdh23. Molecular analysis of the Cdh23 gene in multiple mouse strains showed a strong correspondence of Cdh23 haplotypes with mdfw and ahl phenotypes (Noben-Trauth et al., 2003). The presence of A rather than G at exon 7 position 753 (G753A) of Cdh23 occurs in inbred strains with increased susceptibility to AHL and leads to an increased frequency of exon 7 skipping.
3.2. Other genetic factors contribute to AHL in inbred strains
Homozygosity for the ahl susceptibility allele predisposes mice to AHL, but other genetic factors also contribute to the different onset times and severities of hearing loss that are observed among inbred strains. A single nucleotide insertion in the tRNA-Arg gene (mt-Tr) in the mitochondrial DNA (mtDNA) of A/J mice was shown to contribute to their early onset hearing loss (Johnson et al., 2001). A statistically significant association of A/J-derived mitochondria with AHL was detected in an analysis of (A/J × CAST/Ei) × A/J backcrosses involving reciprocal F1 hybrids. The increase in hearing loss was shown to correspond with A/J maternal lineage and mtDNA from this strain was shown to differ from that of other inbred strains. The mitochondrial effect on AHL, however, was seen only in N2 mice that were homozygous for the ahl susceptibility gene.
Although mice of the C57BL/6J and NOD/LtJ strains share the same ahl susceptibility gene, they differ greatly in age of hearing loss onset. Most C57BL/6J mice do not exhibit AHL until they are more than 12 months of age, whereas most NOD/LtJ mice are significantly hearing impaired before 3 months of age (Johnson et al., 2000). Analysis of (NOD × B6) × NOD backcross mice detected a strong association of ABR thresholds with a locus on Chr 5 (Johnson and Zheng, 2002). The new locus was designated ahl2 because it represents the second locus for age-related hearing loss to be mapped in mice. ABR thresholds of mice that were homozygous for the NOD-derived allele at this locus were significantly higher than in heterozygous mice. Analysis of a (CAST/Ei × NOD/LtJ) × NOD/LtJ back-cross, which segregates strain-specific alleles at both ahl2 and the ahl locus on Chr 10, confirmed an association of hearing loss with ahl2 genotypes. Like the mt-Tr mutation of A/J mice, the effect of ahl2 was dependent on the predisposing ahl/ahl genotype.
Another AHL locus (ahl3) in mice was mapped to the mid-region of Chr 17 by Nemoto et al. (2004) by analysis of crosses between C57BL/6J and Japanese MSM strain mice, and five additional AHL loci (ahl4–ahl8) subsequently have been mapped. One of these new loci (ahl4) increases the severity of AHL in A/J mice and was mapped to distal Chr 10 by linkage analysis of AXB and BXA recombinant inbred strains and a (A/J × CAST) × A/J backcross, and by congenic segment analysis of a B6.A Chr 10 substitution strain (our unpublished data). Two additional mouse AHL loci, ahl5 on Chr 10 and ahl6 on Chr 18, were recently mapped by analysis of crosses involving the Black Swiss and CAST/Ei strains (Drayton and Noben-Trauth, 2006). Yet another new AHL locus (ahl8), which accelerates hearing loss in DBA/2J mice, was mapped to distal Chr 11 by linkage analysis of BXD recombinant inbred strains and a (C57BL/6J × DBA/2J) × DBA/2J backcross (our unpublished data). Table 1 lists the mouse AHL loci that have been mapped so far and the strains that have been examined for their effects. Table 2 lists cases of genetic modifiers and digenic inheritance of hearing loss that have been reported in mice and humans. These tables will be accessible at the Hereditary Hearing Impairment in Mice web site (http://www.jax.org/hmr/index.html) and periodically updated.
Table 1.
Genetic factors that underlie age-related hearing loss in inbred mouse strains
| AHL locus | Chr | Most likely location | Mapping methodsa | Known strains with susceptibility allele | Known strains with resistance allele | References | Underlying genes |
|---|---|---|---|---|---|---|---|
| ahl (mdfw) | 10 | 30 cM | Backcross, haplotypes | Many | Many | Johnson et al., 1997, 2000; Noben-Trauth et al., 2003 | Cdh23753A/G |
| ahl2 | 5 | 40–55 cM | Backcross | NOD/LtJ | C57BL/6J, CAST/Ei | Johnson and Zheng, 2002 | |
| ahl3 | 17 | 20–45 cM | CS strains, backcross | C57BL/6J | MSM (molossinus) | Nemoto et al., 2004 | |
| ahl4 | 10 | Distal | RI, CS strains, backcross | A/J | C57BL/6J, CAST/Ei | Unpublished | |
| ahl5 | 10 | 35–42 cM | Backcross, intercross | Black Swiss | CAST/Ei | Drayton and Noben-Trauth, 2006 | |
| ahl6 | 18 | 38–44 cM | Backcross | Black Swiss | CAST/Ei | Drayton and Noben-Trauth, 2006 | |
| ahl7 | Reserved | ||||||
| ahl8 | 11 | Distal | RI strains, backcross | DBA/2J | C57BL/6J | Unpublished | |
| mtDNA | Mitochondria | Backcross | A/J | Johnson et al., 2001 | mt-Tr | ||
CS = chromosome substitution; RI = recombinant inbred.
