

Abstract
(R)- and (S)-2-hydroxypropyl-CoM (R-HPC and S-HPC) are produced as intermediates in bacterial propylene metabolism from the nucleophilic addition of coenzyme M to (R)- and (S)-epoxypropane, respectively. Two highly enantioselective dehydrogenases (R-HPCDH and S-HPCDH) belonging to the short-chain dehydrogenase/reductase family catalyze the conversion of R-HPC and S-HPC to 2-ketopropyl-CoM (2-KPC), which undergoes reductive cleavage and carboxylation to produce acetoacetate. In the present study, one of three copies of S-HPCDH enzymes present on a linear megaplasmid in Xanthobacter autotrophicus strain Py2 has been cloned and overexpressed, allowing the first detailed side by side characterization of the R-HPCDH and S-HPCDH enzymes. The catalytic triad of S-HPCDH was found to consist of Y156, K160, and S143. R211 and K214 were identified as the amino acid residues coordinating the sulfonate of CoM in S-HPC. R211A and K214A mutants were severely impaired in the oxidation of R-HPC or reduction of 2-KPC but were largely unaffected in the oxidation and reduction of aliphatic alcohols and ketones. Kinetic analyses using (R)- and (S)-HPC as substrates revealed that enantioselectivity in R-HPCDH (value, 944) was dictated largely by differences in kcat while enantioselectivity for S-HPCDH (value, 658) was dictated largely by changes in Km. S-HPCDH had an inherent high enantioselectivity for producing (S)-2-butanol from 2-butanone that was unaffected by modulators that interact with the sulfonate binding site. The tertiary alcohol 2-methyl-2-hydroxypropyl-CoM (M-HPC) was a competitive inhibitor of R-HPCDH-catalyzed R-HPC oxidation, with a Kis similar to the Km for R-HPC, but was not an inhibitor of S-HPCDH. The primary alcohol 2-hydroxyethyl-CoM was a substrate for both R-HPCDH and S-HPCDH with identical Km values. The pH dependence of kinetic parameters suggests that the hydroxyl group is a larger contributor to S-HPC binding to S-HPCDH than for R-HPC binding to R-HPCDH. It is proposed that active site constraints within the S-HPCDH prevent proper binding of R-HPC and M-HPC due to steric clashes with the improperly aligned methyl group on the C2 carbon, resulting in a different mechanism for controlling substrate specificity and enantioselectivity than present in the R-HPCDH.
Alcohol dehydrogenases that catalyze the interconversion of secondary alcohols and ketones are often highly stereoselective with regard to the enantiomer of the alcohol oxidized or produced during the course of catalysis (1-3). However, there are very few examples of pairs of alcohol dehydrogenases synthesized simultaneously to deal with both the (R)- and (S)-enantiomers of an alcohol produced during a metabolic pathway. One such unique pair was discovered in the pathway of propylene oxidation by the hydrocarbon-oxidizing bacteria Xanthobacter autotrophicus Py2 and Rhodococcus rhodochrous B276 (4-8). As shown in Figure 1, these bacteria initiate propylene oxidation by epoxidation to produce a mixture of the (R)- and (S)-enantiomers of epoxypropane, which are further metabolized by a coenzyme M (2-mercaptoethanesulfonate, CoM1)-mediated sequence of reactions involving nucleophilic addition, dehydrogenation, reductive cleavage, and carboxylation. (R)- and (S)-2-hydroxypropyl-CoM (R- and S-HPC) are the substrates for the unique pair of dehydrogenases that form 2-ketopropyl-CoM (2-KPC), which is reductive cleaved and carboxylated (4).
Figure 1.
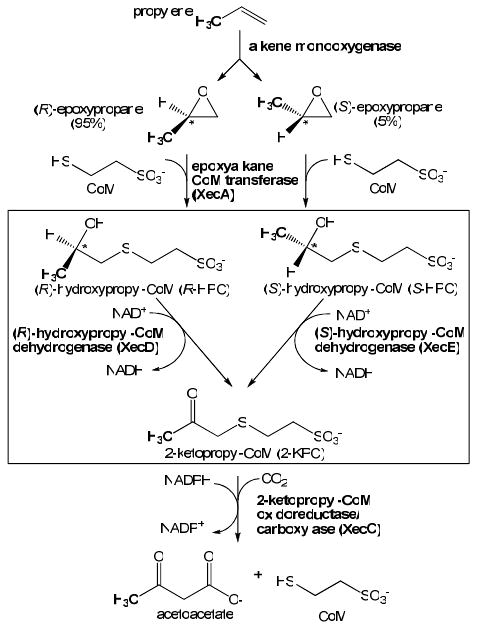
Pathway of propylene oxidation in X. autotrophicus Py2. The reactions catalyzed by R-HPCDH and S-HPCDH are shown in the boxed region.
Biochemical and molecular characterization of the R- and S-HPCDH enzymes showed that they are members of the “classical” short-chain dehydrogenase/reductase (SDR) family of alcohol dehydrogenases (4-6). Classical SDR enzymes are defined by a conserved NAD+-binding motif in the N-terminal portion of the protein and a catalytic triad (or tetrad) within the central portion (9-13). The C-terminal domains of the SDR families diverge to confer specificity for different substrates. The R- and S-HPCDH enzymes from X. autotrophicus are homologous enzymes that share high sequence identity but with notable sequence differences within their C-terminal regions (5-8). These enzymes are highly specific for their respective enantiomers of HPC, exhibiting less that 0.5% activity with the opposite isomer (4).
The R-HPCDH from X. autotrophicus has been extensively characterized mechanistically and structurally, culminating in the formulation of a mechanism of catalysis and stereoselectivity shown on the left side of Scheme 1 (14-16). Stereoselectivity is conferred in large part by two positively charged arginine residues that form salt bridges with the CoM moiety of the substrate within the CoM-binding pocket of the enzyme (14, 16). This strong ionic interaction orients the hydroxyl group and hydrogen atom of the alcohol functional group in the proper orientation for general base abstraction and hydride transfer (Scheme 1) (16). By analogy, a similar mechanistic strategy is believed to operate in the S-HPCDH, wherein the spatial orientations of the CoM- and methyl-binding pockets are reversed relative to the hydroxyl group and hydrogen atom to be transferred (Scheme 1, right side) (14, 16, 17). To date, this model has not been tested for the S-HPCDH due to difficulties encountered in attempting to express the enzyme in an active form in Eschericia coli (14, 15).
Scheme 1.
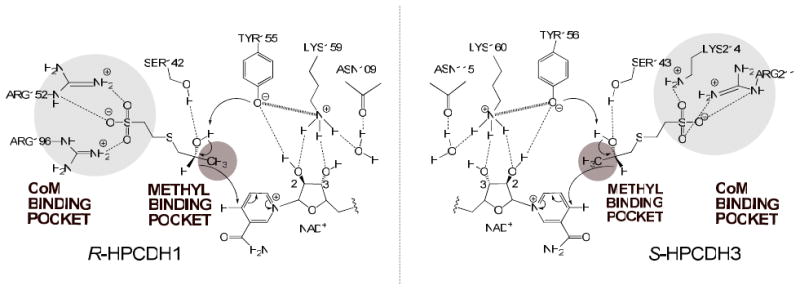
In the present work, one of three copies of S-HPCDH homologs present in X. autotrophicus has been successfully cloned and over-expressed, providing the necessary system for testing the tenets of Scheme 1. These results show that the R- and S-HPCDH enzymes share some common mechanistic features, but differ significantly in the strategies for controlling enantioselectivity for the S- and R-HPC natural substrates, as well as nonphysiological aliphatic alcohols and ketones.
Experimental Procedures
Materials
All commercially available chemicals were purchased from Sigma-Aldrich Chemicals, Acros Organics or Fisher Scientific, and were of analytical grade. 2-KPC, R-HPC S-HPC, HEC and M-HPC were synthesized as described previously (15). Chemical structures of the compounds were confirmed using 1H NMR. The spectra of HPC enantiomers and 2-KPC were identical to those reported previously (4, 15). Purity of the synthesized chemicals as determined by reverse-phase HPLC was estimated to be ≥98%.
