

Abstract
High-risk human papillomaviruses (HPV) encode two oncogenes, E6 and E7, expressed in nearly all cervical cancers. In vivo, HPV-16 E7 has been shown to induce multiple phenotypes in the context of transgenic mice, including cervical cancer. E7 is a multifunctional protein known best for its ability to inactivate the tumor suppressor pRb. To determine the importance of pRb inactivation by E7 in cervical cancer, we pursued studies with genetically engineered mice. E7 expression in estrogen-treated murine cervix induced dysplasia and invasive cancers as reported previously, but targeted Rb inactivation in cervical epithelium was not sufficient to induce any cervical dysplasia or neoplasia. Furthermore, E7 induced cervical cancer formation even when the E7-pRb interaction was disrupted by the use of a knock-in mouse carrying an E7-resistant mutant Rb allele. pRb inactivation was necessary but not sufficient for E7 to overcome differentiation-induced or DNA damage–induced cell cycle arrest, and expression patterns of the E2F-responsive genes Mcm7 and cyclin E indicate that other E2F regulators besides pRb are important targets of E7. Together, these data indicate that non-pRb targets of E7 play critical roles in cervical carcinogenesis.
Introduction
Human papillomaviruses (HPV) are small DNA viruses and the causative agents of epithelial warts. The so-called “high-risk” HPVs infect the anogenital tract epithelium and are associated with the appearance of cervical dysplasia, almost all cases of cervical cancer, many other genital cancers, and a subset of oral cancers (1-3). HPV type 16 accounts for ~60% of all cervical cancers as well as >90% of HPV-positive oral cancers (2, 3). The HPV genome usually exists extrachromosomally, but many HPV-associated cancers contain HPV genomes integrated into the host DNA (4). These integrated genomes invariably contain intact viral E6 and E7 genes, and integration causes their increased expression (5). These data suggest that E6 and E7 contribute to the development of cervical cancers.
E7 binds to >20 cellular proteins (6). The most well characterized target of E7 is the retinoblastoma tumor suppressor, pRb. Interaction between E7 and pRb disrupts the ability of pRb to bind cellular E2F transcription factors, and this inhibits pRb-mediated repression of E2F-responsive genes (7, 8) and results in proteasomal degradation of pRb in cultured cells (9-11). E7 may modulate E2F activity by other mechanisms; E7 also targets the pRb-related p107 and p130 proteins for degradation, inhibits the cyclin-dependent kinase (cdk) inhibitors p21 and p27, and may directly activate both cyclin A/cdk2 and E2F1 (12-14). Furthermore, E7 binds a wide variety of other cellular targets, although the implications of these interactions are largely unclear (14).
Many studies have suggested that pRb inactivation is critical for E7 function. pRb has been connected to all of the processes disrupted by E7 In vivo, including cell cycle regulation, differentiation, DNA damage responses, centrosome synthesis, and tumorigenesis (15-18). Additionally, pRb inactivation is necessary and sufficient for nearly all of the acute effects of E7 on the skin of transgenic mice (17, 19). However, some studies have indicated that targets of E7 other than pRb may also be important for the phenotypes of E7. Two E7 mutants deficient in binding to pRb can cooperate with E6 in the immortalization of primary human keratinocytes (20), and the ability of E7 to transactivate certain E2F-responsive promoters may depend on binding to p107 rather than binding to pRb as shown with the B-myb promoter (21). Furthermore, the E7Δ79-83 mutant exhibits a decreased ability to transform baby rat kidney cells, although it efficiently binds and degrades pRb (22-25). Another E7 mutant, E7CVQ68-70AAA, which is also able to bind and destabilize pRb but is deficient in p21 inactivation, fails to overcome differentiation-dependent cell cycle withdrawal or DNA damage–induced cell cycle arrest in human keratinocytes (24, 26). Additionally, E7 may contribute to transformation of rat embryo fibroblasts via activation of c-Jun independently of pRb inactivation (27), and E7 induces centrosome abnormalities in pRb-deficient cells (28). Finally, E7 is able to produce some phenotypes in murine skin independently of pRb inactivation (17, 19). Thus, multiple studies indicate that the effects of E7 on targets other than pRb make important contributions to E7 function, although no consensus has emerged as to which targets affect which phenotypes.
HPV-16 E7 expression alone is sufficient to induce invasive cervical cancers in transgenic mice treated with chronic low doses of estrogen (29, 30). In the present study, we assessed whether pRb inactivation by E7 is necessary and/or sufficient for cervical phenotypes in mice. The results show that E7 makes critical contributions to cervical carcinogenesis independently of pRb inactivation. Non-pRb targets of E7 were also required to overcome differentiation or DNA damage–induced cell cycle arrest. Expression patterns of the E2F-responsive genes Mcm7 and cyclin E showed that E7 modulates E2F-responsive gene expression through multiple pathways in cervix, suggesting that other E2F regulators besides pRb are important targets of E7.
A final interesting observation is that, when the E7-pRb interaction was abolished in the context of the E7 transgenic mice, cervical cancers arose in the absence of the dysplastic cervical intraepithelial neoplasia (CIN) lesions that are otherwise observed throughout the cervix in estrogen-treated E7 transgenic mice on a wild-type (WT) Rb background. The implications of this finding in terms of the genesis of cervical cancers are discussed.
Materials and Methods
Transgenic mice
K14E7, K14CreRbflox, and RbΔL mice have been described previously (17, 19, 31, 32). All mice were bred and maintained in the American Association for Accreditation of Laboratory Animal Care–approved McArdle Laboratory Cancer Center Animal Care Facility (Madison, WI). All Rbflox mice were analyzed as FVB backcross 6 from a 129-C57/BL6 background; all RbΔLxCxE mice were analyzed as FVB backcross 2 from a 129-C57/BL6 background. All mice were genotyped by PCR as described previously (17, 19). One hour before sacrifice, all mice were i.p. injected with bromodeoxyuridine (BrdUrd; 10 μL per g body weight of 12.5 mg/mL solution). For the irradiation studies, mice were exposed to 0 or 12 Gy ionizing radiation from a 137Cs source 24 hours before BrdUrd administration.
