

Abstract
Background
Identification of infants at risk for sudden arrhythmic death remains one of the leading challenges of modern medicine. We present a family in which a common polymorphism (SNP) inherited from the father combined with a stop codon mutation inherited from the mother, both asymptomatic, leads to two cases of sudden infant death.
Methods and Results
KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, CACNA1c, CACNB2b, and KCNJ2 genes were amplified and analyzed by direct sequencing. Functional electrophysiological studies were performed with the SNP and mutation expressed singly and in combination in Chinese ovary (CHO-K1) and COS-1 cells. An asymptomatic female presenting after death of her 2 day old infant and spontaneous abortion of a second baby in the first trimester was referred for genetic analysis. The newborn had nearly incessant had nearly incessant ventricular tachycardia while inn-utero and prolonged QTc (560ms) The mother was asymptomatic but displayed a prolonged QTc. Genetic screening of the mother revealed a heterozygous nonsense mutation (P926AfsX14) in KCNH2, predicting a stop codon. The father was asymptomatic with a normal QTc, but had a heterozygous polymorphism (K897T) in KCNH2. The baby who died at 2 days of age and the aborted fetus inherited both K897T and P926AfsX14. Heterologous co-expression of K897T and P926AfsX14 led to loss of function of HERG current much greater than expression of K897T or P926AfsX14 alone.
Conclusions
Our data suggest that a common polymorphism (K897T) can markedly accentuate the loss of function of mildly defective HERG channels leading to long QT syndrome-mediated arrhythmias and sudden infant death.
Keywords: Genetics, Arrhythmias, Sudden Cardiac Death, Electrophysiology, HERG
Introduction
Long QT syndrome (LQTS) is a congenital disorder that predisposes affected individuals to sudden cardiac death (SCD)1, 2. To date, mutations in 12 genes have been identified1–5 although some affected LQT patients have symptoms ranging from syncope to severe arrhythmias such as torsade de pointes (TdP), in most cases patients are asymptomatic3–5. In some, the QT interval is within normal range6. LQTS has also been linked to sudden infant death syndrome. In a study done by Arnestad et al.,6 9.5% of the cases diagnosed as sudden infant death syndrome (SIDS) had functional genetic variants in one of the LQT genes. Other studies have also found cardiac ion channels mutation in SIDS cases.7–13 Although evaluation of family members clearly demonstrated that the parents were in some cases not affected, in most studies genetic or electrocardiogram (ECG) screening of the parents was not available and therefore there was no definitive evidence as to whether these mutations were inherited or de novo. Interestingly, in one study by Maron and colleagues,14 ECG data of family members of SIDS victims showed QT prolongation in> 25% of the 1st degree relatives. Thus, there are different phenotypes and varying degrees of QT prolongation in LQTS patients.
Variations of phenotype expression are thought to be attributable to the severity of the disease-causing mutation as well as the possible co- existence of other genetic variations,15, 16 including single nucleotide polymorphisms (SNP) which are not disease-causing by definition but which can alter arrhythmia susceptibility. This has been demonstrated with several SNPs such as D85N (in KCNE1),16 K897T (in KCNH2)17 and H558R (in SCN5A) 18–20. In all cases, the patients also had mutations in the same gene causing the disease phenotype. SNPs have been shown to modify clinical expression either by aggravating the clinical phenotype16, 18, 21 or by attenuating the clinical phenotype19, 20. We present a family in which an inherited common polymorphism in KCNH2 when combined with a loss of function mutation on separate alleles of the same gene lead to infant death. Family members with the only polymorphism or mutation alone did not have any events of syncope or sudden cardiac death.
Methods
ECG analysis
QT interval was measured and adjusted to heart rate (QTc), according to Bazett’s formula22. The end of the T wave was defined as the intersection with the isoelectric line of a tangent drawn to the descending portion of the T wave.
Genetic evaluation
After informed consent was obtained, blood was collected from family members. Tissue obtained from the stillborn fetus was provided with written consent of the parents. Genomic DNA was extracted from peripheral blood leukocytes and from fresh and frozen tissue with a commercial kit (Puregene, Gentra Systems, Inc. Minneapolis, MN). The genomic DNA was amplified by polymerase chain reaction ((PCR) on GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA). All exons and intron borders of the KCNQ1, KCNH2, SCN5A, KCNE1 KCNE2, CACNA1c, CACNB2b, KCNJ2 genes were amplified and analyzed by direct sequencing. PCR products were purified with a commercial reagent (ExoSAP- IT, USB Corporation, Cleveland, OH) and directly sequenced from both directions using ABI PRISM 3100 Automatic DNA sequencer (Applied Biosystems, Foster City, CA). Electropherograms were visually examined for heterozygous peaks and compared with reference sequences for homozygous variations (GenBank accession number NM_000219) using the CodonCode Aligner Ver. 2.0.4 (CodonCode Corporation, Dedham, MA).
Mutagenesis
KCNH2 cDNA (accession No. NM_000238) in a bicistronic vector encoding green fluorescent protein (GFP) (GFIrHerg) was a kind gift from Dr. Connie Bezzina. The P926AfsX14 mutation and the K897T polymorphism (rs1801523) were introduced using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and the primers: sense: GTAGCCGGGGCCGGCCGGGGGGGGCCGTGGGGGGAGAGCCCGTC antisense: GACGGGCTCTCCCCCCACGGCCCCCCCCGGCCGGCCCCGGCTAC The mutated plasmid was sequenced to ensure the presence of the P926AfsX14 mutation as well as the absence of other substitutions introduced by the DNA polymerase.