Cloning of the S-HPCDH Genes (xecE1, xecE2, and xecE3)
Total genomic DNA was isolated from propylene-grown cells of X. autotrophicus strain Py2 using the Epicentre MasterPure DNA purification kit. Each of the three xecE genes was PCR amplified using the FailSafe PCR PreMix Selection Kit (Epicentre). Primers were designed such that the forward and reverse primers contained BamHI and SacI restriction site overhangs, respectively. The primers were as follows: xecE1: forward, GCAGGATCCAATGCTGGACGCAGAGG; reverse, CGTGAGCTCTCATATGGCGGTCATCC; xecE2: forward, TAGGATCCAGTGGCGCGCGCCGCGGT; reverse, ATGAGCTCTCATATGGCGGTCATCCCTC; xecE3: forward, GCAGGATCCAATGTCGAATCGCTTGAAG; reverse, GCAGAGCTCTCATATCGCCGTCATC. The reactions contained 200 ng of X. autotrophicus genomic DNA, 1.0 μM primers, FailSafe PCR Enzyme Mix (1.25 Units), and the manufacturers buffer H. PCR was performed using the following cycling parameters: stage 1, (95 °C × 5 min) × 1; stage 2, (94 °C × 1min, 60 °C × 1 min, 72 °C × 50 s) × 35; stage 3, (72 °C × 10 min) × 1; The PCR products were analyzed on 1% (w/v) agarose gels, and xecE1, xecE2 and xecE3 DNA (0.75 kb, 0.69 kb, and 0.77 kb respectively) were purified using the QIAquick gel extraction kit (Qiagen). Purified DNA along with pET28-b (Novagen) and pRSFDuet-1 expression vectors were simultaneously subjected to a double digest with BamHI and SacI, resolved on 1% agarose gel and purified with the QIAquick gel extraction kit. Digested xecE genes and pET28-b DNA were mixed and ligated with T4 DNA ligase for 2h at 25°C. The resulting plasmids were designated: pDS41, pDS42 and pDS43, and carried the xecE1, xecE2 and xecE3 genes, respectively. Likewise, digested xecE genes and pRSFDuet-1 DNA were mixed and ligated with T4 DNA ligase for 2h at 25°C, to result in pDS51, pDS52 and pDS53 plasmids with xecE1, xecE2 and xecE3 genes, respectively. Subsequent to DNA sequencing, all constructs were transformed into E.coli DH5α for plasmid maintenance and into E. coli BL21 - (DE3) CodonPlus (Stratagene) cells for protein expression.
Site-Directed Mutagenesis (SDM)
All primers used to introduce point mutations were purchased from IDT. SDM of pDS53 was carried out utilizing the QuickChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer's protocols. All mutations were confirmed by DNA sequencing. The sequences of the mutagenic primer pairs used for each codon substitution in xecE3 are as follows: S143A, GATCGTCAATTTTGGCGCCGTCGCTGGCCTC and GAGGCCAGCGACGGCGCCAAAATTGACGATC; Y156A, GACCATGGCGGCCGCCTGCGCAGCCAAG and CTTGGCTGCGCAGGCGGCCGCCATGGTC; Y156F, CCATGGCGGCCTTCTGCGCAGCCAAGG and CCTTGGCTGCGCAGAAGGCCGCCATGG; K160A, CCTACTGCGCAGCCGCGGGCGCAATCGTCA and TGACGATTGCGCCCGCGGCTGCGCAGTAGG; R211A, GAAGTTCAGGCTCGCGCGCTGGCGAAATATCC and GGATATTTCGCCAGCGCGCGAGCCTGAACTTC; K214A, CTCGCCGGCTGGCGGCATATCCGATCGGGC and GCCCGATCGGATATGCCGCCAGCCGGCGAG.
DNA Sequencing
Sequencing was performed on an AB 3730 DNA Analyzer at the Utah State University Biotechnology Center DNA sequencing laboratory. The following sequencing primers were used to confirm all mutations to pDS53; ACYCDuetUP1 Primer (Novagen), GGATCTCGACGCTCTCCCT and DuetDOWN1 Primer (Novagen), GATTATGCGGCCGTGTACAA.
Media and Growth of Bacteria
X. autotrophicus was grown on propylene (10% (v/v) gas phase) in a 15L semicontinuous microferm fermenter as described previously (18). E.coli DH5α was grown in standard Luria-Bertani (LB) broth supplemented with kanamycin (50 μg·mL-1). E. coli BL21- (DE3) CodonPlus was grown in LB media that contained both kanamycin (50 μg· mL-1) and chloramphenicol (50 μg·mL-1). All other procedures were performed as described previously (15).
Preparation of Cell-Free Extracts
About 60 grams of frozen cell paste was thawed at room temperature in 2 volumes of lysis buffer (20 mM Tris chloride, 20% glycerol, 500 mM NaCl, 25 mM imidazole, 0.1% (w/v) Tween20, 0.03 mg·mL-1 DNase I, at pH 8.0). Homogenized cell suspension was passed twice through a French press at 15000 psi and the crude lysate was clarified by centrifugation at 244,717g for 45 min.
Purification of S-HPCDH1, rS-HPCDH1, rS-HPCDH3 and rS-HPCDH3 Mutants
S-HPCDH was purified from propylene-grown X. autotrophicus as described previously (5). For the purification of rS-HPCDH1, rS-HPCDH3 and rS-HPCDH3 mutants, clarified cell extract was applied to a 2.6 × 4.7 cm column of Ni Sepharose 6 Fast Flow (Amersham) at 3.0 mL·min-1 (33.9 cm·h-1). The column was rinsed at 6.0 mL·min-1 (67.8 cm·h-1) with 2 column volumes of lysis buffer, followed by 80 column volumes of rinse buffer (20 mM Tris, 20% glycerol, 500 mM NaCl, 50 mM imidazole, 0.1% (w/v) Tween20, pH 8.0). rS-HPCDH was eluted at 4.0 mL·min-1 (45.2 cm·h-1) with a linear gradient of 19.2 column volumes of 0-100% elution buffer (20 mM Tris, 20% glycerol, 500 mM NaCl, 500 mM imidazole, pH 8.0). Fractions containing rS-HPCDH were concentrated over a 30000 MW cutoff membrane, diluted in a 1:20 ratio with dialysis buffer (20 mM Tris, 10% glycerol, 100 mM NaCl, pH 8.2) and concentrated again (this procedure was repeated twice). Dialyzed rS-HPCDH was flash frozen and stored in liquid nitrogen. All steps were performed at 4 °C, and all buffers used had the pH adjusted at 4 °C. Protein concentrations were determined on a NanoDrop spectrophotometer using theoretical extinction coefficients (∈280 = 10033 M-1 cm-1 for S-HPCDH3, ∈280 = 15595 M-1 cm-1 for S-HPCDH1, and ∈280 = 18512.5 M-1 cm-1 for R-HPCDH1), with dialysis flow-through buffers as blanks.
SDS-PAGE and Native PAGE Analysis
SDS-PAGE (12% T) and native PAGE (4-20% T BioRad) were performed according to the Laemmli procedure (19). The apparent molecular masses of polypeptides on a SDS-PAGE gel were determined by comparison to Rf values of standard proteins. Migration of rS-HPCDH3 mutants was also compared directly to wild-type rS-HPCDH3, wild-type rR-HPCDH1, native S-HPCDH and R-HPCDH (isolated from X. autotrophicus). The apparent molecular masses of polypeptides on a native PAGE gel were determined from a standard curve constructed by plotting Rf values against the log of the native molecular mass for the following standards: β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa) and ovalbumin (43 kDa).
Gel Filtration Chromatography
The native molecular masses of rS-HPCDH3 and rS-HPCDH3 mutants were estimated by gel filtration chromatography using HPLC (Shimadzu SLC-10A) with a fluorescence detector (Shimadzu RF-10AXL) set up for excitation and emission wavelengths at 280nm and 350nm, respectively. The gel filtration column (BioSep-SEC S-2000, 300 × 7.8 mm, Phenomenex) was equilibrated in 50 mM Na2HPO4, 200 mM NaCl, pH 7.5, containing 10% glycerol and 1 mM NAD+. The following molecular mass standards were used for calibration: β-amylase (200 kDa), aldolase (158 kDa), conalbumin (75 kDa), bovine serum albumin (66 kDa), ovalbumin (43 kDa), carbonic anhydrase (29 kDa), and myoglobin (18.8 kDa). All separations were performed at 4°C, at a flow rate of 0.5 mL min-1. A calibration curve was constructed by plotting the retention time against the log of the native molecular mass for standards. This plot was fit using a second order polynomial, and the equation of this line was used to determine the log native molecular masses for rS-HPCDH3 and rS-HPCDH3 mutants using their experimentally determined retention times.
Circular Dichroism (CD)
CD spectra were recorded at 25 °C on an AVIV Model 410 CD Spectrophotometer, using 1 nm spectral bandwidth and a 0.1 cm path length. Stock peptide solutions were desalted on Sephadex G-25 (PD-10) columns and diluted in 10× buffer to a final concentration of 10% glycerol, 10 mM KH2PO4 and 100 mM KF at pH 7.0. Total enzyme concentration was approximately 0.3 mg·mL-1 (10 μM). Typically, five scans were acquired over the wavelength range 190-260 nm. The residue ellipticity (θ) was calculated using rS-HPCDH3 molecular weight of 27085.5 Da and NA (number of amino acid in the protein) of 268 residues.