Estrogen treatment and cervical carcinogenesis
All analyses were done in estrogen-treated mice as described previously (29). Each mouse was assigned a diagnosis according to the worst cervical lesion detected. Statistical analyses of the incidence of dysplasia and invasive cancer were done using two-sided Fisher’s exact tests. Cross-sectional areas of invasive cancers were determined using Zeiss Axiovision 3.1 software on a Zeiss Axioskop microscope (Thornwood, NY). Cancer areas were compared using a two-sided Wilcoxon rank sum test.
Immunohistochemical and immunofluorescence analysis of epidermis
Immunohistochemical stains were done for BrdUrd, pRb, cyclin E, and Mcm7 as described previously (29) with the following modifications. For BrdUrd, pRb, or cyclin E staining, further unmasking was achieved with 20 minutes of immersion in 2 N HCl. Primary antibodies were diluted in blocking buffer and applied for 2.5 hours at room temperature: anti-pRb (1:50, clone G3-245; BD PharMingen, San Diego, CA), anti-BrdUrd (1:40; Oncogene, Carpinteria, CA), anti-Mcm7 (1:200; LabVision Neomarkers, Fremont, CA), or anti–cyclin E (1:50, clone M-20; Santa Cruz Biotechnology, Santa Cruz, CA).
Quantitation of pRb loss and cell proliferation
BrdUrd incorporation into newly synthesized DNA was used as a measure of keratinocyte proliferation by counting BrdUrd-stained cervical sections. All keratinocyte nuclei in eight visual fields were scored as either positive (brown) or negative (blue) for BrdUrd incorporation in both basal and suprabasal layers of the epidermis. For counting purposes, even slightly brown cells were counted as positive. Three to five mice were counted per genotype, and results are presented as the mean ± SD. Similarly, to quantify loss of pRb expression, sections of epidermis were stained for pRb and counted. Because pRb stains most robustly in basal layer cells, only basal keratinocytes were counted in eight visual fields of both endocervical and exocervical epithelium. Three to four mice were counted per genotype. Statistical analyses of results were done using the two-sided Wilcoxon rank sum test.
Results
Rb inactivation in murine cervix
To determine if pRb inactivation by E7 was necessary for cervical phenotypes, mice expressing HPV-16 E7 in the cervical stratified squamous epithelium (K14E7 mice; ref. 31) were mated to mice carrying a mutant Rb knock-in allele, RbΔLxCxE (RbΔL), containing three alanine mutations in pRb that interact with E7, thereby producing a mutant pRb protein that fails to bind E7 (33). pRbΔL retains the ability to bind E2Fs, induces G1 arrest in pRb-negative SAOS2 cells, and is phosphorylated and inactivated by cyclin D/cdk4 complexes similarly to WT pRb (34). pRbΔL also represses gene expression from E2F-responsive promoter constructs, although incrementally less effectively than WT pRb. Unlike WT pRb, pRbΔL fails to bind cellular LxCxE motif-containing proteins, and pRbΔL-induced G1 arrest cannot be reversed by expression of HPV-16 or HPV-18 E7 (32, 34).
To determine if pRb inactivation is sufficient to reproduce any of the phenotypes of E7, we used an Rb allele containing loxP sites surrounding the third exon (Rbflox) and expressed Cre using the human keratin 14 promoter to abrogate pRb expression selectively in stratified squamous epithelia. The unrecombined Rbflox allele behaves exactly as the WT Rb allele in all tested assays. After recombination, the now frameshifted Rbflox allele produces no detectable protein and mimics a germ-line Rb-null allele (17, 35). To verify the loss of pRb expression in K14CreRbflox/flox cervical epidermis, pRb immunohistochemical stains were done on either Rbflox/flox or K14CreRbflox/flox cervical epidermis from mice treated with estrogen for either 6 weeks or 6 months. At both time points, Cre expression resulted in a dramatic 99% loss of detectable pRb (95% confidence interval, 99.0-100; Fig. 1A; data not shown).
Figure 1.
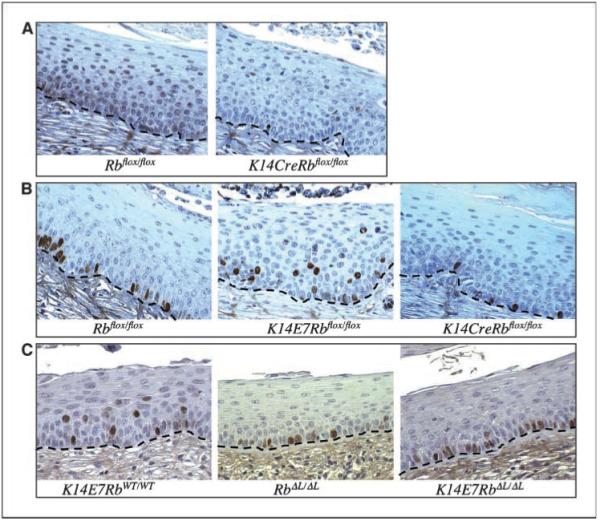
Early cervical phenotypes. Histologic cross sections of cervical epithelium from mice treated for 6 weeks with estrogen. Brown, positive staining; blue, hematoxylin counterstain. Dashed line, location of the basement membrane. A, pRb inactivation in murine cervix. pRb staining. B and C, BrdUrd incorporation in cervical epithelia. BrdUrd staining labels newly synthesized DNA. B, DNA synthesis is restricted to the lowest two cell layers in control epithelium, but E7 expression induces suprabasal DNA synthesis. Rb deletion in K14CreRbflox/flox cervix fails to reproduce the effect of E7. C, E7 fails to induce suprabasal DNA synthesis in the presence of the RbΔL allele. BrdUrd staining pattern in K14E7RbWT/WT cervix is indistinguishable from K14E7Rbflox/flox cervix (data not shown).