Transient expression in CHO- K1 cells
Chinese hamster ovary (CHO-K1) cells were grown in GIBCO F12 nutrient mixture (GIBCO, Invitrogen, Carlsbad, CA) in 35-mm culture dishes and placed in a 5% CO2 incubator at 37°C. The cells were transfected using FuGene6 (Roche Diagnostics, Indianapolis, IN). To assess the influence of wild type (WT) on expression of the mutant channels, CHO-K1 cells were co-transfected with various combinations of WT, K897T and P926AfsX14 KCNH2 in 1:1 molar ratios. Electrophysiological studies were performed 48 to of 72 hours after transfection on cells expressing fluorescence.
Transient expression in African green monkey kidney derived cell line (COS-1) cells
Cells were grown in DMEM (GIBCO) supplemented with 10% FCS (GIBCO) at 37°C, in 5% CO2. 24 hours prior to transfection, cells were plated on square coverslips inside a petri dish and kept in the same culture conditions as before. Cells were transfected with 2 μg of plasmid, using FuGene6 (Roche Diagnostics, Indianapolis, IN) according to the manufacturer’s instructions.
Electrophysiology
Voltage clamp recordings were made as previously described23 using patch pipettes fabricated from borosilicate glass capillaries (1.5 mm O.D., Fisher Scientific, Pittsburgh, PA). The pipettes were pulled using a gravity puller (Narishige Co. Ltd, Tokyo, Japan) and filled with pipette solution of the following composition (mmol/L): 10 KCl, 125 K-aspartate, 1.0 MgCl2, 10 HEPES, 10 NaCl, 5 MgATP and 10 EGTA, pH 7.2 (KOH). The pipette resistance ranged from 1–4 MΩ when filled with the internal solution. The perfusion solution contained (mmol/L): 130 NaCl, 5 KCl, 1.8 CaCl2, 1. MgCl2, 2.8 Na acetate, 10 HEPES, pH 7.3 with NaOH. Current signals were recorded using MultiClamp 700A and Axopatch 200B amplifiers (Axon Instruments Inc., Foster City, CA) and series resistance errors were reduced by about 60–70% with electronic compensation. All signals were acquired at 10–50 kHz (Digidata 1322, Axon Instruments, Foster City, CA) and analyzed with microcomputer running pClamp 9 software (Axon Instruments, Foster City, CA). All recordings were made at room temperature.
Immunofluorescence and confocal analysis
24–48 hours after transfection cells were washed with Phosphate Buffered Saline (PBS) and then fixed with 4% formaldehyde in PBS for 10 minutes. Cells were then permeabilized with 0.1% Triton-X for 5 minutes. Quenching was performed by 30-minute incubation with 0.1% Bovine Serum Albumin (BSA) in PBS. The cells were then incubated for 1–2 hours with primary antibodies diluted in a 0.1% BSA solution in PBS. Cells were then washed with PBS-BSA followed by 1 hour incubation with the fluorophore conjugated-secondary antibody at room temperature. After the final wash, the coverslips were mounted with Prolong Antifade (Molecular Probes, Eugene, OR). XYZ images of labeled cells were collected as previously described16, 21 with a Fluoview confocal microscope. An argon or krypton-argon laser (dependent on fluorophore) provided the excitation light. Fluorescence signals were collected with a 40× oil-immersion objective lens. XY frame was set to 512×512 pixels and laser intensity was set to 6–10% power. The Z-axis was changed in approximately 0.50 μm increments by computer control through the entire volume of the cell. Analysis of labeled cells was performed using both Fluoview and Image J software.
The primary antibodies used in this study were: rabbit polyclonal anti-human Ether-à-go-go Related Gene (HERG) recognizing an extracellular epitope (1:100, Alomone Labs., Jerusalem, Israel). For fluorescence detection a secondary donkey anti-rabbit antibody, conjugated with Alexa Fluor 594 (1:1000; Invitrogen, Carlsbad, CA) was added.
Statistical analysis
Electrophysiological data are presented as mean±SEM, and statistical comparisons were made with ANOVA followed by a Student-Newman-Keuls test or with a Student t test, as appropriate. Significance was defined as P<0.05.
Results
Clinical findings
A 24 year old woman was evaluated due to the loss of a female baby shortly after her birth and a first trimester loss of a male fetus in her second pregnancy (Fig. 1). Her first pregnancy resulted in a female baby born at 33 weeks who had incessant ventricular tachycardia (VT) throughout her last trimester as evidenced by fetal echocardiography showing VA dissociation with a V-rate of 190–270/min (mainly ~200/min) and an atrial rate of 110–120/min. The arrhythmia was treated with sotalol; propranolol; propranolol plus mexiletine and propranolol plus flecainide, all in attempt to treat the fetal tachycardia but without significant benefit to the fetus. The fetus’ arrhythmia responded only to magnesium therapy administered in the last 2 weeks of pregnancy. Post delivery, the baby had no arrhythmia but had severe hydrops and ultimately died. Her 12 lead ECG demonstrated a QTc of 560 ms (QT of 200 ms corrected for an HR of 130 bpm). Post-mortem autopsy revealed no evidence of any congenital or any structural heart disease.
Figure 1.
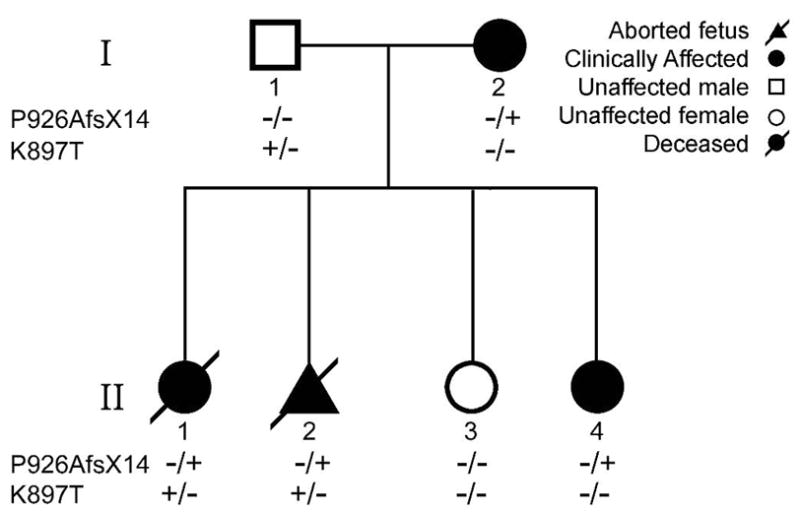
Pedigree of the Family.