Spectrophotometric Enzyme Assays
Assays with 2-KPC, S-HPC, R-HPC and 2-propanol as substrates were performed in 50 mM glycine, 50 mM NaH2PO4, and 50 mM Tris base (GPT buffer mix) at a pH of 7.5 (adjusted at 30 °C), as described previously (15). Assays with all other substrates (C4 to C6 in carbon chain length) were carried out in 50 mM GPT buffer containing 15% (v/v) glycerol. Stock solutions of synthesized substrates were standardized, as described previously (15) All enzyme assays were performed in triplicates at 30 °C in a Shimadzu model UV160U spectrophotometer containing a water-jacketed cell holder for thermal control. Alcohol or ketone production was monitored by measuring the change in absorbance at 340 nm using the extinction coefficient for NADH (∈340 = 6.22 mM-1cm-1). For alcohol oxidation assays, the following ranges of alcohol concentrations were used in determining kinetic constants for R-HPCDH1: R-HPC, 0.020 – 1.0 mM; S-HPC, 0.074-11 mM; 2-propanol, 65-3900 mM; (S)-2-butanol, 10.9-381 mM; (R)-2-butanol, 43.6-490 mM. For S-HPCDH3 the concentrations were: R-HPC, 0.038-48 mM; S-HPC, 0.016 – 0.310 mM; 2-propanol, (S)-2-butanol and (R)-2-butanol, the same as for R-HPCDH1. The concentration of NAD+ for all assays was 10 mM (26 × value of KmNAD+). For assays of ketone reduction, the following concentration ranges were used in determining kinetic constants: 2-KPC, 0.050-2.6 mM; 2-butanone, 10-300 mM. The concentration of NADH for these assays was 0.16 mM, (4.4 × value of KmNADH). On average, seven concentrations of substrates within the ranges indicated were chosen for the kinetic analyses. All samples were degassed/flushed with nitrogen and incubated at 30 °C water-bath for 5 min prior to the enzyme addition. To determine kinetic parameters (apparent Km and Vmax) initial rate values were plotted as a function of substrate concentration and data points were fitted to the Michaelis-Menten equation using SigmaPlot 11.0.
pH Dependence of Kinetic Parameters
All assays were performed in GPT buffer mix or APT buffer mix (50 mM CH3COONa, 50 mM NaH2PO4, and 50 mM Tris base) at ten different pH values (5.0 – 10.0) adjusted to the desired pH at 30 °C, as described previously (15). The assay results were used to construct a plot of kcat/Km vs pH, which was then fit to Equation 1:
| Equation (1) |
where C is the maximal log kcat/Km value and Ka is the acid dissociation constant of the catalytically important ionizing residue. Plots of log kcat and log Km vs pH were constructed in a point-to-point manner. Stability of the enzyme at a pH 5.0 and 10.0 was tested according to the pH-jump method (20). The stock solution of rS-HPCDH3 was diluted to 0.1 mg·mL-1 in 10 mM APT or GPT buffer mix containing 15% glycerol, at pH values of 5.0 and 10.0, respectively. The enzyme was equilibrated on ice at the desired pH (adjusted at 4 °C) for 5 min prior to its addition to the reaction mixture (total of 0.3 μg of rS-HPCDH3). Activity assays were performed for 30 sec in 100 mM GPT buffer mix, pH 7.5, at 30 °C.
Inhibition Studies
All assays were performed in GPT buffer mix at pH 7.5 with saturating concentration of NAD+ (5 mM). Inhibition assays for R-HPCDH1 were performed at variable concentrations of R-HPC (31, 72, 144, 287 and 615 μM) or HEC (0.8 mM, 2.0 mM, 4.0 mM, 8.0 mM, 1.6 mM). Each assay was performed at several fixed concentrations of M-HPC: (0 mM, 0.2 mM, 0.4 mM, 0.8 mM, and 1.6 mM). Initial rate data for rR-HPCDH1 were fit to Equation 2 describing competitive inhibition of enzymatic activity:
| Equation (2) |
where S is the substrate, I is the inhibitor, α = 1 + ([I]/Kis) and Kis is the inhibition constant for the competitive component of enzyme inhibition. The type of inhibition exhibited by M-HPC was determined by graphing initial rate data in the form of double reciprocal plots (1/υ vs 1/[S] at various [I]). Numerical value of Kis was calculated from equation 2 using SIGMA-PLOT. Inhibition assays for S-HPCDH3 were performed at varying concentrations of S-HPC (16, 31, 62, 155 and 310 μM) or HEC (0.4, 0.8, 2.0, 4.0, 8.0, and 1.6 mM) and three fixed concentrations of M-HPC: (0 mM, 1.2 mM, and 4.9 mM). Initial rate data were fit to a rectangular hyperbola described by the standard form of the Michaelis-Menten equation.
Chiral Gas Chromatographic Assay for 2-Butanone Reduction
All assays were performed in GPT buffer mix, pH 7.5 (adjusted at 30 °C), containing 15% (v/v) glycerol. Assay components in 1 mL reaction volume were: NADH (15 mM), 2-butanone (56 mM), and 0.64 mg of enzyme. Serum vials (3 mL) were crimp-sealed and degassed/flushed with nitrogen three times and allowed to equilibrate for 20 min at 30 °C in a shaking water-bath (200 rpm). The assay was initiated by adding enzyme. After 1 h incubation 250 μL of headspace gas was removed and injected into a Shimadzu GC-8A gas chromatograph outfitted with a Supelco β-Dex 225 (30 m × 0.53 mm) column. GC parameters used were as previously described (14).
Results and Discussion
Genetic and bioinformatic analysis of the enzymes of epoxide metabolism
A previous study demonstrated that the genes of alkene and epoxide metabolism are present on a 320 kb linear megaplasmid in X. autotrophicus Py2 (21). Furthermore, the genes encoding the enzymes of epoxide carboxylation were found to be clustered in an operon where xecA, xecC, xecD, and xecE encode the key enzymes shown in Figure 1 (5, 22). The xecD gene (encoding R-HPCDH) was successfully cloned, over-expressed and purified in a fully active state, allowing for detailed mechanistic and structural studies to be performed (14-16). Unfortunately, all attempts to clone and express the counterpart S-HPCDH in a soluble and active form were unsuccessful despite numerous attempts (15), and the mechanistic and structural characterization of this enzyme has thus lagged behind that of the R-HPCDH.
Recently, the genome of X. autotrophicus Py2 was sequenced, assembled, and annotated (http://genome.jgi-psf.org/xanau/xanau.home.html). Unexpectedly, an analysis of the linear megaplasmid of X. autotrophicus shows the presence of multiple copies of the xec genes, as well as for the putative enzymes of coenzyme M biosynthesis. The xec paralogs are distributed among three apparent operons and share high identities. The xecD and xecE genes whose protein sequences were reported previously (5, 22) are those located in an operon approximately 10.4 kbp downstream of the xamo operon, while the additional copies are located about 197 and 223 kbp downstream of this first operon. For naming purposes, these copies are now referred to as xecD1 and xecE1, to designate they are found in the first gene cluster, while the additional copies are referred to as xecD2, xecE2, xecD3 and xecE3 based on which of the additional two clusters they are found within. Since the abbreviations R- and S-HPCDH have been used previously, these proteins will also be affixed with the numbers 1, 2, and 3 for naming purposes (i.e. XecD1 is R-HPCDH1 while XecD2 is R-HPCDH2 and so on).
Cloning, expression, and biochemical characterization of xecE homologs
Each of the three S-HPCDH homologs identified on the linear megaplasmid was expressed using an optimized expression system with the inclusion of N-terminal six-histidine tags. Only small amounts (<0.1 mg/g cell paste) of the XecE1 (S-HPCDH1) protein were present in the soluble fraction, with most of the protein being in inclusion bodies. The small amount of soluble protein, when purified, had a specific activity nearly identical to the native enzyme, demonstrating that the enzyme can be expressed in an active form, albeit at levels too low for detailed biochemical and structural characterization. The second copy, XecE2 (S-HPCDH2), contained apparent mutations in the N-terminal region, most notably the lack of the classic GXXXGXG cofactor sequence motif that is found within the Rossman fold (10). This protein was expressed with more protein in the soluble fraction (∼50%), but was completely inactive, as expected due to the lack of key NAD+-binding residues. The third copy, XecE3 (S-HPCDH3), was very similar to xecE1, with 74% identity, and with all of the conserved SDR sequences intact (Figure 2). XecE3 was expressed in high yields in a soluble form (>4 mg enzyme/g cell paste), and when purified, had a specific activity nearly identical to that of S-HPCDH1 and S-HPCDH purified from X. autotrophicus.
Figure 2.