Cervical carcinogenesis studies
Treatment of mice with chronic, low doses of estrogen for 6 months results in severe cervical dysplasia or invasive cervical cancers in 100% of treated K14E7 mice, whereas little to no dysplasia and no cervical cancers are seen in nontransgenic controls (30). We asked if Rb inactivation was sufficient to reproduce this carcinogenic effect of E7 by treating Rbflox/flox, K14CreRbflox/flox, or K14E7Rbflox/flox mice with estrogen for 6 months and examining for the appearance of cervical abnormalities (see Materials and Methods). As expected, Rbflox/flox mice did not develop dysplasia or cervical cancers, whereas K14E7Rbflox/flox mice always developed multiple CIN2 and CIN3 lesions often accompanied by carcinoma in situ (CIS), microinvasive cancer (MIC), or large invasive cancer (LIC; Fig. 2A; Table 1). All invasive cervical cancers detected were squamous cell carcinomas.
Figure 2.

Cervical carcinogenesis studies. Histologic cross sections of cervical epithelium from mice treated with estrogen for 6 months. A, E7 expression induces severe cervical phenotypes, including cancer. Control (Rbflox/flox) cervix and pRb-deficient K14CreRbflox/flox cervix show no dysplasia or cancers. B, RbWT/WT and RbΔL/ΔL cervix do not develop dysplasia or invasive cancer, but both K14E7RbΔL/ΔL and K14E7RbWT/WT cervixes develop invasive cancers. C, pRb immunohistochemical stain (brown) of invasive cancers from K14E7RbWT/WT and K14RbΔL/ΔL mice. Arrows, nuclei of invasive cells. Blue, counterstained with hematoxylin.
Table 1.
Cervical histopathology summary for Rbflox/flox mice
| NH | CIN1 | CIN2 | CIN3 | CIS | MIC | LIC | |
|---|---|---|---|---|---|---|---|
| Rbflox/flox | 15 | ||||||
| K14E7Rbflox/flox | 1 | 8 | 3 | 2 | |||
| K14CreRbflox/flox | 12 | 2 |
NOTE: Criteria for diagnosis are described in Materials and Methods. Each number indicates the number of mice of the indicated genotype for which the indicated lesion is the worst cervical lesion detected. The lack of cervical dysplasia and cancers in K14CreRbflox/flox mice is highly significant compared with K14E7Rbflox/flox mice (P < 0.05). Abbreviation: NH, normal hyperplasia.
Surprisingly, the cervical epithelia of estrogen-treated pRb-deficient K14CreRbflox/flox mice were visually indistinguishable from that of estrogen-treated pRb-sufficient Rbflox/flox mice (Fig. 2A; Table 1). As controls, we also examined estrogen-treated K14E7K14CreRbWT/WT mice to control for the possibility that Cre has some unexpected effect on carcinogenesis independent of Rb inactivation. These mice developed CIN2, CIN3, CIS, MIC, and LIC lesions similarly to K14E7RbWT/WT mice, indicating that the Cre protein does not itself inhibit cervical cancer formation (data not shown).
To determine if pRb inactivation by E7 was necessary for the carcinogenic effects of E7 in cervix, female RbWT/WT, K14E7RbWT/WT, RbΔL/ΔL, and K14E7RbΔL/ΔL mice were also treated with estrogen for 6 months. As expected, RbWT/WT epithelium exhibited little to no dysplasia, whereas K14E7RbWT/WT epithelium always developed multiple CIN2 and/or CIN3 lesions often with additional CIS, MIC, or LIC (Fig. 2B; Table 2). The only difference seen between the RbWT/WT and RbΔL/ΔL epithelia was the occurrence of microscopic ulcerated areas in the cervical epithelium in some (~50%) of the RbΔL/ΔL mice, indicating that the RbΔL/ΔL mice may have decreased epithelial integrity and/or a defect in wound healing (data not shown). K14E7RbΔL/ΔL mice developed invasive cancers with a similar frequency to that seen in K14E7RbWT/WT mice (4 of 12 versus 5 of 14, respectively; Table 2). To determine if there was any difference in the size of the invasive cancers in K14E7RbΔL/ΔL versus K14E7RbWT/WT mice, the largest cross-sectional area of each cancer was measured. The median cancer area in K14E7RbWT/WT mice (0.12 mm2) was larger than that in the K14E7RbΔL/ΔL mice (0.067 mm2), but this difference was not statistically significant due to large variation within each genotype and the small number of cancers observed (P = 0.1416). Furthermore, we did not observe any consistent difference in tumor morphology or the degree of differentiation in tumor cells between these two genotypes.
Table 2.
Cervical histopathology summary for RbWT and RbΔL mice
| NH | CIN1 | CIN2 | CIN3 | CIS | MIC | LIC | |
|---|---|---|---|---|---|---|---|
| RbWT/WT | 14 | 1 | |||||
| K14E7RbWT/WT | 5 | 4 | 4 | 1 | |||
| RbΔL/ΔL | 11 | 1 | |||||
| K14E7RbΔLΔL | 6 | 2 | 4 |
NOTE: Criteria for diagnosis are described in Materials and Methods. Each number indicates the number of mice of the indicated genotype for which the indicated lesion is the worst cervical lesion detected. Note that all K14E7RbWT/WT mice developed multiple CIN2 and CIN3 lesions. The lack of CIN2 and CIN3 in K14E7RbΔL/ΔL mice is significant compared with K14E7RbWT/WT mice (P < 0.0001). The incidence of invasive cancers in K14E7RbΔL/ΔL mice is significantly higher than in control mice (P < 0.05) and similar to that in K14E7RbWT/WT mice (P = 0.555).
pRb immunohistochemistry was done to determine if pRb expression had been lost in the K14E7RbΔL/ΔL cancers. pRb staining was seen in the nuclei of invasive cells from all four K14E7RbΔL/ΔL cancers, whereas little to no pRb staining was seen in K14E7RbWT/WT cancers, presumably reflective of the ability of E7 to degrade pRb (Fig. 2C). This indicates that the RbΔL alleles were not lost during the formation of K14E7RbΔL/ΔL cancers. Furthermore, the fact that pRb levels are retained in the K14E7RbΔL/ΔL cancers confirms the prior observations that E7 cannot bind the mutant pRbΔL protein. Immunohistochemical detection of p16INK4a was also done on all cancers to determine if pRb function was indirectly compromised by p16 loss in the K14E7RbΔL/ΔL cancers. All cancers in both the K14E7RbWT/WT and K14E7RbΔL/ΔL mice exhibited similar levels of p16 staining, indicating that p16 loss did not substitute for pRb inactivation by E7 in the K14E7RbΔL/ΔL cancers (data not shown).