Table 2.
Genetic modifiers and digenic inheritance of hearing loss reported in mice and humans
| Mouse | References | Human | References | ||
|---|---|---|---|---|---|
| Strain modifier and mutation | Genetic modifier and mutation | ||||
| Modifier gene | Mutant gene | Modifier gene | Mutant gene | ||
| Cdh23753A/G (mdfw) | Atp2b2dfw-2J | Noben-Trauth et al., 1997, 2003 | ATP2B2 | CDH23 | Schultz et al., 2005 |
| Cdh23753A/G | Mass1frings | Johnson et al., 2005 | DFNM1 | DFNB26 | Riazuddin et al., 2000 |
| Mtap1a (Moth1) | Tubtub | Ikeda et al., 1999, 2002 | Unknown, Chr 8 | mtDNA A1555G | Bykhovskaya et al., 2001 |
| Unknown, Chr 6 (?TFB1M) | mtDNA A1555G | Bykhovskaya et al., 2004 | |||
| Inferred | MYO7A | Street et al., 2004 | |||
| Digenic inheritance | Digenic inheritance | ||||
| Mutant gene | Mutant gene | Mutant gene | Mutant gene | ||
| Cdh23v | Pcdh15av | Zheng et al., 2005 | CDH23 | PCDH15 | Zheng et al., 2005 |
| Gjb2 | Gjb6 | Michel et al., 2003 | GJB2 | GJB6 | Del Castillo et al., 2002; Pallares-Ruiz et al., 2002 |
| Thratm2 | Thrbtm1 | Ng et al., 2001 | GJB3 | TECTA | Balciuniene et al., 1998 |
| GJB2 | mtDNA A1555G | Abe et al., 2001 | |||
| USH3 | MYO7A | Adato et al., 1999 | |||
4. Cdh23 variants as modifiers of hearing in mutant mice
The same Cdh23 variants that underlie the ahl locus and contribute to differences in AHL among inbred mouse strains also have been shown to modify hearing loss in Atp2b2dfw-2J mutant mice (the mdfw locus) (Noben-Trauth et al., 2003) and in Mass1frings mutant mice (Johnson et al., 2005). Cdh23 variants also may underlie differences in hearing loss among 129-related strains, which may have important implications for interpreting strain background effects in genetically engineered mutant mice. In all of these cases, the Cdh23753A variant is generally associated with increased susceptibility to hearing loss, and the Cdh23753G variant with enhanced resistance.
4.1. Modifier of deaf waddler (mdfw)
The mdfw modifier was first recognized in the coisogenic BALB/cBy-Atp2b2dfw-2J strain by causing a moderate hearing loss in dfw2J heterozygotes (Noben-Trauth et al., 1997). Subsequent ABR threshold measurements in F2 heterozygotes derived from (BALB/cBy-Atp2b2dfw-2J/dfw-2J × CAST/Ei) F1xF1 and (BALB/cBy-Atp2b2dfw-2J/dfw-2J × MOLF/Ei) F1xF1 intercrosses showed a 3:1 segregation ratio of the hearing loss, suggestive of the effect of a recessive modifier allele. Genome-wide linkage analyses on normal and hearing impaired heterozygotes identified with a high significance a genetic modifier locus (mdfw) on Chr 10. The recessive mdfw allele is fully penetrant, shows little variation in expressivity, and fully accounts for the heterozygous effect on the isogenic background and in the F2 intercrosses. In the comparable age group, the individual effect of mdfw itself without the Atp2b2dfw-2J mutation (Atp2b2+/+ mdfw/mdfw mice) is negligible. As described previously, the molecular basis of mdfw and ahl was shown to be a single nucleotide polymorphism in Cdh23 (Noben-Trauth et al., 2003). Cdh23753A-segregating strains preferentially skip exon 7 leading to an in-frame deletion of 43 amino acids at the extracellular amino-terminus. The effect of the deletion on the function of cadherin 23 is unknown, but the altered conformation of the N-terminal cadherin domains likely leads to either early degradation or impaired adhesion function. The hearing loss in BALB/cBy-Atp2b2+/dfw-2J heterozygotes may result from the combined effect of haploinsufficiency of Cdh23 and Atp2b2 (Wood et al., 2004).