Multiple-sequence alignment of S-HPCDH1, S-HPCDH3 and R-HPCDH1 enzymes from X. autotrophicus Py2. Abbreviations: Cons.S1&3, consensus amino acid alignment for S-HPCDH1 and S-HPCDH3; Cons.all, consensus of S-HPCDH1, S-HPCDH3 and R-HPCDH1 Letter designations: a, Classic GXXXGXG glycine-rich NAD+ binding motif. b, Catalytic tetrad residues of Asn, Ser, Tyr and Lys. c, Positively charged residues that have been shown (R-HPCDH1) or are proposed (S-HPCDH) to interact with the sulfonate group of CoM in the substrate. The alignment was generated using MULTALIN with default parameters, while the consensus was derived using ClustalW2. The following symbols mean that the residues are: (*) identical; (:) conserved; (.) semi-conserved.
SDS- and native PAGE, gel filtration chromatography, and CD spectrapolarimetric analyses were performed with wild-type rS-HPCDH1-3 and the rS-HPCDH3 mutants described subsequently (S143A, Y156A, Y156F, K160A, R211A, K214A). All of the proteins behaved with properties similar to that of S-HPCDH purified from the native bacterium, indicating that the oligomerization state is unaffected for the recombinant enzymes. CD analysis of the rS-HPCDH3 mutants generated spectra that are indistinguishable from that of the wild-type enzyme suggesting that reduced activities in mutants are not due to major structural changes, but a result of a change in the chemical environment surrounding substituted residues.
Kinetic parameters for rS-HPCDH1, rS-HPCDH3, and rR-HPCDH with physiological substrates
In the present work, the kinetic analysis of rR-HPCDH1 performed previously (14) was repeated in side by side experiments with the two active XecE homologs so that the three enzymes can be compared directly. Although expressed only at very low levels, rS-HPCDH1 was included in these analyses in order to see if the two XecE homologs had any significant differences in catalytic properties. These results are presented in Table 1.
Table 1.
Kinetic Parameters for R- and S-HPCDH with physiological substrates in the forward and reverse directionsa
| rR-HPCDH1 | rS-HPCDH1 | rS-HPCDH3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Substrate | Km (μM) | kcat (s-1) | kcat/Km (M-1 s-1) | Km (μM) | kcat (s-1) | kcat/Km (M-1 s-1) | Km (μM) | kcat (s-1) | kcat/Km (M-1 s-1) |
| R-HPC | 96 ± 7 | 48 ± 1 | 5.0 × 105 | 5200 ± 300 | 2.4 ± 0.06 | 4.6 × 102 | 9100 ± 1100 | 5.5 ± 0.2 | 1.2 × 103 |
| S-HPC | 220 ± 70 | 0.12 ± 0.01 | 5.3 × 102 | 220 ± 20 | 28 ± 0.8 | 1.3 × 105 | 31 ± 0.9 | 25 ± 0.2 | 7.9 × 105 |
| 2-KPC | 68 ± 10 | 29 ± 2 | 4.2 × 105 | 240 ± 10 | 20 ± 0.2 | 8.3 × 104 | 270 ± 13 | 11 ± 0.2 | 3.9 × 104 |
Assay for R-HPC and S-HPC oxidation by rR-HPCDH1 contained 1.0 μg and 46 μg of enzyme, respectively. Assay for R-HPC and S-HPC oxidation by rS-HPCDH1 and rS-HPCDH3 contained 5.0 μg and 1.0 μg of enzyme, respectively. Assay for 2-KPC reduction contained 1 μg of enzyme. Apparent kcat and Km values are reported as means ± standard deviations. All assays were performed in triplicates at 30 °C with fixed concentrations of NAD+ (10 mM) or NADH (0.17 mM). Apparent kinetic constants were determined by fitting experimental data to the standard form of the Michaelis-Menten equation.
The apparent kcat and Km values determined for R-HPC and S-HPC oxidation, and 2-KPC reduction, by rR-HPCDH1 are similar to those reported previously (14). With regard to rS-HPCDH1 and rS-HPCDH3, the apparent kcat values for S-HPC oxidation are nearly identical to each other, while the apparent Km is seven-fold lower for rS-HPCDH3. For 2-KPC reduction, rS-HPCDH1 exhibits a two-fold higher kcat but nearly identical Km relative to rS-HPCDH3. As noted above, S-HPCDH1 and S-HPCDH3 share high identity (74%) but are not identical enzymes, so it is not surprising that some differences in kinetic parameters are observed.
The most surprising results of the kinetic analyses are the large relative differences in kcat and Km for the opposite enantiomers when characterized as substrates for the R- and S-dehydrogenases. As shown in Table 1, rR-HPCDH1 catalyzed the oxidation of S-HPC with a kcat that is 402-times less than that for R-HPC. In contrast, rS-HPCDH1 and rS-HPCDH3 catalyzed the oxidation of R-HPC with kcat values only 11 and 4.5 times less than that for S-HPC, but with substantially higher Km values (24 and 290-times higher) (Table 1).
A comparison of the catalytic efficiencies (kcat/Km) of the three enzymes for the (R)- and (S)-enantiomers provides additional insights. The catalytic efficiencies for the “natural” enantiomers for each enzyme are in the range of 1 to 8 × 105. By comparison, the catalytic efficiencies for the opposite enantiomers are about three orders of magnitude lower. When enantioselectivity (E) is defined as the ratio of kcat/Km for the natural enantiomer to kcat/Km for the opposite enantiomer, the following values are obtained: ERHPCDH1 = 944, ESHPCDH1 = 283, and ESHPCDH3 = 658. Thus, the HPCDH enzymes are highly efficient at discriminating between the HPC enantiomers, with enantioselectivity controlled largely by differences in kcat for R-HPCDH, and predominantly by differences in Km for the two S-HPCDH enzymes.
rS-HPCDH1 and rS-HPCDH3 are similar but not identical in terms of their catalytic properties, verifying that the homologs are redundant S-HPC specific enzymes. The subsequent studies of S-HPCDH are focused on rS-HPCDH3, since only this protein could be expressed in sufficient amounts for the detailed analyses described below.
The apparent Km values for NAD+ and NADH with R-HPC and 2-KPC as substrates were determined for rS-HPCDH3 and gave the following values: Km for NAD+, 191 ± 21 μM; Km for NADH, 8.42 ± 1.58 μM. By comparison, the Km values for rR-HPCDH1 using a complete bisubstrate kinetic analysis were previously reported to be 457 μM and 36.6 μM for NAD+ and NADH, respectively (15).
pH dependence of the kinetic parameters for rS-HPCDH3
Kinetic parameters were determined for the oxidation of S-HPC by rS-HPCDH3 at a range of pH values and plotted as log kcat vs pH, log kcat /Km vs pH and log Km vs pH as was done previously for oxidation of R-HPC by rR-HPCDH1 (15). These results are presented in Figure 3. The overall trends are similar to those reported for rR-HPCDH1 but with some important differences (15) suggesting a similar fundamental chemical mechanism for substrate oxidation. The differences in the pH analyses for the two enzymes are highlighted below.
Figure 3.

Changes of kinetic parameters with pH for rS-HPCDH3 catalyzed oxidation of S-HPC. (A) kcat vs. pH, (B) kcat/KmS-HPC vs. pH, (C) KmS-HPC vs. pH, represented in log scale. The plots in (A) and (C) are shown as simple line plots. The line in plot (B) was generated by a fit of the experimental data to equation 1.
Figure 3A shows a fairly steady increase in log kcat as the pH increases (slope 0.08) suggesting that isomerization of the enzyme-NAD+ complex could be a major rate-determining step, as described for other dehydrogenases (23). The same effect was seen for rR-HPCDH1 (15), although the change in kcat was more pronounced (over 6-fold increase vs. 3-fold increase for rS-HPCDH3).
Linear regression of the plot of log kcat/Km vs. pH from pH 5.0 – 8.0 (Figure 3B) gave a slope of 0.998 with an R2 value of 0.980, suggesting the importance of a single ionizable residue that must be deprotonated for catalysis. A fit of the data in Figure 3B to Equation 1 provides a pKa value for this residue of 7.9. Based on pH studies of other SDR enzymes, the ionizable residue most likely represents the tyrosine of the catalytic triad, which serves as the general acid/base for catalysis. For comparison, the pKa value for the corresponding ionizable group of rR-HPCDH1 was reported to be a lower value of 6.9 (15), while the prototype alcohol dehydrogenases from Drosophila melanogaster and D. lebanonensis were reported to be 7.1 and 7.6, respectively (24, 25).
The data presented in Figure 3C show a dramatic decrease in Km (321-fold decrease from pH 5.0 to pH 9.0) as the pH is increased. Taken together with the above results, this suggests that the deprotonated tyrosine general base plays an important role in both alcohol binding (Km effect) and catalysis (kcat and kcat/Km effects). The increase in Km from pH 9 to pH 10 could reflect deprotonation of another functional group important in coordinating the sulfonate of CoM (K214, as described below). The decrease in Km for rS-HPCDH3 was much greater than that observed for rR-HPCDH1, where only a 14-fold decrease in Km was observed (15). This suggests that the general acid/base of S-HPCDH3 is more important for binding of the alcohol substrate in the (S)-dehydrogenase than the (R)-dehydrogenase, an idea that is supported by the further studies described below.