The mother has no structural heart disease and had no apparent clinical history of syncope, near syncope or palpitations. She had been on telemetry in a high risk unit during her pregnancy for weeks without any documented arrhythmia of her own (or during and after her c-section). Twelve lead ECG of the mother showed QTc prolongation at baseline and displayed repolarization abnormalities with a markedly prolonged QTc following administration of sotalol to treat the fetal arrhythmias. She did not develop Torsade de Pointes (TdP) (Fig. 2). There is no known family history of long QT or syncope in the family. Of note, one cousin had SIDS.
Figure 2.

Twelve lead ECG of mother (I- 2 in Figure 1) and daughter (II- 4 in Figure 1) showing repolarization abnormalities with a prolonged QTc. The mother’s T wave morphology is suggestive of LQT2.
The mother’s second pregnancy ended on the ninth week. The fetal tissue was sent for genetic screening. The mother remained asymptomatic throughout without any complaints of palpitations, dizziness or syncope. She was subsequently maintained low dose of propranolol and a decision was made by the treating physician to implant an automatic implantable cardioverter-defibrillator (AICD). The mother’s third and fourth pregnancies resulted in delivery of a full term healthy baby with no documented arrhythmias however the fourth child had a prolonged QTc of 505 (Fig. 2). The father is asymptomatic with a normal QTc interval of 380 ms.
Genetic studies
Genetic analysis of the mother revealed a heterozygous nucleotide insertion of G at position 2775 of KCNH2 predicting a substitution of proline for alanine acid at position 926 of HERG and leading to a frame shift resulting in a stop codon (TAG) 14 amino acids later (P926AfsX14) (Figs. 3 and 4). Genetic analysis of the father (number I-1 in pedigree; Fig. 1) revealed a heterozygous nucleotide change from A to C at position 2690 of KCNH2 predicting a substitution of lysine with threonine at position 897 of HERG (K897T) (Fig. 3). K897T is a common polymorphism found in approximately 32% of the general Caucasian population. Genetic analysis of the first child and the second child (fetal tissue) (II-1 and II-2 in pedigree) demonstrated that both children inherited both P926AfsX14 and K897T. The third child (II-3) inherited neither the mutation nor the K897T polymorphism. She had a normal ECG with a QTc of 406 ms. The fourth child (II-4) who was found to have a QTc of 505 ms, inherited only the P926AfsX14 mutation and not the K897T polymorphism.
Figure 3.
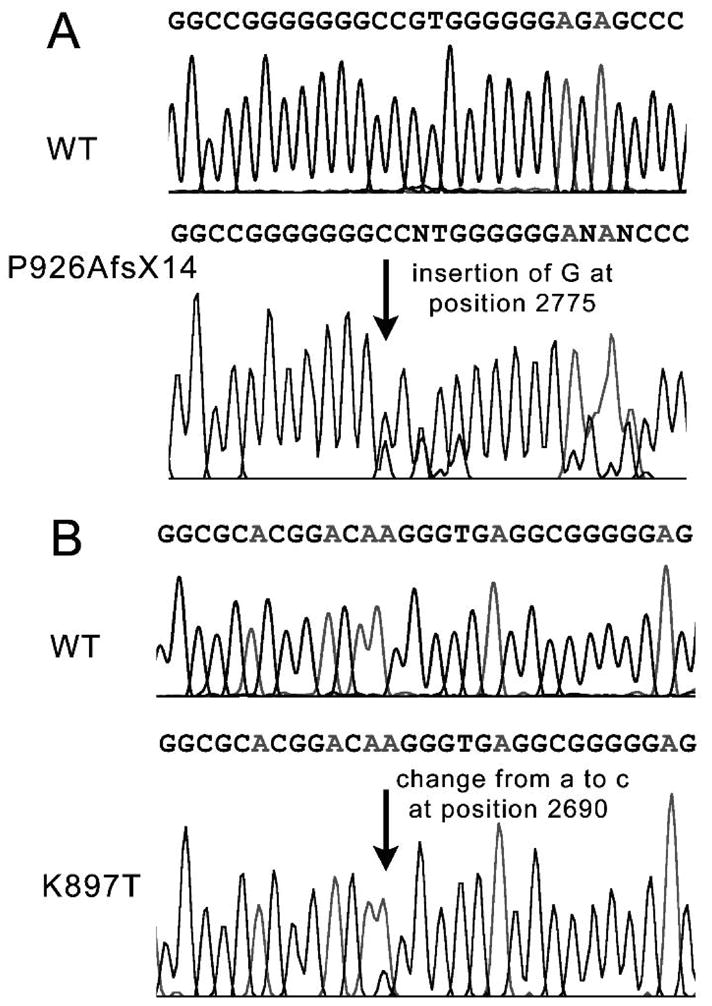
Electropherograms demonstrating. A: Insertion of G at position 2775 of KCNH2 causing a frame shift that leads to a stop codon 14 amino acids downstream (P926AfsX14). B: Heterozygous nucleotide change from A to C at position 2690 of KCNH2 predicts a substitution of lysine with threonine at position 897 of HERG (K897T).
Figure 4.