4.2. Hearing modifier of Mass1frings mutant mice
Recently, we have shown that Cdh23 variants also modify the hearing loss caused by the Mass1frings mutation in BUB/BnJ strain mice (Johnson et al., 2005). The Mass1 gene was originally identified as a susceptibility locus for audiogenic seizures in Frings and BUB/BnJ strain mice (Skradski et al., 2001), but this phenotype may be secondary to the hearing impairment exhibited by mice of these strains. BUB/BnJ mice are hearing impaired already by 3 weeks of age and become deaf by 20 weeks. We analyzed two backcrosses involving the BUB/BnJ inbred strain with mice of strains CAST/EiJ and MOLD/RkJ and found highly significant linkage of the Chr 13 Mass1 locus with ABR threshold variation in young mice from both backcrosses. We found that Cdh23 and Mass1 genotypes act in an additive fashion to modulate the progression of hearing loss in the backcross mice. These modifying effects of the Cdh23 locus can account for the hearing and seizure differences observed between Frings (Cdh23753G) and BUB/BnJ (Cdh23753A) mice, which share the Mass1frings mutation. Strain-specific Cdh23 variants thus have been shown to modify the hearing of mice with two different gene mutations, Atp2b2dfw-2J and Mass1frings; and the potential modifying effect of the ahl locus should be considered when evaluating the hearing phenotypes of other mutant genes.
4.3. Cdh23 variants may contribute to 129-strain background effects on hearing
The importance of controlling for strain background effects when assessing phenotypes of genetically engineered mice has been stressed in previous reviews (Barthold, 2004; Gao et al., 2004; Gerlai, 1996; Linder, 2001; Wolfer et al., 2002). Of particular importance in the analysis of knockout mice is the origin of the embryonic stem (ES) cell lines that are used in gene targeting. Most ES cell lines were derived from 129 mouse substrains. Although all of these strains include 129 in their names, they exhibit extensive genetic variation because of accidental or deliberate admixtures with other strains during their derivations and subsequent genetic divergence (Simpson et al., 1997). Investigators should be aware of these differences when designing targeted mutagenesis experiments and evaluating the phenotypes of mutant mice produced from them.
To address the possible modifying effects that different 129 strain backgrounds might have on hearing assessments of mutant mice, we compared the progression of hearing loss in six of these strains (Fig. 2). Average ABR thresholds were determined at multiple ages for each of the strains. To test whether the ahl gene might underlie the observed hearing loss differences among strains, we genotyped mice of each strain for the Cdh23 753A/G variant. All three of the strains that exhibited early hearing loss by 20 weeks of age had the Cdh23753A variant, whereas all three of the strains that retained normal hearing levels beyond 30 weeks of age had the Cdh23753G allele. These results are consistent with the effects that these variants have on hearing in other inbred strains (Noben-Trauth et al., 2003), and demonstrate the importance of knowing which particular 129-related strain is represented in the strain background on which the mutant phenotype is being evaluated. Consistent with their proposed historical derivations (Simpson et al., 1997), the strains with the Cdh23753A allele (129P1/ReJ, 129P3/J, 129X1/SvJ) belong to a different lineage than strains with the Cdh23753G allele (129S1/SvImJ, 129S6/SvEv-Mostm1Ev/J, and 129T2/SvEmsJ).
Fig. 2.
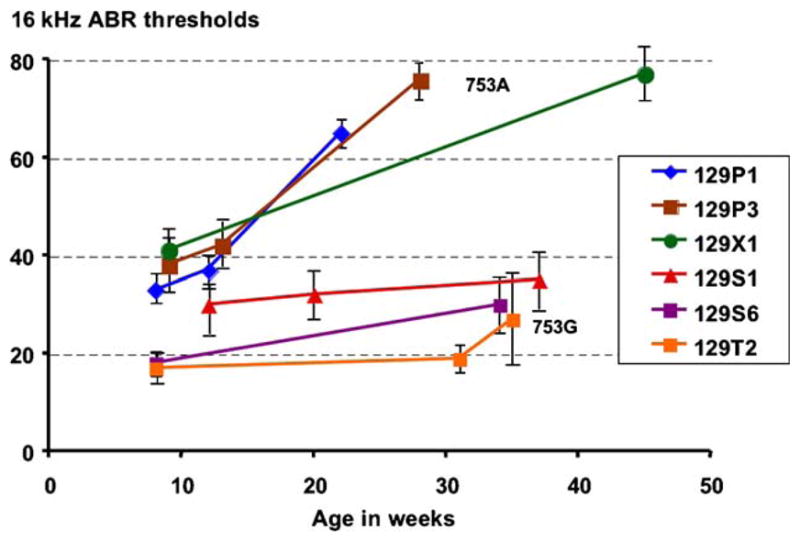
Hearing loss progression and Cdh23 alleles among six different 129-related inbred strains. The average ABR thresholds for a 16 kHz pure tone stimulus and their standard error bars are shown for each strain and age when tested. The full strain names for the six abbreviations shown in the figure are as follows: 129P1/ReJ (129P1), 129P3/J (129P3), 129X1/SvJ (129X1), 129S1/SvImJ (129S1), 129S6/SvEv-Mostm1Ev/J (129S6), 129T2/SvEmsJ (129T2). 753A and 753G are allelic variants of the Cdh23 gene.