Site-directed mutagenesis of the catalytic triad
As shown in Figure 2, the positions of the residues in the primary sequences believed to constitute the catalytic triad are conserved between the R- and S-HPCDH enzymes. To confirm the importance of the key residues, site-directed mutants were constructed. As shown in Table 2, the Y156F and K160A mutants were completely inactive. The S143A mutant exhibited a small amount of activity but with a catalytic efficiency that was reduced by more than five orders of magnitude relative to wild-type. Interestingly, the Y156A mutant exhibited a kcat that was 2.6% of the activity of the wild-type enzyme, with a Km value 60-fold higher than wild-type. The observation that the Y156A, but not Y156F mutant retains some activity suggests that the removal of the bulky phenyl group allows the enzyme to facilitate hydrogen atom abstraction from the hydroxyl group by another mechanism. An analysis of the primary sequences of the HPCDH enzymes shows that a cysteine residue is next to the tyrosine base in the S-enzymes but not the R-enzyme (Figure 2). A cysteine is also adjacent to the catalytic triad in the SDR 17β-hydroxysteroid dehydrogenase, and the Y to A substitution in that enzyme also exhibits a small amount of activity (26). It is conceivable that C157 substitutes as the general base in the Y156A mutant, albeit with a reduced catalytic efficiency, although this possibility has not been pursued further in this study.
Table 2.
Summary of Amino Acid Substitutions Made to the Putative rS-HPCDH35 Catalytic Residues Using Site-Directed Mutagenesis a
| amino acid | postulated catalytic role(s)b | substitution | Km (μM S-HPC) | kcat (s-1) | kcat/Km (M-1 s-1) |
|---|---|---|---|---|---|
| none | 31 ± 0.9 | 25 ± 0.2 | 7.5 × 105 | ||
| S143 | H-bond donor to substrate/charge stabilization of the transition state | S143A | 9500 ± 2500 | 0.013 ± 0.001 | 1.4 × 100 |
| Y156 | general acid/base | Y156A | 1800 ± 200 | 0.65 ± 0.02 | 3.5 × 102 |
| Y156F | no activityc | ||||
| K160 | lowers pKa of Y156/coenzyme binding | K160A | no activityc |
Assays contained 66 μg of S143A or 20 μg of Y156A, 5 mM NAD+ and variable concentration of S-HPC.
Postulated catalytic roles are based on a general trend found in most SDR enzymes (including R-HPCDH).
No activity was defined as no measurable change in the absorbance at 340 nm (for NADH) in assays containing 100 μg protein, 2.5 mM S-HPC and 20 mM NAD+. Apparent kcat and Km values are reported as means ± standard errors. All other values are reported as means only. All assays were performed in triplicate at 30 °C with fixed concentrations of NAD+. Apparent kinetic constants were determined by fitting experimental data to the standard form of the Michaelis-Menten equation.
Identification and Site-Directed Mutagenesis of the Sulfonate Binding Residues
For rR-HPCDH1, two arginine residues, R156 and R196, were shown to interact with the sulfonate of CoM via ionic interactions (Scheme 1, left side) (14, 16). Site-directed mutagenesis of either of these residues to alanine resulted in substantially reduced kcat values and increased Km values for 2-KPC reduction. Importantly, the same amino acid substitutions did not significantly alter kcat or Km when 2-butanone, a non-physiological substrate lacking the sulfonate, was used as the substrate (14). Thus, the sulfonate-coordinating arginine residues of R-HPCDH1 are only required for effective catalysis with the natural substrate.
By analogy, positively charged residues within a pocket are believed to interact with the sulfonate moiety of 2-KPC and S-HPC in S-HPCDH, but with differential placement relative to the methyl binding pocket such that the hydroxyl group and hydrogen atom of the substrate are oriented properly for catalysis (14, 16). To facilitate the identification of these residues, a homology model was constructed for S-HPCDH3 with substrates modeled in using the R-HPCDH1 structure with 2-KPC bound as a guide (16).
As shown in Figure 4A, the positioning of the catalytic triad residues, verified for S-HPCDH3 by the mutational analyses in Table 2, are conserved between the two S-HPCDH enzymes. As predicted, the active sites of the enzymes differ in the spatial orientations of the CoM and methyl groups, with the positions of the hydrogen atom and hydroxyl group being fixed relative to NAD+ and the tyrosine general base. Figure 4B shows a different view of the active sites, highlighting the interactions known (for rR-HPCDH1) and proposed (for rS-HPCDH3) to be important in binding the sulfonate of CoM. While two arginines coordinate the sulfonate within R-HPCDH1, the model suggests coordination by an arginine and a lysine for S-HPCDH3 (as well as S-HPCDH1 as seen in the multiple sequence alignment of Figure 2).
Figure 4.
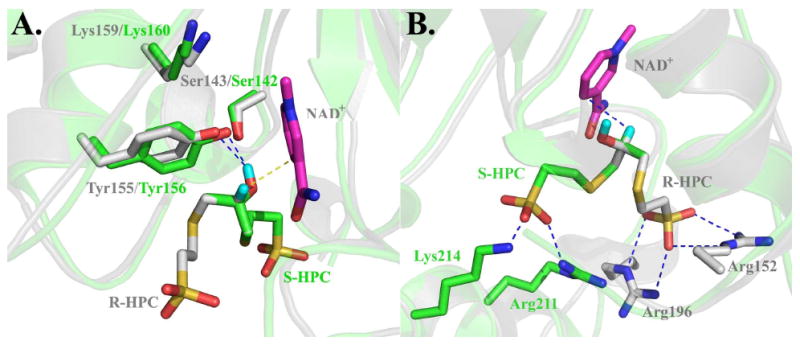
Superimposed active sites of R-HPCDH1 and S-HPCDH3 based on the crystal structure of R-HPCDH1 and a homology model of S-HPCDH3. The cartoon structures and carbon atoms of amino acid residues of R-HPCDH1 (pdb ID 2cfc) and the homology model for S-HPCDH3 are colored grey and green, respectively. NAD+ is shown in magenta. R-HPC and S-HPC were modeled using the crystal structure for S-HPC bound at the active site of R-HPCDH1 as described previously (16). In both views, R-HPC (grey carbon atoms) and S-HPC (green carbon atoms) are modeled at the active sites such that the positions of the hydroxyl group and hydrogen atom occupy the same positions. The methyl groups and the methylene groups linking the hydroxypropyl groups to CoM are overlayed on top of each other to highlight the different spatial orientations of these groups in R- and S-HPC. Panel A, Superimposed structures highlighting the interactions of substrates with the catalytic triads. Panel B, Superimposed structures highlighting the interactions of substrates with the amino acids that coordinate the sulfonate of CoM.
As shown in Table 3, substitution of either R211 or K214 by alanine resulted in substantially reduced catalytic efficiencies for S-HPCDH3 with the natural substrates in both the forward (oxidative, S-HPC as substrate) and reverse (reductive, KPC as substrate) directions. Interestingly, the most dramatic effects of the mutations were on Km values, which were 50-74 times higher in the oxidative direction and 41-43 times higher in the reductive direction. There was a significant (40-80%) decrease in kcat due to the mutations for the forward reaction, but very little change for the reductive direction. By comparison, 2-KPC reduction by the R152A and R196A mutants of R-HPCDH1 were impaired sizably in both Km and kcat values (14).
Table 3.
Kinetic Parameters for wild-type rS-HPCDH3 and R211A and K214A with various substratesa
| Enzyme | Km (mM) | Change in Km (x-fold) | kcat (s-1) | Change in kcat (x-fold) | kcat/Km (M-1 s-1) |
|---|---|---|---|---|---|
| Substrate: S-HPC | |||||
| wild-type | 0.031 ± 0.001 | 1.0 | 25 ± 0.2 | 1.0 | 7.9 × 105 |
| R211A | 1.6 ± 0.1 | 49 | 16 ± 0.4 | 0.64 | 1.0 × 104 |
| K214A | 2.3 ± 0.1 | 73 | 5.8 ± 0.1 | 0.23 | 2.5 × 103 |
| Substrate: 2-KPC | |||||
| wild-type | 0.28 ± 0.01 | 1.0 | 11 ± 0.2 | 1.0 | 3.9 × 104 |
| R211A | 11 ± 0.4 | 41 | 8.6 ± 0.2 | 0.80 | 7.5 × 102 |
| K214A | 12 ± 0. 9 | 43 | 9.6 ± 0.3 | 0.89 | 8.1 × 102 |
| Substrate: 2-butanone | |||||
| wild-type | 120 ± 5 | 1.0 | 0.044 ± 0.001 | 1.0 | 3.7 × 102 |
| R211A | 180 ± 30 | 1.5 | 0.072 ± 0.05 | 1.6 | 4.0 × 102 |
| K214A | 72 ± 6 | 0.61 | 0.039 ± 0.001 | 0.90 | 5.4 × 102 |
| Substrate: 2-propanol | |||||
| wild-type | 1400 ± 60 | 1.0 | 2.0 ± 0.5 | 1.0 | 1.4 × 103 |
| R211A | 720 ± 40 | 0.51 | 1.7 ± 0.03 | 0.85 | 2.4 × 103 |
| K214A | 950 ± 20 | 0.67 | 1.7 ± 0.2 | 0.86 | 1.8 × 103 |
Assay for S-HPC oxidation contained 0.20 μg of wild-type rS-HPCDH3, or 4.0 μg of R211A and K214A while assay for 2-KPC reduction contained 0.20 μg of wild-type rS-HPCDH3, 5.0 μg of R211A and 2.0 μg of K214A. Assay for 2-butanone reduction contained 197 μg of enzyme while assay for 2-propanol oxidation contained 8.0 μg of enzyme. Apparent kcat and Km values are reported as means ± standard deviations. All other values are reported as means only. All assays were performed in triplicates at 30 °C with fixed concentrations of NAD+ (10 mM) or NADH (0.17 mM). Apparent kinetic constants were determined by fitting experimental data to the standard form of the Michaelis-Menten equation.