Combined, these cervical carcinogenesis studies show that pRb inactivation is not sufficient to induce cervical cancers in estrogentreated mice and that E7 is able to induce cervical cancer formation independently of pRb inactivation in the K14E7RbΔL/ΔL mice, indicating that the effects of E7 on targets other than pRb are critical for cervical carcinogenesis.
Acute cervical phenotypes
In previous work, we showed that pRb inactivation is necessary and sufficient for most of the short-term phenotypes of E7 in the cutaneous epithelium of mice (17, 19). Given the surprising results described above, we examined the cervical epithelia of younger mice to determine if pRb inactivation is able to reproduce the short-term effects of E7 in cervix as it is in cutaneous epithelia. For these analyses, mice were treated with estrogen for 6 weeks to ensure an estrus-like state in all mice, thus avoiding background variation in the degree of epithelial hyperplasia due to estrus cycling.
Suprabasal DNA synthesis
A hallmark of E7 is its ability to reprogram suprabasal cells within stratified epithelia to support DNA synthesis (36-38). To determine if pRb inactivation is necessary or sufficient for this effect, we injected mice with the nucleotide analogue BrdUrd 1 hour before sacrifice to label newly synthesized DNA. As observed in cutaneous epithelia, BrdUrd incorporation in E7-negative RbWT/WT, Rbflox/flox, or RbΔL/ΔL cervical epithelia was mostly confined to the basal cell layer, with a few BrdUrd-positive cells also observed in the parabasal layer (Fig. 1B and C, quantified in Fig. 3A and B). As expected, E7 expression in RbWT/WT or Rbflox/flox epithelia increased the frequency of BrdUrd incorporation in suprabasal cells, including cells many layers above the basement membrane (P = 0.021; Fig. 1B and C, quantified in Fig. 3A and B). In contrast, Rb inactivation by Cre in K14CreRbflox/flox epithelium did not induce any increase in suprabasal DNA synthesis compared with control mice (Figs. 1A and 3A), and E7 failed to induce suprabasal DNA synthesis in K14E7RbΔL/ΔL epithelium (Figs. 1C and 3B). These observations indicate that pRb inactivation is necessary but not sufficient for E7 to induce suprabasal DNA synthesis.
Figure 3.
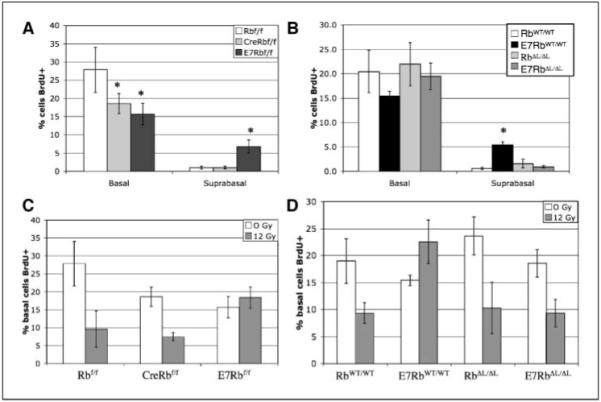
Quantitative effects on DNA synthesis. Basal and suprabasal cells from BrdUrd-stained cervical epithelia were counted as BrdUrd positive or BrdUrd negative, and the frequency of BrdUrd-positive cells was graphed. A, BrdUrd incorporation in Rbflox mice. *, P < 0.05, statistically significant change compared with Rbflox/flox. B, BrdUrd incorporation in RbWT and RbΔL mice. *, P < 0.05, statistically significant increase in suprabasal DNA synthesis in K14E7RbWT/WT mice compared with all other genotypes. C, E7 expression, but not Rb inactivation, prevents DNA damage–induced cell cycle arrest. Mice were exposed to 0 or 12 Gy ionizing radiation 24 hours before BrdUrd administration. BrdUrd staining frequency in basal cells is graphed. BrdUrd incorporation frequency is significantly reduced in irradiated Rbflox/flox and K14CreRbflox/flox mice versus unirradiated mice (P < 0.05). D, pRb inactivation is necessary for E7 to disrupt DNA damage–induced arrest. Mice were irradiated and analyzed as in (C). BrdUrd incorporation frequency is significantly reduced in irradiated RbWT/WT, RbΔL/ΔL, and K14E7RbΔL/ΔL mice versus unirradiated mice (P < 0.05).
Surprisingly, E7 expression in the K14E7Rbflox/flox mice also had the unexpected effect of reducing the frequency of BrdUrd incorporation in the basal layer cells by ~40% (P = 0.021; Fig. 3A). A similar effect was produced by Rb inactivation in the K14CreRbflox/flox mice (P = 0.021) or by E7 expression in K14E7RbWT/WT mice (P = 0.055; Fig. 3A and B). However, E7 had no effect on basal cell proliferation in the K14E7RbΔL/ΔL epithelium (P = 0.56; Fig. 3B). This counterintuitive result shows that pRb inactivation by Cre or E7 inhibits proliferation in basal cervical keratinocytes.