5. Other modifiers of hearing in mutant mice
Not all hearing modifiers contribute to inbred strain AHL as does the Cdh23753A/G variant described above. Some modifiers may not express a phenotype by themselves and thus can be detected only by their influence on the phenotypes caused by other gene mutations. The modifier of tubby hearing (Moth1) is a dominant modifier that can be detected only by its effect on the hearing phenotype of mice homozygous for the tubby mutation (Tubtub). On the coisogenic C57BL/6J background, tub/tub mice have a profound hearing loss. When crossed onto the AKR/J and CAST/Ei backgrounds, tub homozygotes show a wide scatter of hearing thresholds ranging from deafness to a complete rescue. Given the quantitative nature of the phenotype, Ikeda et al. (1999) performed a QTL linkage analyses and mapped a significant locus to Chr 2. A positional-candidate gene approach combined with transgenic rescue provided convincing evidence that amino acid polymorphisms in the MTAP1 protein mediate the modifying effect, which was suggested to occur at synaptic terminals between hair cells and cochlear neurons (Ikeda et al., 2002). Like mdfw, the Moth1 modifier affects only the hearing phenotype, whereas other phenotypic manifestations of the tubby mutation, such as retinal degeneration and obesity, are unaffected.
6. Genetic modifiers of hearing loss in human disorders
Whereas mutant deafness alleles in the mouse are often isogenic on inbred strains, mutations in humans occur on a genetically diverse and segregating background providing a greater chance to detect the effect of a modifying polymorphism but presenting a more difficult challenge to identify the underlying gene. Linkage studies on human pedigrees segregating mitochondrial deafness and recessive, nonsyndromic hearing loss DFNB26 identified and mapped two loci modifying hearing impairment. In a Pakistani family, individuals homozygous for the disease causing DFNB26 haplotype but with apparently normal hearing carry a dominant allele of the DFNM1 locus on Chr 1 (near D1S2850) (Riazuddin et al., 2000). Linkage analyses in large Spanish and Italian families with the mitochondrial A1555G mutation and sensorineural deafness identified a modifier locus on Chr 8 (near D8S277) with suggestive genome-wide significance (lod score of 3.1) (Bykhovskaya et al., 2000). There is suggestive evidence that the TFB1M gene on Chr 6, which is involved in mitochondrial RNA metabolism, may be another nuclear modifier of the mitochondrial A1555G mutation, suggesting that modification of 12s rRNA is a carefully regulated process (Bykhovskaya et al., 2004). In a large American family, severity of MYO7A-associated hearing loss varied in an apparent homomorphic environment, suggesting the effect of genetic modifier(s) (Street et al., 2004).
A genetic interaction between alleles of CDH23 and ATP2B2, similar to the Cdh23mdfw–Atp2b2dfw-2J paradigm in the mouse, was recently described in a human pedigree (Schultz et al., 2005). In this family, affected members expressed variable degrees of hearing loss differentially affecting the higher frequencies. While all individuals with hearing loss were homozygous for a missense mutation (F1888S) in CDH23, those individuals with the hearing loss affecting all frequencies also carried a missense mutation (V586M) in ATP2B2. Individuals with hearing loss in the higher frequencies but with normal thresholds in the lower range had the normal 586V allele. The 586M allele of ATP2B2 seems to have a clear modifying effect when combined with the CDH23 mutation, but the allele per se has no independent pathological effect.
7. Digenic inheritance and hearing loss in mice and humans
Additive or interactive effects can occur between two mutant genes (digenic inheritance) or between a mutant gene and a benign polymorphism (modifier gene). The boundary between these distinctions is not always clear. The difference between a mutation and a polymorphism is relative to its frequency of occurrence in a population, and some DNA alterations that appear to be benign actually may have subtle phenotypic effects if examined more carefully. Because modifier genes and digenic inheritance are similar phenomena that are not always distinguishable, we have included in this review several examples of digenic inheritance of hearing loss that have been identified in both mice and humans.
The general mechanism underlying modulation of hearing loss appears to be specific for each digenic interaction. In the case of a reported interaction of targeted alleles of the mouse thyroid hormone receptor genes Thra and Thrb, over-expression of a Thra splice form seems to mediate a modifying effect on the Thrb hearing phenotype (Ng et al., 2001). Mice deficient for the Thrb gene (Thrbtm1/tm1) show hearing loss and thyroid dysfunction, but mice with a targeted deletion in the Thra locus are phenotypically normal. Interestingly, in Thrbtm1/tm1 homozygotes carrying one or two copies of the targeted Thra mutation (Thratm2), hearing function was completely restored. It was shown that the Thratm2 mutation, which deletes part of the Thra locus, results in over-expression of a splice form (Thra1) that is capable of compensating for the lack of Thrb (Ng et al., 2001).