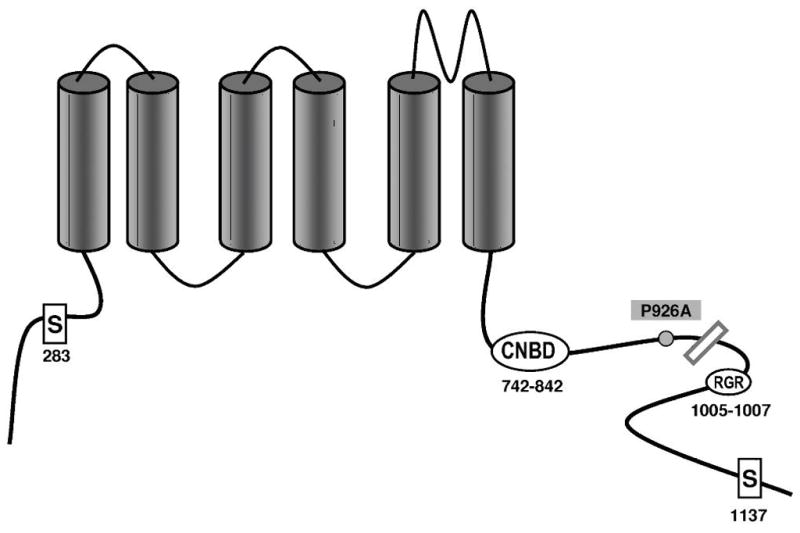
The P926AfsX14 mutation and the K897T variant are located in the C terminus. The site of the deletion is indicated by the bar. RGR is an endoplasmic reticulum (ER) retention signal and 1137S is PKA phosphorylation site. Note that all transmembrane segments and the cyclic nucleotide binding domain (cNBD) are preserved in the truncated channel.
Expression studies
To determine how the mutation P926AfsX14 and K897T altered the biophysical properties of HERG current and contributed to the clinical phenotype, we expressed HERG channels (WT and mutants) in CHO-K1 cells and performed patch clamp experiments. Currents activated rapidly during step depolarizations to positive potentials and displayed the characteristic tail current generated by channels recovering upon repolarization (Figure 5). Homozygous K897T and P926AfsX14 both demonstrated a reduction in tail current density to 59.0±8.5% (n=9) and 60.2±14.4% (n=11) respectively compared to WT (Fig. 5B). The P926AfsX14 mutant yielded current even though a large portion of the C-terminal was truncated. In order to mimic the heterozygote state of family members I-1 and I- 2 and II-4, K897T and P926AfsX14 were co-expressed with WT. Co-expression of either K897T or P926AfsX14 with WT resulted in no significant difference in current density compared to WT alone. In contrast, co-expression of K897T with P926AfsX14 as found in family member II-2, the deceased infant, severely reduced tail current density to 38.7±4.7% of WT (n=10; p<0.05).
Figure 5. Heterologous co-expression of K897T and mutation P926AfsX14 results in a severe loss of function of HERG channel current.
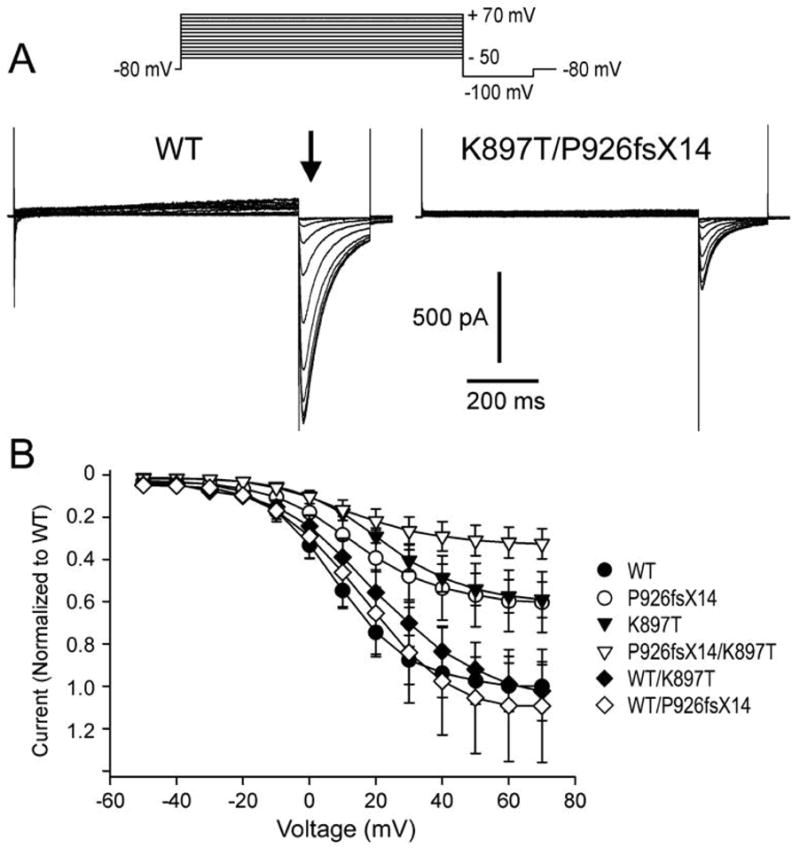
A: Representative current traces from CHO-K1 cells transfected with either WT or K897T/P926AfsX14. HERG currents were elicited using the pulse protocol depicted at the top of the trace. Following an 800 ms depolarizing pulse and upon repolarization to −100 mV, channels recovering from inactivation produced large inward tail currents. Peak tail currents were measured at the time indicated by the arrow. B: Normalized tail current-voltage relationship obtained as in panel A as a function of the step potential. Normalized currents from WT/K897T and WT/P926AfsX14 transfected cells reach a maximum value of 102.4±13.9 % (n=8) and 109.3±26.7% of WT (n=13), respectively. At the same voltage, the current resulting from the heterozygous expression of K897T/P926AfsX14 channels shows a reduction to 38.7±4.7% (n=10) vs WT. Intermediate current values can be observed for the homozygous expression of K897T and P926AfsX14 (59.0±8.5%, n=9; and 60.2±14.4%, n=8, respectively).