Inhibition of DNA damage response
E7 expression is able to drive the synthesis of damaged DNA, which may contribute to the accumulation of mutations and progression to cancer (39, 40). In the epidermis of mouse skin, pRb inactivation is necessary and sufficient to inhibit DNA damage–induced cell cycle arrest (17, 19). To determine if pRb inactivation is sufficient to block DNA damage responses in the cervix, K14CreRbflox/flox mice were exposed to ionizing radiation from a 137Cs source, injected with BrdUrd 24 hours later, and sacrificed 1 hour after injection. As expected, irradiation induced a prominent decrease in the frequency of DNA synthesis in the cervical epithelium of Rbflox/flox mice, but E7 expression prevented this response (Fig. 3C). By contrast, Rb inactivation in K14CreRbflox/flox epithelia did not inhibit this DNA damage response in the cervical epithelium (Fig. 3C). Thus, pRb inactivation is not sufficient to explain the ability of E7 to inhibit DNA damage responses in the cervix.
K14E7RbΔL/ΔL and RbΔL/ΔL mice were irradiated to determine if pRb inactivation is necessary for E7 to disrupt DNA damage–induced arrest. Irradiation decreased the frequency of BrdUrd incorporation in both of these genotypes, indicating that pRb inactivation is necessary for E7 to disrupt the DNA damage–induced cell cycle arrest response (Fig. 3D).
Induction of E2F-responsive genes
In several studies, other groups have observed functional overlap between pRb and the related pocket protein p107. These studies showed that pRb inactivation alone was necessary but not sufficient to induce phenotypes, including increased cell cycle progression, apoptosis, resistance to senescence, and retinoblastoma. Only when both pRb and p107 were inactivated were these phenotypes observed (33, 35, 41-43). Because pRb inactivation was also necessary but not sufficient for multiple phenotypes in our mice, we hypothesized that pRb loss in the cervix may be functionally compensated for by the action of other E2F regulators, resulting in little effect of Rb inactivation alone. By contrast, the effects of E7 on a variety of E2F pathway proteins, including p107, p130, p21, p27, cyclin A, and E2F1, could overcome this compensatory effect (12-14).
If Rb loss in K14CreRbflox/flox cervix is compensated for by the action of other E2F regulators that are inactivated in K14E7 cervix, greater E2F-responsive gene expression should be seen in K14E7 cervix than in K14CreRbflox/flox cervix. To test this, we looked for protein expression from two E2F-responsive genes, Mcm7 and cyclin E, by immunohistochemistry. These were also chosen because both Mcm7 and cyclin E are reported biomarkers for the detection and diagnosis of cervical abnormalities (29).
Very little cyclin E was detected in Rbflox/flox cervix, but cyclin E staining was greatly up-regulated in K14E7Rbflox/flox cervix in both basal and suprabasal cells (Fig. 4A). By contrast, loss of pRb in K14CreRbflox/flox cervix did not detectably induce cyclin E. This result shows that E7 induces cyclin E to a greater extent than can be explained by pRb inactivation alone.
Figure 4.

Expression of E2F-responsive genes. A, cervical sections were stained for cyclin E. Brown, positive staining; blue, hematoxylin counterstain. Dashed line, location of the basement membrane. B, cervical cross sections of the indicated genotypes were stained for Mcm7. K14E7RbWT/WT cervix was identical to K14E7Rbflox/flox, so is not shown.
Mcm7 staining in Rbflox/flox cervix was robust in nearly all of the basal cells and in many of the parabasal cells immediately above the basal layer. As cells differentiated further and became quiescent, Mcm7 staining diminished rapidly (Fig. 4B). In K14E7Rbflox/flox and K14E7RbWT/WT cervix, strong Mcm7 staining was observed in all suprabasal cell layers. Interestingly, though, E7 also increased the frequency of negatively staining basal cells, consistent with E7 having caused a reduction in the proliferative index in this compartment as scored by BrdUrd incorporation (Fig. 4B). In K14CreRbflox/flox cervix, an intermediate phenotype was observed. Mcm7 staining was clearly increased in suprabasal cells compared with Rbflox/flox mice, and sporadic Mcm7-negative basal cells were observed. However, the intensity of suprabasal staining was consistently diminished compared with K14E7Rbflox/flox mice in the uppermost epithelial cell layers (Fig. 4B). This suggests that Mcm7 induction by E7 may result largely from pRb inactivation, in contrast to the result with cyclin E. To test this further, we examined Mcm7 staining in K14E7RbΔL/ΔL cervix. In RbΔL/ΔL epithelium, Mcm7 staining was very similar to that seen in RbWT/WT or Rbflox/flox epithelium (Fig. 4B). K14E7RbΔL/ΔL cervix showed much greater suprabasal Mcm7 staining than that seen without E7, although staining intensity in the uppermost layers was still less than in K14E7RbWT/WT (Fig. 4B). This indicates that E7 can induce Mcm7 expression both via pRb inactivation and by another mechanism. In sum, the Mcm7 and cyclin E expression data show that different E2F-responsive promoters can be activated by different mechanisms and that E7 activates E2F-responsive gene expression both via pRb inactivation and via another mechanism.
Cervical cancer in the absence of CIN
One other striking observation was made in the course of these studies. In the K14E7RbΔL/ΔL mice treated with estrogen for 6 months, we observed invasive cervical cancers in the absence of noninvasive dysplastic CIN or CIS lesions, which are commonly thought to be the precursors of invasive cancer. Although four of the 12 K14E7RbΔL/ΔL mice developed invasive cancers, we did not detect any CIN2, CIN3, or CIS lesions in any of the mice in spite of analyzing every tenth 5 μmol/L section throughout the entire thickness of each cervix (Fig. 5; Table 2). In addition, we found no evidence for dysplasia proximal to the tumors. No dysplasias could be observed either in the regions directly neighboring the invasive cancers in sections, in which the cancers were present, or in serial sections surrounding the cancers (Fig. 5; data not shown). In contrast, 100% of K14E7RbWT/WT mice treated with estrogen for 6 months developed multiple CIN2 and CIN3 lesions in the cervix, including those mice that developed invasive carcinoma (Fig. 5; Table 2). Thus, whereas cervical cancers in the K14E7RbWT/WT mice arose in the context of highly dysplastic epithelium, cancers in the K14E7RbΔL/ΔL mice arose in the context of otherwise histopathologically normal epithelium. The implications of this finding are discussed below.