Given the high degree of conservation between mouse and human genomes, it is not surprising that some forms of digenic inheritance and genetic modifications are found in both species. Recessive mutations in both Cdh23 and Pcdh15 cause hearing loss in mice and humans. Single heterozygotes in both species are normal, but double heterozygotes show elevated hearing thresholds (Zheng et al., 2005). In double heterozygous mice (Cdh23+/v-2J Pcdh15+/av-3J), hearing loss is age-related and associated with stereocilia disorganization and degeneration of organ of Corti and spiral ganglion cells, all of which are typical manifestations of the waltzer (Cdh23v2-J) and Ames waltzer (Pcdh15av-3J) mutations. Rather than modifying an existing phenotype, both alleles synergistically interact to produce trans dominant effects. Although the auditory effect is rather strong, it does not affect vestibular function in double heterozygotes. The genetic programs in these tissues are apparently buffering against the partial loss of Cdh23 and Pcdh15.
Digenic inheritance of deafness has been reported for the closely linked genes encoding the gap junction proteins GJB2 (connexin 26) and GJB6 (connexin 30). Mutations in GJB2 are the most common cause of recessive nonsyndromic deafness; however, many deaf patients have only a single GJB2 mutant allele, suggesting the presence of accompanying mutations in other genes. The deafness of individuals heterozygous for GJB2 mutations has been associated with in trans heterozygosity for GJB6 gene deletions in both Spanish (Del Castillo et al., 2002) and French (Pallares-Ruiz et al., 2002) populations. Mice that are heterozygous for both Gjb2 and Gjb6 knockout mutations (Gjb2+/− Gjb6+/− double heterozygotes) exhibit a moderate hearing impairment and reduced endocochlear potential compared with single heterozygotes (Michel et al., 2003), a result consistent with the cases of digenic inheritance reported for the corresponding human genes.
Several additional cases of digenic inheritance underlying hearing loss have been reported in human families. A digenic interaction has been reported between GJB2 and a mitochondrial gene, in which the hearing loss associated with the 1555A>G mitochondrial mutation is more severe in patients who also are heterozygous for the GJB2 mutation (Abe et al., 2001). An additive effect of mutant alleles at the DFNA12 and DFNA2 loci was proposed to explain the variation in severity of a progressive nonsyndromic hearing loss observed in a Swedish family, wherein each separate locus was associated with a mild and sometimes undiagnosed phenotype, but both loci together were associated with a more severe phenotype (Balciuniene et al., 1998). Subsequently, mutations of the alpha tectorin gene TECTA were shown to underlie DFNA12 in this family, but no mutations were found in the gap junction gene GJB3 or the potassium channel gene KCNQ4, which were shown to be responsible for deafness at the DFNA2 locus in other families (Balciuniene et al., 1999). These results do not rule out digenic inheritance, because the DFNA2 locus may include an additional, as yet unidentified, gene that modulates the hearing loss associated with TECTA mutations. A different pattern of digenic inheritance was reported for the Usher syndrome loci USH1B and USH3A in a family of Jewish Yemenite origin, where the presence of a single defective MYO7A allele appeared to increase the severity of deafness in individuals with two defective USH3 alleles, suggesting an interaction between the gene products (Adato et al., 1999). The defective USH3 allele in this family was subsequently identified as a deletion in the USH3A gene, which encodes an integral membrane protein expressed in cochlear hair cells and ganglion cells, suggesting that the USH3A–MYO7A interaction may play a role in hair cell synapses (Adato et al., 2002).
8. Conclusion
Genetic background is an important consideration when assessing mutant phenotypes. In this review, we have shown how genes affecting AHL in inbred strains can also act as strain-specific modifiers of hearing in mutant mice. Cdh23 variants, which underlie the ahl locus effects on AHL in many inbred strains, are particularly important in this regard (Table 1). Other modifier genes and digenic interactions have been identified that contribute to genetic background effects on hearing in both mice and humans (Table 2). Advances in genomic resources and technologies have increased our capability to discover genes underlying complex traits such as hearing (Dipetrillo et al., 2005). Identification and molecular characterization of hearing modifiers and analyses of their protein products will provide insights into pathways and mechanisms of gene and protein interactions involved in auditory development and function. It may also further our understanding of clinically relevant phenotypic variation and predisposition to hearing loss.
9. Note added in proof
The Eya1bor mutation is a hypomorphic allele that causes inner ear and kidney abnormalities in homozygous mice, with severity dependent on strain background. A recent publication describes the genetic mapping of two modifier loci, Mead1 on Chr 4 and Mead2 on Chr 12, that suppress hearing loss and cochlear dysmorphogenesis in Eya1bor/bor mice produced from an intercross of (C3HeB/FeJ-Eya1bor/+ × C57BL/6J) F1 hybrids: (Niu et al., in press).
Acknowledgments
This work was in part supported by NIH grants DC005827 and DC62108 and by the intramural research program at NIDCD/NIH. We thank Heping Yu for ABR threshold measurements presented in Fig. 2 and Alain Dabdoub and Feng Qian for helpful comments on the manuscript.
