

Summary
The BCL6 transcriptional repressor is the most frequently involved oncogene in diffuse large B cell lymphoma (DLBCL). We combined computer-aided drug design with functional assays to identify low molecular weight compounds that bind to the corepressor binding groove of the BCL6 BTB domain. One such compound disrupted BCL6/corepressor complexes in vitro and in vivo, and was observed by X-ray crystallography and NMR to bind the critical site within the BTB groove. This compound could induce expression of BCL6 target genes and kill BCL6-positive DLBCL cell lines. In xenotransplantation experiments, the compound was non-toxic and potently suppressed DLBCL tumors in vivo. The compound also killed primary DLBCLs from human patients.
Introduction
BCL6 is the most frequently involved oncogene in diffuse large B cell lymphomas (DLBCLs) (Ye, 2000). In normal lymphoid biology BCL6 is required for mature B-cells to form germinal centers (GCs), which are cellular compartments dedicated to the affinity maturation of antibodies (Dent et al., 1997; Ye et al., 1997). In order to generate clonal diversity of cells expressing the greatest possible variations in their immunoglobulin coding sequence, B cells must acquire the ability to simultaneously tolerate rapid proliferation and genomic recombination. BCL6 facilitates this phenotype of physiological genomic instability by repressing genes involved in sensing DNA damage or their downstream checkpoints (Phan and Dalla-Favera, 2004; Phan et al., 2005; Ranuncolo et al., 2007; Ranuncolo et al., 2008). Mice engineered to constitutively express BCL6 in GC B cells develop DLBCL similar to the human disease (Baron et al., 2004; Cattoretti et al., 2005). BCL6 is also expressed constitutively in the majority of patients with aggressive B cell lymphomas most often due to translocations of heterologous promoter elements or promoter point mutations in the BCL6 locus (Ye, 2000). BCL6 loss of function, mediated by delivery of shRNA or peptide inhibitors, can kill DLBCL cells, demonstrating that BCL6 is required for survival of lymphoma cells and could be an excellent therapeutic target (Cerchietti et al., 2008; Cerchietti et al., 2009; Phan and Dalla-Favera, 2004; Polo et al., 2004; Polo et al., 2007).
BCL6 is a member of the BTB/POZ family of transcription factors (Stogios et al., 2005). The BCL6 BTB domain has potent autonomous repressor activity, which is dependent on its ability to recruit the SMRT, N-CoR and BCOR corepressor to an exposed surface groove formed at the interface of the two chains in the BTB dimer (Ahmad et al., 2003; Ghetu et al., 2008). These corepressors interact with micromolar affinity to the lateral groove of the BCL6 BTB domain via a 17-residue BCL6 binding domain (BBD) (Ahmad et al., 2003; Ghetu et al., 2008). The residues that define the BCL6 lateral groove surface are not conserved in other transcription factors from the BTB family (Ahmad et al., 2003; Ghetu et al., 2008; Stogios et al., 2007). Notably, a cell-penetrating BCL6 peptide inhibitor (BPI) containing the SMRT BBD inhibits the transcriptional repressor activity of BCL6 but had no effect on other BTB repressors (Polo et al., 2004). Moreover, BPI induces the upregulation of critical BCL6 target genes including ATR, CHEK1 and TP53 in DLBCL cell lines (Cerchietti et al., 2008; Cerchietti et al., 2009; Ranuncolo et al., 2007; Ranuncolo et al., 2008).
Our goal was to use structure-based strategies to identify small molecules that specifically disrupt the activity of BCL6 by blocking its interaction with its corepressors BCor, N-CoR and SMRT. In addition, we wanted to determine whether these compounds would reactivate BCL6 target genes and selectively kill BCL6-dependent lymphoma cells in vitro and in vivo.