Figure 5.
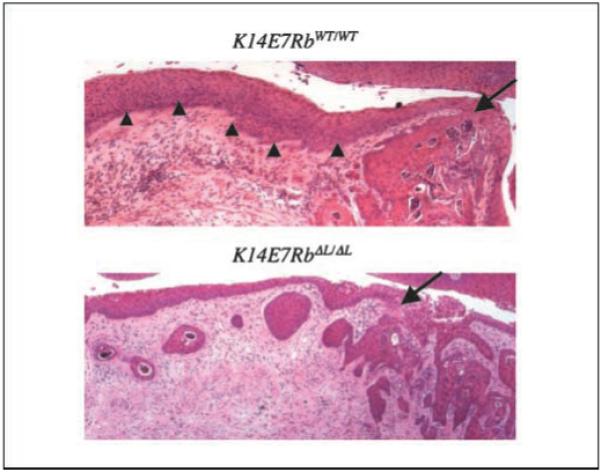
Absence of CIN lesions in K14E7RbΔL/ΔL cervix. Representative sections of K14E7RbWT/WT and K14E7RbΔL/ΔL cervixes. Arrows, sites of cancer invasion into the dermis. In K14E7RbWT/WT cervix, cancer arises from epithelium containing many dysplastic changes (arrowheads), whereas in K14E7RbΔL/ΔL cervix, cancer is observed without any dysplastic changes elsewhere in the cervical epithelium.
Discussion
In this study, we conducted the first genetic analysis of the molecular mechanisms of E7 function in intact cervix. Our analysis indicates that the effects of E7 on targets other than pRb are critical for both the short-term and tumorigenic consequences of E7 expression and that cervical cancers can develop independently of CIN lesions.
pRb inactivation in cervical dysplasia and neoplasia
As described previously (30), we saw that, after 6 months of estrogen treatment, 100% of K14E7 mice developed numerous CIN2 and CIN3 lesions and many developed CIS and/or invasive cancers (Fig. 2; Tables 1 and 2). Surprisingly, Rb deletion in the cervix of estrogen-treated K14CreRbflox/flox did not induce any CIN2, CIN3, CIS, or invasive lesions (Fig. 2A; Table 1). Thus, pRb inactivation alone cannot explain the ability of E7 to induce cervical dysplasia or invasive cancers.
Invasive cancers were observed in K14E7RbΔL/ΔL cervix at a similar frequency to that seen in K14E7RbWT/WT cervix (Table 2), and there were no consistent differences in cancer size or morphology between these genotypes (Fig. 2; data not shown). pRb expression was maintained in the K14E7RbΔL/ΔL cancers, showing that carcinogenesis in these mice did not require mutational loss of the RbΔL alleles (Fig. 2C). Furthermore, immunohistochemical staining for p16INK4a detected p16 in all the cancers in both K14E7RbWT/WT and K14E7RbΔL/ΔL mice (data not shown), indicating that pRb function was not compromised indirectly in these cancers via loss of p16 expression. These data do not imply that pRb inactivation is dispensable for cervical carcinogenesis because partial defects in pRb function in the mutant RbΔL allele potentially cooperate with the effects of E7 on other targets to induce cancers in K14E7RbΔL/ΔL cervix. This likelihood is reinforced by both In vivo and in vitro observations. In vivo, we observed that RbΔL/ΔL mice often developed ulcerations of the cervical and vaginal epithelium in estrogen-treated mice, implying that the RbΔL/ΔL cervix may have reduced epithelial integrity, defective wound healing, or some other defect. in vitro, pRbΔL exhibited diminished transcriptional repression function compared with WT pRb, showing that this mutant allele is defective in some aspects of pRb function, likely due to failure to bind LxCxE motif-containing cellular proteins (34). More recently RbΔL/ΔL mouse embryo fibroblasts were found to exhibit aneuploidy associated with defective centromere methylation and lagging mitotic chromosomes (32). Thus, it remains reasonable to hypothesize that defects in at least some functions of pRb contribute to cervical carcinogenesis. Regardless, our data with the conditional null Rb mice clearly show that other molecular activities of E7 make critical contributions to cervical carcinogenesis.
pRb in acute cervical phenotypes
In previous work, we showed that pRb inactivation is necessary and sufficient for most of the short-term effects of E7 on murine skin and ear epithelium (17, 19). In contrast, our analysis of K14E7RbΔL/ΔL and K14CreRbflox/flox mice showed that pRb inactivation is necessary but not sufficient to induce suprabasal DNA synthesis and to block DNA damage–induced cell cycle arrest in the cervical epithelium (Figs. 1 and 3). Thus, the consequences of Rb inactivation are very different in cervix than in cutaneous epithelia, indicating that the mechanisms of E7 function may differ in one versus another epithelial tissue. This may be important to human carcinogenesis because HPV-16 is associated with cancers in multiple epithelial tissues (3, 44). Future studies of E7 function in other physiologically relevant sites, such as the oral cavity, will determine if the mechanisms of E7 function also vary between different mucosal epithelia.
Multiple mechanisms of E2F regulation by E7
Analysis of the E2F-responsive cyclin E and Mcm7 genes showed that E7 induces E2F-responsive genes via multiple mechanisms, such that full induction of E2F-responsive genes requires both pRb inactivation and some other activity or activities of E7 (Fig. 4). Furthermore, Mcm7 was induced in K14CreRbflox/flox cervix but cyclin E was not (Fig. 4), implying that different E2F-responsive genes can be differentially regulated, which may explain why multiple mechanisms of E2F activation are necessary for full E7 function. Other components of the E2F pathway bound by E7 in vitro include p107, p130, p21, p27, cyclin A/cdk2, and E2F1 itself (12-14), making these excellent candidates for future studies of E7 function.