Channel availability was assessed using a standard triple-pulse protocol (Fig. 6). Compared to WT, K897T currents showed a small positive shift in the voltage dependence of mid-inactivation values (−60.7±1.15 mV (n=13) and −53.1±1.13mV (n=9) respectively, p<0.05). Co-expression with P926AfsX14 resulted in a tendency toward a leftward shift of mid-inactivation voltage (−63.3±1.13mV (n=12, p=N.S.)), which was most pronounced when P926AfsX14 was co-expressed with K897T, resulting in a mid-inactivation potential of −69.2±1.91mV (n=8, p<0.05).
Figure 6.
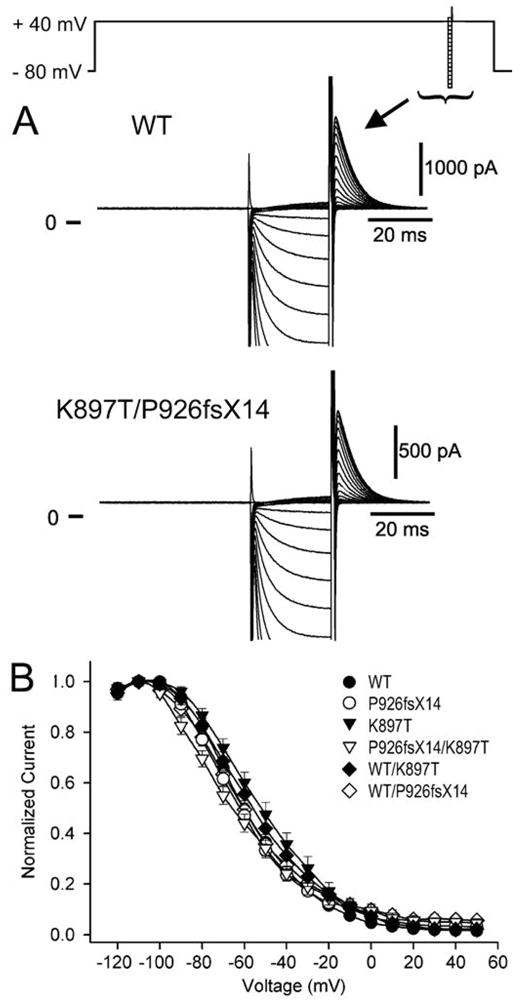
Figure 6A: Representative current recordings from WT and K897T/P926AfsX14 channels recovering using a standard triple pulse protocol (top of figure). During the 25 ms pulse the potentials were varied between −140 and +50 mV. B: Averaged steady-state inactivation curves for homozygous expression of WT, K897T, and P926AfsX14 and, heterozygous expression of K897T/WT, P926AfsX14/WT and K897T/P926AfsX14. A Boltzmann distribution function fitted to the normalized tail current amplitudes yielded mid-inactivation potentials (in mV) of −60.7±1.15, −57.1±1.35, −63.3±1.13, −53.1±1.13, −63.8±1.39, and −69.2±1.91, for WT/WT, WT/K897T, WT/P926AfsX14, K897T/K897T, P926AfsX14/P926AfsX14, and K897T/P926AfsX14,respectively.
Hence, K897T together with P926AfsX14 reduced HERG channel function to a greater degree than homozygous expression of P926AfsX14, both with regard to tail current density and the steady state availability of rapidly activating delayed rectifier potassium current (IKr) channels.
Because the truncated section of the mutant channel contains the C- termini endoplasmic reticulum retention signal (Fig. 4)18 we sought to determine whether the loss of function is due in part to a trafficking defect. We transfected COS cells with all possible combinations: WT, K897T, P926AfsX14, WT/K897T, WT/P926AfsX14 and K897T/P926AfsX14. The cells were probed with rabbit antibodies against HERG. XYZ scans of the cells using confocal microscopy, showed normal distribution of HERG channels with no evidence of abnormal trafficking of the K897T and P926AfsX14 mutated channels (alone or combined) to the cell membrane. Fig. 7 shows normal distribution of HERG resulting from transfecting K897T/P926AfsX14. Similar images were obtained when other combinations of WT, K897T and P926AfsX14 were used for transfection (not shown).
Figure 7.
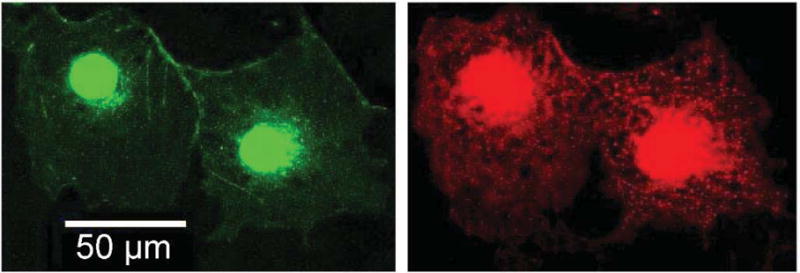
Representative confocal images showing subcellular localization of K897T and P926AfsX14 human Ether-à-go-go Related Gene (HERG) channels in transiently transfected COS-1 cells. Cells were probed with rabbit antibodies directed against HERG. Green fluorescene protein (GFP) identifies cells that were effectively transfected with K897T and P926AfsX14 KCNH2. Red shows positive staining against HERG channel protein revealed with an Alexa 594-conjugated anti-rabbit antibody.