The kcat and Km values measured for the reduction of the aliphatic ketone 2-butanone and the oxidation of the aliphatic alcohol 2-propanol were largely unaffected by the amino acid changes in S-HPCDH3 (Table 3). These aliphatic substrates had dramatically higher Km values and lower kcat values relative to the natural substrates. The fact that the kinetic parameters did not change substantially in the site-directed mutants demonstrates that the sulfonate-binding residues are not important for aliphatic substrates where no favorable interaction with the positively charged residues would occur. Similar results were obtained when 2-butanone was analyzed as a substrate for wild-type R-HPCDH1 and the R152A and R196A mutants (14).
S-HPCDH3 has an inherent stereoselectivity for 2-butanone reduction not present in R-HPCDH1
As shown in Table 4, S-HPCDH3 has a very high inherent stereoselectivity, producing (S)-2-butanol with a 98.4% enantioexcess (ee). These results are dramatically different than those observed for R-HPCDH1, where (S)-2-butanol is also produced in higher amounts than (R)-2-butanol, but in only a 44% enantioexcess (Table 4 and (14)). Thus, even though the R- and S-HPCDH enzymes are highly specific for production of R-HPC and S-HPC from 2-KPC reduction, they both form excesses of (S)-2-butanol from 2-butanone.
Table 4.
Enantioselectivity of 2-butanone reduction by wild-type rS-HPCDH3, rS-HPCDH3 mutants, and rR-HPCDH1a
| Enzyme | no additions | + 1 mM ethanesulfonate | ||||
|---|---|---|---|---|---|---|
| % (S)-2-butanol | % (R)-2-Butanol | ee (S)-2-butanol | % (S)-2-butanol | % (R)-2-butanol | ee (S)-2-butanol | |
| rS-HPCDH3 | 99.2 ± 0.2 | 0.82 ± 0.15 | 98.36 | 98.0 ± 0.08 | 1.97 ± 0.08 | 96.06 |
| rS-HPCDH3 R211A | 99.4 ± 0.2 | 0.58 ± 0.22 | 98.84 | 99.4 ± 0.2 | 0.64 ± 0.15 | 98.72 |
| rS-HPCDH3 K214A | 91.6 ± 0.3 | 8.4 ± 0.31 | 83.18 | 90.9 ± 0.2 | 9.12 ± 0.20 | 81.76 |
| rR-HPCDH1 | 71.9 ± 1.3 | 28.0 ± 1.3 | 43.88 | 92.7 ± 0.1 | 7.33 ± 0.05 | 85.34 |
All assays were performed in triplicate at 30 °C using 0.64 mg enzyme, 15 mM NADH, and 56 mM 2-butanone. Percent yields are reported as means ± standard deviations.
An interesting feature of the R-HPCDH1 is that the enantioselectivity of 2-butanone reduction can be “modulated” to produce higher amounts of (S)-2-butanol by including CoM or alkylsulfonates in the assay (14). These “enantioiselective modulators” modulated stereospecificity in a saturable fashion, with a theoretical yield of 100% (S)-2-butanol at the saturation points (14). The effects of these enantioselective modulators were abolished in mutants in the sulfonate-coordinating arginines (R152A and R196A) (14). These results were interpreted as follows: the alkylsulfonates bind in the CoM binding pocket, constraining 2-butanone to bind in the active site with the methyl group rather than the ethyl group oriented towards the bound alkylsulfonate to prevent steric clashes. In this orientation, the sp2 hybridized carbonyl of 2-butanone is necessarily constrained for hydride transfer to the plane that produces (S)-2-butanol (14). As shown in Table 4, the addition of ethanesulfonate to wild-type rS-HPCDH3 and the R211A and K214A mutants had very little effect on the stereochemical outcome of 2-butanone reduction relative to the effect observed with rR-HPCDH, demonstrating that the modulating effect is a phenomenon only seen with R-HPCDH.
Dehydrogenation of 2-butanol enantiomers by rR- and rS-HPCDH enzymes
The studies of 2-butanone reduction were expanded to examine the kinetic parameters for the reverse reaction, i.e. (R)- and (S)-2-butanol oxidation to 2-butanone. (Table 5). Both enzymes exhibited a preference for (S)-2-butanol as the substrate, with enantioselectivity values of 6.23 and 3.04 for rS-HPCDH3 and rR-HPCDH1, respectively. The Km values for 2-butanone reduction were about 1000-times higher than for 2-KPC.
Table 5.
Kinetic Parameters for rS-HPCDH3 and rR-HPCDH1 catalyzed oxidation of 2-butanola
| Substrate | Km (mM) | Vmax (units/mg) | kcat (s-1) | kcat/Km (M-1 s-1) | Change in kcat (x-fold) | Change in Km (x-fold) | enantioselectivity (E) |
|---|---|---|---|---|---|---|---|
| rS-HPCDH3 | |||||||
| (R)-2-butanol | 68 ± 3 | 2.2 ± 0.03 | 1.0 | 14.8 | 1.0 | 1.0 | 0.16 |
| (S)-2-butanol | 28 ± 2 | 5.8 ± 0.07 | 2.6 | 92.8 | 2.6 | 0.4 | 6.3 |
| rR-HPCDH1 | |||||||
| (R)-2-butanol | 220 ± 10 | 3.77 ± 0.06 | 1.9 | 8.69 | 1.0 | 1.0 | 0.33 |
| (S)-2-butanol | 350 ± 50 | 18.8 ± 0.9 | 9.3 | 26.4 | 5.0 | 1.6 | 3.0 |
aAll assays were performed in triplicate at 30 °C with fixed concentrations of NAD+ (10 mM). Assays of 2-butanol oxidation catalyzed by rS-HPCDH3 and rR-HPCDH1 contained 25 and 21 μg of enzyme, respectively. Apparent kinetic constants were determined by fitting experimental data to the standard form of the Michaelis-Menten equation. Apparent Vmax and Km values are reported as means ± standard deviations. All other values are reported as means only. Enantioselectivity was defined as (kcat/Km)R-enantiomer/(kcat/Km)S-enantiomer for (R)-2-butanol oxidation and as (kcat/Km)S-enantiomer/(kcat/Km)R-enantiomer for (S)-2-butanol oxidation.
2-(2-methyl-2-hydroxypropylthio)ethanesulfonate is a competitive inhibitor of natural substrate oxidation by rR-HPCD1H but not rS-HPCDH3
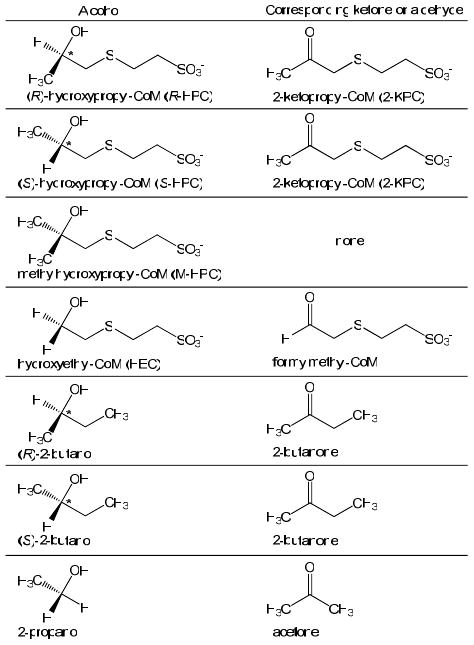
As shown in Table 1, rR-HPCDH1 exhibits a Km for S-HPC in the same range as R-HPC, while rS-HPCDH3 exhibits a Km for R-HPC that is nearly 300-times higher than for S-HPC. These results indicate that rR-HPCDH1 is able to bind either enantiomer with high affinity, although for the improper enantiomer, the hydroxyl and hydrogen of the substrate are misaligned, resulting in lower turnover (Table 1). In contrast, the high Km value for R-HPC with rS-HPCDH3 suggests that the other enantiomer does not bind well in the first place, possibly due to steric constraints when the methyl group is misaligned. As shown in the Structures, 2-(2-methyl-2-hydroxypropylthio)ethanesulfonate (M-HPC) is an achiral analog of both R-HPC and S-HPC where the hydrogen atoms of each alcohol are replaced by a methyl group, resulting in a tertiary alcohol that cannot undergo oxidation. M-HPC is thus an equivalent mimic of both HPC enantiomers and can be studied as a possible inhibitor of both enzymes to shed light on the Km differences for the opposite enantiomers discussed above.