More specifically, published studies of p107 function suggest that p107 may be of particular importance. The observation that pRb inactivation is necessary but not sufficient for many cervical phenotypes is strongly reminiscent of studies, which have shown that p107 can functionally compensate for the loss of pRb under some circumstances. These studies identified multiple phenotypes, which do not occur following pRb or p107 inactivation alone but are observed only when both genes are simultaneously inactivated (35, 41-43). If p107 is similarly compensating for the loss of pRb in K14CreRbflox/flox cervix, then E7-mediated p107 inactivation could explain why pRb inactivation is not sufficient to induce phenotypes in the cervix. Unfortunately, K14CreRbflox/floxp107−/− mice do not survive to adulthood (S.B. and P.F.L.; data not shown; ref. 45), so more refined mouse model studies will be necessary to test this hypothesis.
Cervical carcinogenesis in the absence of detectable CIN
In carrying out this study, we made the unexpected observation that cervical cancers can develop in the absence of any detectable, coexisting CIN or CIS lesions. Specifically, whereas cancers in K14E7RbWT/WT mice always developed in the context of a severely dysplastic epithelium, cancers in K14E7RbΔL/ΔL mice developed in the absence of detectable, coexisting CIN2, CIN3, or CIS lesions (Fig. 5; Table 2). Importantly, the incidence and size of invasive cancers were similar in K14E7RbΔL/ΔL and K14E7RbWT/WT mice, arguing that the rate at which the cancers arose is similar. These unexpected results suggest that the profuse cervical dysplasia and the formation of cervical cancer seen in K14E7 mice on the WT Rb background are separable events.
Although both cervical dysplasia and cervical cancer are associated with HPV infection (46, 47) and the likelihood of both increase over time (47), there is no direct evidence that the clinically apparent dysplastic lesions are obligate intermediates to cervical cancer. Our mouse model data would argue otherwise. That malignant cancer can arise without overt benign disease is not unprecedented; in chemical carcinogenesis studies, invasive skin carcinomas developed in p53-null mice without prior development of detectable benign skin papillomas (48). It remains to be determined in either model how these cancers arise.
One hypothesis is that, in the K14E7RbΔL/ΔL mice, malignant conversion arises so quickly as to obscure the preexistence of dysplastic lesions. However, the fact that the incidence and size of cancers in the K14E7RbΔL/ΔL mice are no greater than that seen in K14E7RbWT/WT mice argues against this possibility. Furthermore, the cancers that arise on the K14E7RbΔL/ΔL mice are not sufficiently large as to obscure an ability to see underlying dysplastic disease elsewhere in the cervix; yet, such dysplasia was not apparent in these mice.
An alternative hypothesis that we favor is that both the profuse dysplasia and the stochastic malignancies observed in the K14E7RbWT/WT mice arise from a common progenitor tumor cell. However, different derivative lineages develop from this common progenitor, some of which lead to dysplasia, whereas others lead to frank cancer without clinically apparent precursor benign disease. Here, one has to argue further that the defects in Rb function caused by the interaction of E7 with WT pRb (and therefore absent in the K14E7RbΔL/ΔL mice) contribute preferentially to the lineages that cause dysplasias that are unable or unlikely to progress to frank malignancy. This hypothesis is consistent with the observation that removal of dysplastic lesions in women reduces significantly the risk of subsequent cancer, assuming that the derivative cells that can cause frank cancers to reside proximal to the dysplasia; this is a reasonable prediction given the focal nature of HPV infections within the cervix.
There are several important implications to the favored hypothesis. First is that treatments shown to eliminate dysplastic lesions may or may not affect frank cancers, depending on whether the therapy targets a feature shared by both the malignant as well as dysplastic tumor lineages or a feature distinct only to the latter. Likewise, genetic/epigenetic alterations found in dysplastic lesions may or may not be predictive of alterations that contribute to frank cancer, depending on whether the change arose at an early stage in the genesis of the common progenitor cell or at a later stage in the genesis of the derivative lineage that gave rise specifically to the dysplasia. Further investigation is needed to determine whether this hypothesis, which is derived from our study of HPV-associated cancers in a mouse model, is relevant to understanding the genesis of human cervical cancer.
Acknowledgments
Grant support: NIH grants CA64402, CA098428, and CA22443; NIH grant T32 GM00721 (S. Balsitis); and University of Wisconsin Comprehensive Cancer Center grant P30 CA014520. F. Dick is a research scientist of the Canadian Cancer Society through an award from the National Cancer Institute of Canada.
We thank the University of Wisconsin Comprehensive Cancer Center histology facility (Madison, WI) for expert histologic support and Ruth Sullivan for assistance with histopathology.