References
- Abe S, Kelley PM, Kimberling WJ, Usami SI. Connexin 26 gene (GJB2) mutation modulates the severity of hearing loss associated with the 1555A→G mitochondrial mutation. Am J Med Genet. 2001;103:334–338. [PubMed] [Google Scholar]
- Adato A, Kalinski H, Weil D, Chaib H, Korostishevsky M, Bonne-Tamir B. Possible interaction between USH1B and USH3 gene products as implied by apparent digenic deafness inheritance. Am J Hum Genet. 1999;65:261–265. doi: 10.1086/302438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millan JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10:339–350. doi: 10.1038/sj.ejhg.5200831. [DOI] [PubMed] [Google Scholar]
- Ahituv N, Avraham K. Mouse models for human deafness: current tools for new fashions. Trends Mol Med. 2002;8:447. doi: 10.1016/s1471-4914(02)02388-2. [DOI] [PubMed] [Google Scholar]
- Balciuniene J, Dahl N, Borg E, Samuelsson E, Koisti MJ, Pettersson U, Jazin EE. Evidence for digenic inheritance of nonsyndromic hereditary hearing loss in a Swedish family. Am J Hum Genet. 1998;63:786–793. doi: 10.1086/302012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciuniene J, Dahl N, Jalonen P, Verhoeven K, Van Camp G, Borg E, Pettersson U, Jazin EE. Alpha-tectorin involvement in hearing disabilities: one gene-two phenotypes. Hum Genet. 1999;105:211–216. doi: 10.1007/s004390051091. [DOI] [PubMed] [Google Scholar]
- Barthold SW. Genetically altered mice: phenotypes, no phenotypes, and Faux phenotypes. Genetica. 2004;122:75–88. doi: 10.1007/s10709-004-1439-3. [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Bryda EC, Kim HJ, Legare ME, Frankel WN, Noben-Trauth K. High-resolution genetic and physical mapping of modifier-of-deaf waddler (mdfw) and Waltzer (Cdh23v) Genomics. 2001;73:338–342. doi: 10.1006/geno.2001.6538. [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y, Estivill X, Taylor K, Hang T, Hamon M, Casano RA, Yang H, Rotter JI, Shohat M, Fischel-Ghodsian N. Candidate locus for a nuclear modifier gene for maternally inherited deafness. Am J Hum Genet. 2000;66:1905–1910. doi: 10.1086/302914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M, Fischel-Ghodsian N. Human mitochondrial transcription factor B1 as a modifier gene for hearing loss associated with the mitochondrial A1555G mutation. Mol Genet Metab. 2004;82:27–32. doi: 10.1016/j.ymgme.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y, Yang H, Taylor K, Hang T, Tun RY, Estivill X, Casano RA, Majamaa K, Shohat M, Fischel-Ghodsian N. Modifier locus for mitochondrial DNA disease: linkage and linkage disequilibrium mapping of a nuclear modifier gene for maternally inherited deafness. Genet Med. 2001;3:177–180. doi: 10.1097/00125817-200105000-00005. [DOI] [PubMed] [Google Scholar]
- Darvasi A. The effect of selective genotyping on QTL mapping accuracy. Mamm Genome. 1997a;8:67–68. doi: 10.1007/s003359900353. [DOI] [PubMed] [Google Scholar]
- Darvasi A. Interval-specific congenic strains (ISCS): an experimental design for mapping a QTL into a 1-centimorgan interval. Mamm Genome. 1997b;8:163–167. doi: 10.1007/s003359900382. [DOI] [PubMed] [Google Scholar]
- Darvasi A. Experimental strategies for the genetic dissection of complex traits in animal models. Nat Genet. 1998;18:19–24. doi: 10.1038/ng0198-19. [DOI] [PubMed] [Google Scholar]
- Davis RR, Newlander JK, Ling X, Cortopassi GA, Krieg EF, Erway LC. Genetic basis for susceptibility to noise-induced hearing loss in mice. Hear Res. 2001;155:82–90. doi: 10.1016/s0378-5955(01)00250-7. [DOI] [PubMed] [Google Scholar]
- del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346:243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- Dipetrillo K, Wang X, Stylianou IM, Paigen B. Bioinformatics toolbox for narrowing rodent quantitative trait loci. Trends Genet. 2005;21:683–692. doi: 10.1016/j.tig.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Drayton M, Noben-Trauth K. Mapping quantitative trait loci for hearing loss in Black Swiss mice. Hear Res. 2006;212:128–139. doi: 10.1016/j.heares.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Erickson RP. Mouse models of human genetic disease: which mouse is more like a man? Bioessays. 1996;18:993–998. doi: 10.1002/bies.950181209. [DOI] [PubMed] [Google Scholar]
- Erway LC, Shiau YW, Davis RR, Krieg EF. Genetics of age-related hearing loss in mice: III. Susceptibility of inbred and F1 hybrid strains to noise-induced hearing loss. Hear Res. 1996;93:181–187. doi: 10.1016/0378-5955(95)00226-x. [DOI] [PubMed] [Google Scholar]