Structures.
As shown in Figure 5A, M-HPC was a competitive inhibitor of R-HPC oxidation by R-HPCDH1, with a Kis of 290 ± 14 μM. This value is in the range of the Km for the natural substrate R-HPC (96 μM) and the opposite enantiomer S-HPC (224 μM). In marked contrast to these results, no detectable inhibition was observed for M-HPC vs. the natural substrate S-HPC with S-HPCDH3 (Figure 5B). This result correlates with the observation that R-HPC has a dramatically higher Km for S-HPCDH3 relative to the natural substrate (9100 vs. 31 μM). This experiment was repeated with rS-HPCDH1 and the same result was obtained: no detectable inhibition was seen with M-HPC (Figure 5C). Thus, the addition of the methyl group in the improper position relative to the CoM and hydroxyl groups has very different effects on binding affinity for the R- and S-specific dehydrogenases.
Figure 5.

Effects of 2-(2-methyl-2-hydroxypropylthio)ethanesulfonate (M-HPC) on R- and S-HPC oxidation by R-HPCDH1, S-HPCDH3, and S-HPCDH1. Panel A, Competitive inhibition of R-HPCDH1-catalyzed R-HPC oxidation by M-HPC. The double reciprocal plots for assays performed in the presence of different concentrations of M-HPC are shown in the main diagram. Data points represent the average of triplicate experiments. The solid lines were generated by nonlinear least-square fits of the ν vs. S data, shown in the inset, to the equation for a rectangular hyperbola using Sigmaplot. M-HPC concentrations: (●) 0 mM, (○) 0.2 mM, (▼) 0.4 mM, (△) 0.8 mM, (■) 1.6 mM. Panels B and C, ν vs. S plots for S-HPC oxidation by S-HPCDH3 and S-HPCDH1, respectively, in the presence of different concentrations of M-HPC. The lines were generated by fitting the data to the standard form of the Michaelis-Menten equation. M-HPC concentrations: (●) 0 mM, (○) 1.2 mM, (▼) 4.9 mM.
2-(2-hydroxyethylthio)ethanesulfonate (HEC) is a substrate for both the R- and S-HPCDH enzymes with identical Km values
HEC is an achiral mimic of both R-HPC and S-HPC in which the methyl group is replaced by a hydrogen (see Structures). HEC was found to be a substrate for both rR-HPCDH1 (kcat = 0.55 ± 0.02 μM, Km = 960 ± 100 μM) and rS-HPCDH3 (kcat = 3.8 ± 0.3 μM, Km = 970 ± 250 μM). Thus, the loss of the methyl groups imparting chirality resulted in an enzyme substrate with identical (within experimental error) Km values for both enzymes, suggesting that for this substrate, the CoM moiety and hydroxyl group have become equal determinants in binding affinity. As a final experiment, M-HPC was investigated as an inhibitor of HEC oxidation to see if the result of Figure 5 would hold with the achiral substrate. M-HPC was a competitive inhibitor of HEC oxidation by R-HPCDH1 with a Kis of 110 ± 10 μM, but was not an inhibitor of HEC oxidation by S-HPCDH3.
Different strategies control enantioselectivity in the R- and S-HPCDH enzymes
The results presented above are interpreted as follows: R-HPCDH1 can bind either enantiomer of HPC with the CoM moiety oriented properly in the sulfonate-binding pocket consisting of R152 and R196 (Figure 4), but S-HPCDH3 is unable to bind R-HPC in this fashion, presumably due to steric clashes imposed by the presence of the misaligned methyl group on the C2 atom. Both enzymes bind HEC with identical affinities since no methyl group is present. Thus, a high affinity ternary complex of S-HPC, NAD+, and R-HPCDH forms, but the misalignment of the hydrogen and hydroxyl groups on C2 relative to NAD+ and the tyrosine general base results in a 403-fold lower turnover rate.
In contrast to R-HPCDH1, S-HPCDH3 apparently cannot bind S-HPC analogs (R-HPC and M-HPC) with CoM oriented properly in the sulfonate-binding pocket that consists of R211 and K214. The most logical explanation for this is that the misaligned methyl groups on C2 required for this high affinity binding are not accommodated due to steric clashes with amino acid side chain(s). Thus, R-HPC binds to S-HPCDH3 with a 290-fold lower affinity, but in an orientation where the hydroxyl and hydrogen on C2 can be more properly aligned with tyrosine156 and NAD+, such that kcat only decreases by 4.5-fold relative to the natural substrate S-HPC.
The results of the pH studies further indicate that the hydroxyl group at C2 is a larger determinant in the binding of S-HPC to S-HPCDH3 than for binding of R-HPC to R-HPCDH1. Note that the Km for the natural substrate is 3-fold lower for S-HPCDH3 than for R-HPCDH1, which could be due to higher affinity binding of the hydroxyl group. However, the Km values for HEC, lacking the methyl group, are identical for both enzymes. Thus, the methyl group of S-HPC may facilitate the alignment of the hydroxyl group in this higher affinity position.
Physiological implications of these studies
The differential control of enantioselectivity in the R- and S-HPCDH enzymes may have evolved to reflect the roles these enzymes play in propylene metabolism. As shown in Figure 1, alkene monooxygenase from X. autotrophicus produces a mixture of (R) and (S)-epoxypropane, although (R)-epoxypropane is produced in a 90% enantioexcess (4). Both (R)- and (S)-epoxypropane are substrates for epoxyalkane:CoM transferase, which results in the production of R- and S-HPC, respectively. Although (S)-epoxypropane (and by extrapolation, S-HPC) are the minor products of the metabolic pathway, it is still essential for these compounds to be further metabolized by the bacterium, since epoxides are highly toxic. While epoxyalkane:CoM transferase detoxifies (S)-epoxypropane by conversion to S-HPC, S-HPC must proceed through the pathway of epoxide carboxylation to regenerate free CoM; if it did not, the CoM pool would be wastefully sequestered. R-HPCDH1 will still operate efficiently even with a similar Km value for the opposite enantiomer, since S-HPC is predicted to be produced at about 20-fold lower concentrations than R-HPC. In contrast, in order for S-HPCDH to be efficient at the lower concentrations of S-HPC encountered in the cell, it needs to have a lower Km for the natural substrate and substantially higher Km for the opposite enantiomer to work efficiently. To summarize, the different amounts of R- and S-HPC that accumulate in the cells due to the inherent stereoselectivity of the alkene monooxygenase is proposed to have led to the evolution of different strategies for controlling efficient substrate flux through the pathway. R-HPC oxidation is controlled by kcat, since R-HPCDH does not need to discriminate substrate binding at the much lower concentrations of the opposite enantiomer present, while control of S-HPC oxidation is controlled by Km, since discrimination of substrate binding is crucial with the higher concentrations of the inhibitory enantiomer present. In this context it should be noted that R- and S-HPCDH were found to have comparable specific activities in cell extracts of X. autotrophicus when grown on propylene, with each predicted to account for about 1% of soluble cell protein (4-6). This observation reiterates that substrate flux is controlled by differences in mechanisms of these enzymes and not by differences in levels of expression.
The fact that X. autotrophicus has redundant copies of the dehydrogenases (as well as the other enzymes of epoxide metabolism) further highlights the importance of these enzymes to the bacterium. The results of Table 1 suggest some differences in the kinetic properties for two of the redundant S-enzymes. The copies we purified from X. autotrophicus in our previous studies (4-6, 18, 27) consisted primarily (or wholly) of the enzymes in the first operon based on yields from the purification schemes and their biochemical properties. Now that we have a greater understanding of the organization of the epoxide carboxylation genes we can apply molecular genetics to determine how important the individual copies are to bacterial growth and survival under different conditions.
Acknowledgments
We thank Dr. Michael Kay and Matthew Weinstock, University of Utah School of Medicine, for technical assistance with the CD spectrometer.