Discussion
The results of our study suggest that a common polymorphism such as K897T-KCNH2 can potently accentuate the effects of a mutation in HERG, thus resulting in a lethal form of long QT syndrome. SNPs have previously been implicated in acquired forms of long QT syndrome leading to the development of drug-induced TdP.19 Westenskow et al.20 and Crotti et al.17 were the first to provide evidence that a common SNP (KCNE1- D85N and HERG- K897T respectively) may modify the disease phenotype of congenital LQTS. The role of K897T in the long QT syndrome, however, is not entirely straightforward. In large scale population studies, the 897T allele has been associated with a shorter QTc interval compared to the 897K allele.24–28 In one study,29 Finnish women with K897T had a longer QTc than those with K897K. In contrast to the population studies, in functional studies performed using cells expressing K897T- HERG channels, most investigators found that K897T alters the channel biophysical properties leading to a reduction in HERG current.17, 30 Our results suggest that the loss of function of K897T is relatively small, consistent with the results of large population studies24–29 and that its manifestation is generally subclinical, as in the case of the father of the family described in this report. CHO-K1 cells expressing K897T HERG channels demonstrated only a mild reduction in tail current compared to WT and the heterozygous expression (WT/K897T) resulted in currents similar to those observed with WT. However when K897T is associated with a KCNH2 mutation, it clearly contributed to a much more severe LQT phenotype. The mother and fourth child (I-2 and II-4 respectively in pedigree; Fig. 1) had a mutation leading to a stop codon in position 940 of the amino acid sequence. They both displayed mild prolonged QT intervals on their ECG but did not have any documented arrhythmias or syncope. Of note, the mother displayed a wider range of QTc intervals most of which were mildly prolonged at baseline, but QTc intervals as long as 542 ms when treated with sotalol in an attempt to suppress the fetal arrhythmias. Her first two children (II-1 and subject II-2 in family pedigree; Fig. 1) inherited the K897T polymorphism from the father and the P926AfsX14 mutation from the mother. The first child had incessant VT and a prolonged QTc on her ECG and eventually died on her second day of life. The second child died in utero in the first trimester. The third child (subject II-3 in family pedigree; Fig. 1) which inherited neither the polymorphism nor the mutation is healthy and has a normal QTc. Consistent with the phenotype, co-expression of K897T and P926AfsX14 produced a major reduction of HERG current (to 38.7±4.7% of WT) whereas expression of homozygous P926AfsX14 produced only a modest reduction in HERG current. Our results suggest that expression of the K897T polymorphism and the stop-codon mutation in KCNH2 on separate alleles leads to a lethal form of LQT2 that can result in in utero VT and sudden infant death.
Our findings are congruent to those of Crotti et al.,17 where K897T combined with a KCNH2 mutation modified the clinical expression of a LQT2 mutation leading to sudden cardiac death. Numerous mechanisms have been proposed to underlie SIDS. Filiano and Kinney31 suggested that SIDS results from the intersection of three overlapping factors. Our study, like that of Crotti et al., suggests the convergence of at least two gentic factors. It is possible that still others such as ischemia, acidosis, or other environmental factors contribute as well.
SNPs have been associated with sudden infant death syndrome (SIDS) in several studies.6–13 Recently, NOS1AP a genetic modifier found to increase the risk of SCD in LQTSC patients 32 was also found to be involved in SIDS.33 However the precise mechanisms by which these SNPs are linked to SIDS are not clear. SNPs have been shown to contribute to drug-induced TdP.19 Genetic variations can create a subclinical form of LQTS, which may manifest only following exposure to drugs with QT prolonging actions. Similarly, SNPs may alter the repolarization reserve of the infants rendering them more vulnerable to LQTS mutations. Triggering event or factors such as sleep (specifically in SIDS cases with SCN5A SNPs),34 sympathetic surge, other “de novo” polymorphisms/mutations6 or drugs used by the mother, may trigger a life threatening arrhythmia. Wang et al.34 demonstrated that certain genetic variants in SCN5A found to be associated with SIDS are not sufficient by themselves to cause pathological manifestations, but may become pathological in vitro when exposed to acidosis.
To our knowledge, this report is the first to demonstrate how a common SNP may contribute to SIDS or lethal infant arrhythmias. Both parents often showed a normal QTc, although careful scrutiny revealed that the QTc of the mother was at times prolonged. Loss of function of HERG channel current was mild when the genetic variants of the individual parents were expressed alone, but resulted in a severe loss of function of HERG current that was associated with as severe clinical phenotype, when the two variants were combined.
It is noteworthy that K897T was reported to be associated with a HERG mutation in a Norwegian SIDS study leading to a 50% reduction in the HERG tail current when co-expressed.35 However, no evidence was presented as to whether the phenotype was the result of the mutation alone or from the combination of K897T and the mutation. Moreover, no evidence was presented showing that K897T accentuates the effect of the mutation to reduce HERG current. Also note is the fact that the allele frequency K897T is similar in SIDS cases and controls.6
Identification of infants at risk for SIDS or for fetal life threatening arrhythmias remains one of the leading challenges of modern medicine. One difficulty is that SIDS is generally not a familial disease. Some of these cases are due to “de novo” mutations7–13, 36. Other possibilities may be a parental mosaicism for LQT associated mutations37 or homozygous missense mutations.38 Our results suggest that even a common SNP may place an infant born to asymptomatic parents at risk, emphasizing the importance of identifying SNPs when screening families for SCD and SIDS.