- Flint J, Valdar W, Shifman S, Mott R. Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev, Genet. 2005;6:271–286. doi: 10.1038/nrg1576. [DOI] [PubMed] [Google Scholar]
- Friedman T, Battey J, Kachar B, Riazuddin S, Noben-Trauth K, Griffith A, Wilcox E. Modifier genes of hereditary hearing loss. Curr Opin Neurobiol. 2000;10:487–493. doi: 10.1016/s0959-4388(00)00120-3. [DOI] [PubMed] [Google Scholar]
- Gao J, Wu X, Zuo J. Targeting hearing genes in mice. Brain Res Mol Brain Res. 2004;132:192–207. doi: 10.1016/j.molbrainres.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- Haider NB, Ikeda A, Naggert JK, Nishina PM. Genetic modifiers of vision and hearing. Hum Mol Genet. 2002;11:1195–1206. doi: 10.1093/hmg/11.10.1195. [DOI] [PubMed] [Google Scholar]
- Harding GW, Bohne BA, Vos JD. The effect of an age-related hearing loss gene (Ahl) on noise-induced hearing loss and cochlear damage from low-frequency noise. Hear Res. 2005;204:90–100. doi: 10.1016/j.heares.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Henry KR, Chole RA. Genotypic differences in behavioral, physiological and anatomical expressions of age-related hearing loss on the laboratory mouse. Audiology. 1980;19:369–383. doi: 10.3109/00206098009070071. [DOI] [PubMed] [Google Scholar]
- Houlston RS, Tomlinson IP. Modifier genes in humans: strategies for identification. Eur J Hum Genet. 1998;6:80–88. doi: 10.1038/sj.ejhg.5200156. [DOI] [PubMed] [Google Scholar]
- Ikeda A, Zheng QY, Rosenstiel P, Maddatu T, Zuberi AR, Roopenian DC, North MA, Naggert JK, Johnson KR, Nishina PM. Genetic modification of hearing in tubby mice: evidence for the existence of a major gene (moth1) which protects tubby mice from hearing loss. Hum Mol Genet. 1999;8:1761–1767. doi: 10.1093/hmg/8.9.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Zheng QY, Zuberi AR, Johnson KR, Naggert JK, Nishina PM. Microtubule-associated protein 1A is a modifier of tubby hearing (moth1) Nat Genet. 2002;30:401–405. doi: 10.1038/ng838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Erway LC, Cook SA, Willott JF, Zheng QY. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res. 1997;114:83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY. Ahl2, a second locus affecting age-related hearing loss in mice. Genomics. 2002;80:461–464. [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70:171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fischel-Ghodsian N. A nuclear-mitochondrial DNA interaction affecting hearing impairment in mice. Nat Genet. 2001;27:191–194. doi: 10.1038/84831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Weston MD, Ptacek LJ, Noben-Trauth K. The Mass1(frings) mutation underlies early onset hearing impairment in BUB/BnJ mice, a model for the auditory pathology of Usher syndrome IIC. Genomics. 2005;85:582–590. doi: 10.1016/j.ygeno.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keithley EM, Canto C, Zheng QY, Fischel-Ghodsian N, Johnson KR. Age-related hearing loss and the ahl locus in mice. Hear Res. 2004;188:21–28. doi: 10.1016/S0378-5955(03)00365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- Linder CC. The influence of genetic background on spontaneous and genetically engineered mouse models of complex diseases. Lab Anim (NY) 2001;30:34–39. [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16:188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- Manly KF, Cudmore RH, Jr, Meer JM. Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome. 2001;12:930–932. doi: 10.1007/s00335-001-1016-3. [DOI] [PubMed] [Google Scholar]
- Mburu P, Liu XZ, Walsh J, Saw D, Jr, Cope MJ, Gibson F, Kendrick-Jones J, Steel KP, Brown SD. Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1997;1:191–203. doi: 10.1046/j.1365-4624.1997.00020.x. [DOI] [PubMed] [Google Scholar]
- Michel V, Hardelin JP, Petit C. Molecular mechanism of a frequent genetic form of deafness. N Engl J Med. 2003;349:716–717. doi: 10.1056/NEJMc030327. [DOI] [PubMed] [Google Scholar]
- Montagutelli X. Effect of the genetic background on the phenotype of mouse mutations. J Am Soc Nephrol. 2000;11 (Suppl 16):S101–S105. [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes in mice and humans. Nat Rev, Genet. 2001;2:165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes and protective alleles in humans and mice. Curr Opin Genet Dev. 2003;13:290–295. doi: 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Singer JB, Matin A, Lander ES. Analysing complex genetic traits with chromosome substitution strains. Nat Genet. 2000;24:221–225. doi: 10.1038/73427. [DOI] [PubMed] [Google Scholar]