References
- 1.zur Hausen H. Papillomavirus infections—a major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 2.Walboomers J, Jacobs M, Manos M, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Gillison ML. Human papillomavirus-associated head and neck cancer is a distinct epidemiologic, clinical, and molecular entity. Semin Oncol. 2004;31:744–54. doi: 10.1053/j.seminoncol.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Durst M, Kleinheinz A, Hotz M, Gissman L. The physical state of human papillomavirus type 16 DNA in benign and malignant genital tumors. J Gen Virol. 1985;66:1515–22. doi: 10.1099/0022-1317-66-7-1515. [DOI] [PubMed] [Google Scholar]
- 5.Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc Natl Acad Sci U S A. 1995;92:1654–8. doi: 10.1073/pnas.92.5.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munger K, Baldwin A, Edwards KM, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phelps WC, Bagchi S, Barnes JA, et al. Analysis of trans activation by human papillomavirus type 16 E7 and adenovirus 12S E1A suggests a common mechanism. J Virol. 1991;65:6922–30. doi: 10.1128/jvi.65.12.6922-6930.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chellappan S, Kraus VB, Kroger B, et al. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A. 1992;89:4549–53. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–91. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones DL, Munger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997b;71:2905–12. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4. [PubMed] [Google Scholar]
- 12.He W, Staples D, Smith C, Fisher C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J Virol. 2003;77:10566–74. doi: 10.1128/JVI.77.19.10566-10574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang SG, Lee D, Kim J, Seo T, Choe J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. J Biol Chem. 2002;277:2923–30. doi: 10.1074/jbc.M109113200. [DOI] [PubMed] [Google Scholar]
- 14.Munger K, Basile JR, Duensing S, et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888–98. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- 15.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 16.Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 1998;14:223–9. doi: 10.1016/s0168-9525(98)01470-x. [DOI] [PubMed] [Google Scholar]
- 17.Balsitis SJ, Sage J, Duensing S, Munger K, Jacks T, Lambert PF. Recapitulation of the effects of the HPV-16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol Cell Biol. 2003;23:9094–103. doi: 10.1128/MCB.23.24.9094-9103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slebos R, Lee MH, Plunkett BS, et al. p53-dependent G1 arrest involves pRb-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc Natl Acad Sci U S A. 1994;91:5320–4. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balsitis SJ, Dick F, Farrell L, et al. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J Virol. 2005;79:11392–402. doi: 10.1128/JVI.79.17.11392-11402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jewers RJ, Hildebrandt P, Ludlow W, Kell B, McCance D. Regions of human papillomavirus type 16 E7 oncoprotein required for immortalization of human keratinocytes. J Virol. 1992;66:1329–35. doi: 10.1128/jvi.66.3.1329-1335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam E, Morris J, Davies R, Crook T, Watson R, Vousden KH. HPV-16 E7 oncoprotein deregulates B-myb expression: correlation with targeting of p107/E2F complexes. EMBO J. 1994;13:871–8. doi: 10.1002/j.1460-2075.1994.tb06330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massimi P, Pim D, Banks L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J Gen Virol. 1997;78:2607–13. doi: 10.1099/0022-1317-78-10-2607. [DOI] [PubMed] [Google Scholar]
- 23.Zwerchke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen-Durr P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci U S A. 1999;96:1291–6. doi: 10.1073/pnas.96.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Helt A, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75:6737–47. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zwerchke W, Mannhardt B, Massimi P, et al. Allosteric activation of acid α-glucosidase by the human papillomavirus E7 protein. J Biol Chem. 2000;275:9534–41. doi: 10.1074/jbc.275.13.9534. [DOI] [PubMed] [Google Scholar]
- 26.Helt A, Funk JO, Galloway DA. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J Virol. 2002;76:10559–68. doi: 10.1128/JVI.76.20.10559-10568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antinore M, Birrer M, Patel D, Nader L, McCance D. The human papillomavirus type 16 E7 gene product interacts with and trans-activates the AP1 family of transcription factors. EMBO J. 1996;15:1950–60. [PMC free article] [PubMed] [Google Scholar]
- 28.Duensing S, Munger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003;77:12331–5. doi: 10.1128/JVI.77.22.12331-12335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brake T, Connor J, Petereit D, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–80. [PubMed] [Google Scholar]
- 30.Riley R, Duensing S, Brake T, Munger K, Lambert PF, Arbeit J. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–71. [PubMed] [Google Scholar]
- 31.Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene. J Virol. 1996;70:1873–81. doi: 10.1128/jvi.70.3.1873-1881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isaac CE, Francis SM, Martens AL, et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol. 2006;26:3659–71. doi: 10.1128/MCB.26.9.3659-3671.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J, Russo A, Pavletich N. Structure of the retinoblastoma tumor-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–65. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 34.Dick F, Sailhamer E, Dyson N. Mutagenesis of the pRb pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol Cell Biol. 2000;20:3715–27. doi: 10.1128/mcb.20.10.3715-3727.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 36.Flores ER, Allen-Hoffman L, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74:6622–31. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gulliver GA, Herber RL, Liem A, Lambert PF. Both conserved region 1 (CR1) and CR2 of the human papillomavirus type 16 E7 oncogene are required for induction of epidermal hyperplasia and tumor formation in transgenic mice. J Virol. 1997;71:5905–14. doi: 10.1128/jvi.71.8.5905-5914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995;9:2335–49. doi: 10.1101/gad.9.19.2335. [DOI] [PubMed] [Google Scholar]
- 39.Song S, Gulliver GA, Lambert PF. Human papillomavirus type 16 E6 and E7 oncogenes abrogate radiation-induced DNA damage responses in vivo through p53-dependent and p53-independent pathways. Proc Natl Acad Sci U S A. 1998;95:2290–5. doi: 10.1073/pnas.95.5.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demers GW, Foster SA, Halbert CL, Galloway DA. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci U S A. 1994;91:4382–6. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robanus-Maandag E, Dekker M, van der Valk M, et al. p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev. 1998;12:1599–609. doi: 10.1101/gad.12.11.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sage J, Mulligan GJ, Attardi LD, et al. Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 2000;14:3037–50. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dannenberg J, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G1 control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 2000;14:3051–64. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steenbergen RD, de Wilde J, Wilting SM, Brink AA, Snijders PJ, Meijer CJ. HPV-mediated transformation of the anogenital tract. J Clin Virol. 2005;32:525–33. doi: 10.1016/j.jcv.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 45.Ruiz S, Santos M, Segrelles C, et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004;131:2737–48. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- 46.Herrero R, Castle P, Schiffman M, et al. Epidemiologic profile of type-specific human papillomavirus infection and cervical neoplasia in Guanacaste, Costa Rica. J Infect Dis. 2005;191:1787–9. doi: 10.1086/428850. [DOI] [PubMed] [Google Scholar]
- 47.Schiffman M, Brinton L. The epidemiology of cervical carcinogenesis. Cancer. 1997;76:1888–901. doi: 10.1002/1097-0142(19951115)76:10+<1888::aid-cncr2820761305>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 48.Kemp C, Donehower L, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813–22. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]