Another interesting finding is that CHO-K1 cells co- expressing K897T and P926AfsX14 had a significant reduction in HERG activity compared to cells expressing homozygous P926AfsX14. Previous studies18, 38, 39 describing mutations resulting in truncation of the HERG C- terminus also did not find major reductions in HERG function when expressed in heterologous systems. Similar to our results the truncated HERG C- terminus proteins reached the cell surface with no apparent trafficking defect.18, 39 It is noteworthy that the truncation removes the endoplasmic retention signal as well as a motif involved in masking the retention18 (Fig. 4). The truncated HERG proteins in all of these reports as in our report contain all 6 transmembrane segments and the cyclic nucleotide binding domain and therefore it is not surprising that the function of these channels is relatively preserved in expression studies. The reason why some of the patients carrying these mutations have a severe phenotype has long been puzzling. Bhuiyan et al.38 suggested that in their case the mutant HERG was destroyed by the nonsense-mediated decay pathway regulating mRNA. Another possibility suggested by Choe et al.39 is that the truncation of the protein leads to the loss of the protein kinase A (PKA) phosphorylation site (Fig. 4) and therefore cannot respond to beta adrenergic stimulation properly. Our report suggests that in some cases the severity of the clinical phenotype may be due to the influence of a common polymorphism such as K897T. When combined with the HERG mutation, K897T alters the functional properties of the channel to reduce HERG current, since trafficking of K897T/P926AfsX14 was unaffected.
Study limitations
While it is tempting to conjecture that the spontaneous abortion of the second sibling was due to a genetic profile similar to that responsible for the death of the neonate (subject II-1), it is possible that causes other than an arrhythmic event associated with LQT2 were responsible.
Our expression studies were performed without co-transfection of KCNE2 along with KCNH2. The role of this subunit in modifying KCNH2 function continues to be debated. Relevant to this issue is the observation that the results of the functional studies correlated well with the clinical phenotypes.
Our results present further compelling evidence implicating a common polymorphism (K897T) in the accentuation of a loss of function of mildly defective HERG channels leading to long QT syndrome-mediated arrhythmias and sudden infant death and suggest that further investigations into similar relationships are warranted.
Acknowledgments
We are grateful to Judith Hefferon with assistance with preparation of the illustrations.
Funding Sources: Supported by grant HL47678 (CA) from NHLBI, and grants from BIOTRONIK® (EN) and the American Physicians Fellowship for Medicine in Israel (EN) and New York State and Florida Grand Lodges
Footnotes
Conflict of Interest Disclosures: None
Reference List
- 1.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori SG, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 2.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, George AL, Horie M, Andelfinger G, Snow GL, Fu YH, Ackerman MJ, Vincent GM. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation. 2005;111:2720–2726. doi: 10.1161/CIRCULATIONAHA.104.472498. [DOI] [PubMed] [Google Scholar]
- 4.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusie-Luna MT, Makielski JC, Ackerman MJ. SCN4B-encoded sodium channel b4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnestad M, Crotti Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL, Jr, Schwartz PJ. Prevalence of long-QT syndrome genen variants in sudden infant death syndrome. Circulation. 2007;115:361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 7.Ackerman MJ, Siu BL, Sturner WQ, Tester DJ, Valdivia CR, Makielski JC, Towbin JA. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286:2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, Richard T, Berti MR, Bloise R. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med. 2000;343:262–267. doi: 10.1056/NEJM200007273430405. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz PJ, Priori SG, Bloise R, Napolitano C, Ronchetti E, Piccinini A, Goj C, Breithardt G, Schulze-Bahr E, Wedekind H, Nastoli J. Molecular diagnosis in a child with sudden infant death syndrome. Lancet. 2001;358:1342–1343. doi: 10.1016/S0140-6736(01)06450-9. [DOI] [PubMed] [Google Scholar]
- 10.Christiansen M, Tonder N, Larsen LA, Andersen PS, Simonsen H, Oyen N, Kanters JK, Jacobsen JR, Fosdal I, Wettrell G, Kjeldsen K. Mutations in the HERG K(+)-ion channel: A novel link between long QT syndrome and sudden infant death syndrome. Am J Cardiol. 2005;95:433–434. doi: 10.1016/j.amjcard.2004.09.054. [DOI] [PubMed] [Google Scholar]
- 11.Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Ackerman MJ. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–166. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 13.Tester DJ, Dura M, Carturan E, Reiken S, Wronska A, Marks AR, Ackerman MJ. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4:733–739. doi: 10.1016/j.hrthm.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maron BJ, Clark CE, Goldstein RE, Epstein SE. Potential role of interval prolongation in sudden infant death syndrome. Circulation. 1976;54:423–430. doi: 10.1161/01.cir.54.3.423. [DOI] [PubMed] [Google Scholar]