- Nemoto M, Morita Y, Mishima Y, Takahashi S, Nomura T, Ushiki T, Shiroishi T, Kikkawa Y, Yonekawa H, Kominami R. Ahl3, a third locus on mouse chromosome 17 affecting age-related hearing loss. Biochem Biophys Res Commun. 2004;324:1283–1288. doi: 10.1016/j.bbrc.2004.09.186. [DOI] [PubMed] [Google Scholar]
- Ng L, Rusch A, Amma LL, Nordstrom K, Erway LC, Vennstrom B, Forrest D. Suppression of the deafness and thyroid dysfunction in Thrb-null mice by an independent mutation in the Thra thyroid hormone receptor alpha gene. Hum Mol Genet. 2001;10:2701–2708. doi: 10.1093/hmg/10.23.2701. [DOI] [PubMed] [Google Scholar]
- Niu H, Makmura L, Shen T, Sheth SS, Blair K, Friedman RA. Identification of two major loci that suppress hearing loss and cochlear dysmorphogenesis in Eya1(bor/bor) mice. Genomics. doi: 10.1016/j.ygeno.2006.01.005. in press. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR, Nishina PM. mdfw: a deafness susceptibility locus that interacts with deaf waddler (dfw) Genomics. 1997;44:266–272. doi: 10.1006/geno.1997.4869. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Gagnon PM. Cellular correlates of progressive hearing loss in 129S6/SvEv mice. J Comp Neurol. 2004;469:377–390. doi: 10.1002/cne.11011. [DOI] [PubMed] [Google Scholar]
- Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet. 2002;10:72–76. doi: 10.1038/sj.ejhg.5200762. [DOI] [PubMed] [Google Scholar]
- Riazuddin S, Castelein CM, Ahmed ZM, Lalwani AK, Mastroianni MA, Naz S, Smith TN, Liburd NA, Friedman TB, Griffith AJ, Wilcox ER. Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat Genet. 2000;26:431–434. doi: 10.1038/82558. [DOI] [PubMed] [Google Scholar]
- Riazuddin S, Ahmed ZM, Friedman TB, Griffith AJ, Wilcox ER. Genetic modifiers of hereditary hearing loss. Adv Oto-Rhino-Laryngol. 2002;61:224–229. doi: 10.1159/000066813. [DOI] [PubMed] [Google Scholar]
- Romeo G, McKusick VA. Phenotypic diversity, allelic series and modifier genes. Nat Genet. 1994;7:451–453. doi: 10.1038/ng0894-451. [DOI] [PubMed] [Google Scholar]
- Schultz JM, Yang Y, Caride AJ, Filoteo AG, Penheiter AR, Lagziel A, Morell RJ, Mohiddin SA, Fananapazir L, Madeo AC, Penniston JT, Griffith AJ. Modification of human hearing loss by plasma-membrane calcium pump PMCA2. N Engl J Med. 2005;352:1557–1564. doi: 10.1056/NEJMoa043899. [DOI] [PubMed] [Google Scholar]
- Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Skradski SL, Clark AM, Jiang H, White HS, Fu YH, Ptacek LJ. A novel gene causing a Mendelian audiogenic mouse epilepsy. Neuron. 2001;31:537–544. doi: 10.1016/s0896-6273(01)00397-x. [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Biesecker LG. Genetic modifiers in human development and malformation syndromes, including chaperone proteins. Hum Mol Genet. 2003;12 (Spec 1):R45–R50. doi: 10.1093/hmg/ddg099. [DOI] [PubMed] [Google Scholar]
- Spongr VP, Flood DG, Frisina RD, Salvi RJ. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J Acoust Soc Am. 1997;101:3546–3553. doi: 10.1121/1.418315. [DOI] [PubMed] [Google Scholar]
- Street VA, Kallman JC, Kiemele KL. Modifier controls severity of a novel dominant low-frequency MyosinVIIA (MYO7A) auditory mutation. J Med Genet. 2004;41:e62. doi: 10.1136/jmg.2003.013557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez AE, Jimenez AM, Martin GK, Luebke AE, Lonsbury-Martin BL. Evaluating cochlear function and the effects of noise exposure in the B6. CAST+Ahl mouse with distortion product otoacoustic emissions. Hear Res. 2004;194:87–96. doi: 10.1016/j.heares.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997;16:191–193. doi: 10.1038/ng0697-191. [DOI] [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2001;2:research 0046.1–research 0046.18. doi: 10.1186/gb-2001-2-11-research0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–340. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- Wood JD, Muchinsky SJ, Filoteo AG, Penniston JT, Tempel BL. Low endolymph calcium concentrations in deafwaddler2J mice suggest that PMCA2 contributes to endolymph calcium maintenance. J Assoc Res Otolaryngol. 2004;5:99–110. doi: 10.1007/s10162-003-4022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR. Hearing loss associated with the modifier of deaf waddler (mdfw) locus corresponds with age-related hearing loss in 12 inbred strains of mice. Hear Res. 2001;154:45–53. doi: 10.1016/s0378-5955(01)00215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Yan D, Ouyang XM, Du LL, Yu H, Chang B, Johnson KR, Liu XZ. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum Mol Genet. 2005;14:103–111. doi: 10.1093/hmg/ddi010. [DOI] [PMC free article] [PubMed] [Google Scholar]