Footnotes
1Abbreviations: R-HPC, 2-[(R)-2-hydroxypropylthio]ethanesulfonate, (R)-2-hydroxypropyl-CoM); S-HPC, 2-[(S)-2-hydroxypropylthio] ethanesulfonate, (S)-2-hydroxypropyl-CoM); S-HPCDH, 2-[(S)-2-hydroxypropylthio]ethanesulfonate dehydrogenase; rS-HPCDH, recombinant 2-[(S)-2-hydroxypropylthio]ethanesulfonate dehydrogenase; R-HPCDH, 2-[(R)-2-hydroxypropylthio]ethanesulfonate dehydrogenase; rR-HPCDH, recombinant 2-[(R)-2-hydroxypropylthio]ethanesulfonate dehydrogenase; M-HPC, 2-(2-methyl-2-hydroxypropylthio)ethanesulfonate; HEC, 2-(2-hydroxyethylthio)ethanesulfonate; 2-KPC, 2-(2-ketopropylthio)ethanesulfonate (2-ketopropyl-CoM); 2-KPCC, 2-(2-ketopropylthio)ethanesulfonate carboxylase/oxidoreductase; CoM, coenzyme M, (2-mercaptoethanesulfonate); SDR, short-chain dehydrogenase/reductase; Tris, tris(hydroxymethyl)aminomethane, CD, circular dichroism; ee, enantiomeric excess; E, enantioselectivity; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis;
This work was supported by National Institutes of Health Grant GM51805 to S.AE. and by Department of Energy Grant DE-FG02-04ER15563 to J.W.P.
References
- 1.Kroutil W, Mang H, Edegger K, Faber K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr Opin Chem Biol. 2004;8:120–126. doi: 10.1016/j.cbpa.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K, Yamanaka R, Matsuda T, Harada T. Recent developments in asymmetric reduction of ketones with biocatalysts. Tetrahedron: Asymmetry. 2003;14:2659–2681. [Google Scholar]
- 3.Goldberg K, Schroer K, Lütz S, Liese A. Biocatalytic ketone reduction— a powerful tool for the production of chiral alcohols—part I: processes with isolated enzymes. Appl Microbiol Biotechnol. 2007;76:237–248. doi: 10.1007/s00253-007-1002-0. [DOI] [PubMed] [Google Scholar]
- 4.Allen JR, Clark DD, Krum JG, Ensign SA. A role for coenzyme M (2-mercaptoethansulfonic acid) in a bacterial pathway of aliphatic epoxide carboxylation. Proc Natl Acad Sci USA. 1999;96:8432–8437. doi: 10.1073/pnas.96.15.8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen JR, Ensign SA. Purification to Homogeneity and Reconstitution of the Individual Components of the Epoxide Carboxylase Multiprotein Enzyme Complex from Xanthobacter strain Py2. J Biol Chem. 1997;272:32121–32128. doi: 10.1074/jbc.272.51.32121. [DOI] [PubMed] [Google Scholar]
- 6.Allen JR, Ensign SA. Two Short-Chain Dehydrogenases Confer Stereoselectivity for Enantiomers of Epoxypropane in the Multiprotein Epoxide Carboxylating Systems of Xanthobacter Strain Py2 and Nocardia corallina B276. Biochemistry. 1999;38:247–256. doi: 10.1021/bi982114h. [DOI] [PubMed] [Google Scholar]
- 7.Ensign SA. Microbial metabolism of aliphatic alkenes. Biochemistry. 2001;40:5845–5853. doi: 10.1021/bi015523d. [DOI] [PubMed] [Google Scholar]
- 8.Ensign SA, Allen JR. Aliphatic Epoxide Carboxylation. Ann Rev Biochem. 2003;72:55–76. doi: 10.1146/annurev.biochem.72.121801.161820. [DOI] [PubMed] [Google Scholar]
- 9.Jörnvall H, Persson B, Krook M, Atrian S, Gonzàlez-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases. Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 10.Kavanagh KL, Jornvall H, Persson B, Oppermann U. The SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci. 2008;65:3895–3906. doi: 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jörnvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem-Biol Interact. 2003;143-144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 12.Persson B, Kallberg Y, Bray JE, Bruford E, Dellaporta SL, Favia AD, Duarte RG, Jornvall H, Kavanagh KL, Kedishvili N, Kisiela M, Maserk E, Mindnich R, Orchard S, Penning TM, Thornton JM, Adamski J, Oppermann U. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem-Biol Interact. 2009;178:94–98. doi: 10.1016/j.cbi.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Persson B, Krook M, Jörnvall H. Characteristics of short-chain alcohol dehydrogenases and related enzymes. Eur J Biochem. 1991;200:537–543. doi: 10.1111/j.1432-1033.1991.tb16215.x. [DOI] [PubMed] [Google Scholar]
- 14.Clark DD, Boyd JM, Ensign SA. The stereoselectivity and catalytic properties of Xanthobacter autotrophicus 2-[(R)-2-Hydroxypropylthio]ethanesulfonate dehydrogenase are controlled by interactions between C-terminal arginine residues and the sulfonate of coenzyme M. Biochemistry. 2004;43:6763–6771. doi: 10.1021/bi049783h. [DOI] [PubMed] [Google Scholar]
- 15.Clark DD, Ensign SA. Characterization of the 2- (R)-2-hydroxypropylthio ethane sulfonate dehydrogenase from Xanthobacter strain Py2: product inhibition, pH dependence of kinetic parameters, site-directed mutagenesis, rapid equilibrium inhibition, and chemical modification. Biochemistry. 2002;41:2727–2740. doi: 10.1021/bi0118005. [DOI] [PubMed] [Google Scholar]
- 16.Krishnakumar AM, Nocek BP, Clark DD, Ensign SA, Peters JW. Structural basis for stereoselectivity in the (R)- and (S)-hydroxypropylthioethanesulfonate dehydrogenases. Biochemistry. 2006;45:8831–8840. doi: 10.1021/bi0603569. [DOI] [PubMed] [Google Scholar]
- 17.Krishnakumar AM, Sliwa D, Endrizzi JA, Boyd ES, Ensign SA, Peters JW. Getting a Handle on the Role of Coenzyme M in Alkene Metabolism. Microbiol Mol Biol Rev. 2008;72:445. doi: 10.1128/MMBR.00005-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen JR, Ensign SA. Characterization of Three Protein Components Required for Functional Reconstitution of the Epoxide Carboxylase Multienzyme Complex from Xanthobacter strain Py2. J Bacteriol. 1997;179:3110–3115. doi: 10.1128/jb.179.10.3110-3115.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Cook PF, Cleland WW. Enzyme kinetics and mechanism. Garland Science; New York: 2007. [Google Scholar]
- 21.Krum JG, Ensign SA. Evidence that a linear megaplasmid encodes enzymes of aliphatic alkene and epoxide metabolism and Coenzyme M (2-mercaptoethanesulfonate) biosynthesis in Xanthobacter strain Py2. J Bacteriol. 2001;183:2172–2177. doi: 10.1128/JB.183.7.2172-2177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swaving J, Weijers CA, van Ooyen AJ, de Bont JAM. Complementation of Xanthobacter Py2 mutants defective in epoxyalkane degradation, and expression and nucleotide sequence of the complementing DNA fragment. Microbiology. 1995;141:477–484. doi: 10.1099/13500872-141-2-477. [DOI] [PubMed] [Google Scholar]
- 23.Grimshaw CE, Cook PF, Cleland WW. Use of isotope effects and pH studies to determine the chemical mechanism of Bacillus subtilis L-alanine dehydrogenase. Biochemistry. 1981;20:5655–5661. doi: 10.1021/bi00523a003. [DOI] [PubMed] [Google Scholar]
- 24.Winberg JO, Brendskag MK, Sylte I, Lindstad RI, McKinley-McKee JS. The catalytic triad in Drosophila alcohol dehydrogenase: pH, temperature and molecular modelling studies. J Mol Biol. 1999;294:601–616. doi: 10.1006/jmbi.1999.3235. [DOI] [PubMed] [Google Scholar]
- 25.Brendskag MK, McKinley-McKee JS, Winberg JO. Drosophila lebanonensis alcohol dehydrogenase: pH dependence of the kinetic coefficients. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology. 1999;1431:74–86. doi: 10.1016/s0167-4838(99)00028-x. [DOI] [PubMed] [Google Scholar]
- 26.Puranen TJ, Poutanen MH, Peltoketo HE, Vihko PT, Vihko RK. Site-directed mutagenesis of the putative active site of human 17β-hydroxysteroid dehydrogenase type. Biochem J. 1994;304:289–293. doi: 10.1042/bj3040289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark DD, Allen JR, Ensign SA. Characterization of five catalytic activities associated with the NADPH : 2-ketopropyl-coenzyme M [2-(2-ketopropylthio)ethanesulfonate] oxidoreductase/carboxylase of the Xanthobacter strain Py2 epoxide carboxylase. Biochemistry. 2000;39:1294–1304. doi: 10.1021/bi992282p. [DOI] [PubMed] [Google Scholar]