- 15.Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, Dumaine R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–127. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 16.Cordeiro JM, Barajas-Martinez H, Hong K, Burashnikov E, Pfeiffer R, Orsino AM, Wu YS, Hu D, Brugada J, Brugada P, Antzelevitch C, Dumaine R, Brugada R. Compound heterozygous mutations P336L and I1660V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation. 2006;114:2026–2033. doi: 10.1161/CIRCULATIONAHA.106.627489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL, Jr, Schwartz PJ. KCNH2-K897T Is a Genetic Modifier of Latent Congenital Long-QT Syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 18.Kupershmidt S, Yang T, Chanthaphaychith S, Wang Z, Towbin JA, Roden DM. Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J Biol Chem. 2002;277:27442–27448. doi: 10.1074/jbc.M112375200. [DOI] [PubMed] [Google Scholar]
- 19.Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, Hohnloser SH, Shimizu W, Schwartz PJ, Stanton M, Murray KT, Norris K, George AL, Jr, Roden DM. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 20.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–1841. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 21.Cordeiro JM, Spitzer KW, Giles WR, Ershler PE, Cannell MB, Bridge JH. Location of the initiation site of calcium transients and sparks in rabbit heart Purkinje cells. J Physiol. 2001;531:301–314. doi: 10.1111/j.1469-7793.2001.0301i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bazett HC. An analysis of the time-relations of electrocardiograms. J Heart. 1920;7:353–370. [Google Scholar]
- 23.Cordeiro JM, Brugada R, Wu YS, Hong K, Dumaine R. Modulation of IKr inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc Res. 2005;67:498–509. doi: 10.1016/j.cardiores.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Laitinen P, Fodstad H, Piippo K, Swan H, Toivonen L, Viitasalo M, Kaprio J, Kontula K. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Hum Mutat. 2000;15:580–581. doi: 10.1002/1098-1004(200006)15:6<580::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Pfeufer A, Jalilzadeh S, Perz S, Mueller JC, Hinterseer M, Illig T, Akyol M, Huth C, Schopfer-Wendels A, Kuch B, Steinbeck G, Holle R, Nabauer M, Wichmann HE, Meitinger T, Kaab S. Common variants in myocardial ion channel genes modify the QT interval in the general population: results from the KORA study. Circ Res. 2005;96:693–701. doi: 10.1161/01.RES.0000161077.53751.e6. [DOI] [PubMed] [Google Scholar]
- 26.Gouas L, Nicaud V, Berthet M, Forhan A, Tiret L, Balkau B, Guicheney P. Association of KCNQ1, KCNE1, KCNH2 and SCN5A polymorphisms with QTc interval length in a healthy population. Eur J Hum Genet. 2005;13:1213–1222. doi: 10.1038/sj.ejhg.5201489. [DOI] [PubMed] [Google Scholar]
- 27.Bezzina CR, Verkerk AO, Busjahn A, Jeron A, Erdmann J, Koopmann TT, Bhuiyan ZA, Wilders R, Mannens MM, Tan HL, Luft FC, Schunkert H, Wilde AA. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res. 2003;59:27–36. doi: 10.1016/s0008-6363(03)00342-0. [DOI] [PubMed] [Google Scholar]
- 28.Newton-Cheh C, Guo CY, Larson MG, Musone SL, Surti A, Camargo AL, Drake JA, Benjamin EJ, Levy D, D’Agostino RB, Sr, Hirschhorn JN, O’Donnell CJ. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation. 2007;116:1128–1136. doi: 10.1161/CIRCULATIONAHA.107.710780. [DOI] [PubMed] [Google Scholar]
- 29.Pietila E, Fodstad H, Niskasaari E, Laitinen PP, Swan H, Savolainen Kesaniemi YA, Kontula K, Huikuri HV. Association between HERG K897T polymorphism and QT interval in middle-aged Finnish women. J Am Coll Cardiol. 2002;40:511–514. doi: 10.1016/s0735-1097(02)01979-4. [DOI] [PubMed] [Google Scholar]
- 30.Anson BD, Ackerman MJ, Tester DJ, Will ML, Delisle BP, Anderson CL, January CT. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. 2004;286:H2434–H2441. doi: 10.1152/ajpheart.00891.2003. [DOI] [PubMed] [Google Scholar]
- 31.Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate. 1994;65:194–197. doi: 10.1159/000244052. [DOI] [PubMed] [Google Scholar]
- 32.Crotti L, Monti MC, Insolia R, Peljto AA, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL., Jr NOS1AP Is a genetic modifier of the long QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osawa M, Kimura R, Hasegawa I, Mukasa N, Satoh F. SNP association and sequence analysis of the NOS1AP gene in SIDS. Leg Med (Tokyo) 2009;11 (Suppl 1):S307–S308. doi: 10.1016/j.legalmed.2009.01.065. [DOI] [PubMed] [Google Scholar]
- 34.Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Rognum T, Schwartz PJ, George AL., Jr Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation. 2007;115:368–376. doi: 10.1161/CIRCULATIONAHA.106.646513. [DOI] [PubMed] [Google Scholar]
- 35.Rhodes TE, Abraham RL, Welch RC, Vanoye CG, Crotti L, Arnestad M, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Rognum T, Roden DM, Schwartz PJ, George AL., Jr Cardiac potassium channel dysfunction in sudden infant death syndrome. J Mol Cell Cardiol. 2008;44:571–581. doi: 10.1016/j.yjmcc.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, Borggrefe M, Brinkmann B, Warnecke I, Funke H, Bhuiyan ZA, Wilde AA, Breithardt G, Haverkamp W. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. 2001;104:1158–1164. doi: 10.1161/hc3501.095361. [DOI] [PubMed] [Google Scholar]
- 37.Miller TE, Estrella E, Myerburg RJ, De Viera JG, Moreno N, Rusconi P, Ahearn ME, Baumbach L, Kurlansky P, Wolff G, Bishopric NH. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation. 2004;109:3029–3034. doi: 10.1161/01.CIR.0000130666.81539.9E. [DOI] [PubMed] [Google Scholar]
- 38.Bhuiyan ZA, Momenah TS, Gong Q, Amin AS, Ghamdi SA, Carvalho JS, Homfray T, Mannens MM, Zhou Z, Wilde AA. Recurrent intrauterine fetal loss due to near absence of HERG: clinical and functional characterization of a homozygous nonsense HERG Q1070X mutation. Heart Rhythm. 2008;5:553–561. doi: 10.1016/j.hrthm.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choe CU, Schulze-Bahr E, Neu A, Xu J, Zhu ZI, Sauter K, Bahring R, Priori S, Guicheney P, Monnig G, Neapolitano C, Heidemann J, Clancy CE, Pongs O, Isbrandt D. C-terminal HERG (LQT2) mutations disrupt IKr channel regulation through 14-3-3epsilon. Hum Mol Genet. 2006;15:2888–2902. doi: 10.1093/hmg/ddl230. [DOI] [PubMed] [Google Scholar]