Results
Computer-aided drug design identifies low-molecular weight compounds with the potential to bind to the BCL6 lateral groove
Structural analyses of the BCL6-BBD complex (Ahmad et al., 2003; Ghetu et al., 2008) indicated that the region of the lateral groove associated with SMRT residues 1423-1428 had a high complexity and density of intermolecular contacts between the protein and BBD peptide (Figure 1A). Alanine scanning mutagenesis confirmed that all six of these BBD residues are required for the stability of the complex (Ghetu et al., 2008). Therefore, this region was selected for the application of computer-aided drug design (CADD) to identify low-molecular weight compounds with the potential to bind in the BCL6 lateral groove. The first step of CADD involved identification of putative small molecule binding sites using the SPHGEN module in the program DOCK (Kuntz, 1992). The resulting sphere set was used to direct a CADD screen of a database of over 1,000,000 commercially available compounds. Screening involved two rounds of docking of the compounds into the putative binding site with final compound selection based on maximizing both chemical diversity (Butina, 1999) and physical properties associated with drug-like characteristics (Lipinski, 2000). In the first round of CADD, 50,000 compounds having the most favorable N-normalized vdW (van der Waals) attractive energy were selected. This normalization procedure(Pan et al., 2003) centers the median of molecular weight (MW) distribution of selected compounds to approximately 300 daltons, consistent with the known MW distribution of drug-like molecules (Figure S1A) (Lipinski, 2000). In addition, the use of the vdW attractive energy for ranking eliminates compounds that do not sterically complement the putative binding pocket (Huang et al., 2004). In the second round of docking, additional conformations of the protein were included to partially account for the conformational flexibility of the binding pocket. These additional conformations were obtained from a molecular dynamics (MD) simulation of the apo (unliganded) BCL6 BTB domain. Included in Table S1 are RMSD values of residues in the binding region for the different conformations. This more rigorous second-step screening yielded a final ranking of 1000 compounds as evaluated using the N2/5-normalized total interaction energy (selected from the initial 50,000 compounds) (Figure S1B). The 1000 compounds were then subjected to chemical diversity clustering which yielded approximately 100 groups, consisting of compounds with related chemical structures. One or two compounds were then selected from each group based on maximizing adherence to Lipinski’s Rule of Five (Lipinski, 2000), yielding a total of 199 compounds. The CADD-identified inhibitors were predicted to bind in the vicinity of the targeted binding site, assuming a variety of orientations within the BCL6 lateral groove. 100 of the final 199 compounds were available from commercial vendors for experimental testing. The Figure 1B shows the CADD-selected compounds docked to the BCL6 lateral groove pocket along with the green transparent spheres used to define the putative binding site.
Figure 1. The structure of the BCL6 BTB domain.
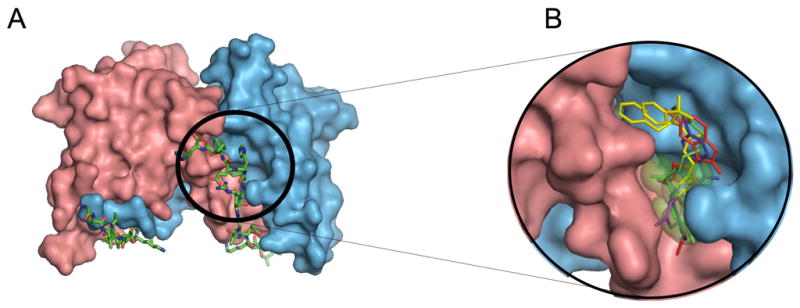
A: Structure of the 2:2 complex between the BCL6 BTB domain and the SMRT BBD peptide. The two chains of the BTB domain are shown in pink and blue and the two SMRT peptides are shown in stick representation with green carbon atoms. The SMRT 1423-1428 region is circled. B: View of the selected compounds docked to the BCL6 lateral groove pocket predicted by the CADD procedure along with the putative binding site represented by green transparent spheres. The compounds are stick representations based on the following color scheme: 28 (blue), 72 (magenta), 79 (red), 53 (yellow) and 55 (green). See also Figure S1 and Table S1.
Compounds identified by CADD can inhibit BCL6 BTB domain repressor activity
To determine whether compounds selected by CADD could inhibit the repressor activity of the BCL6 BTB domain, reporter assays were performed in which a GAL4 DNA binding domain (DBD)-BCL6BTB fusion construct was co-transfected with a luciferase reporter plasmid containing GAL4 DBD binding sites, (GAL4)5TK-Luc. The fold change in repressor activity of the BCL6 BTB domain was determined in the presence of either the CADD-selected compounds at a 50 μM concentration or vehicle, and was controlled for nonspecific effects on transcription by normalizing to the activity of the GAL4-DBD alone. Ten compounds consistently attenuated BCL6BTB-mediated transcriptional repression compared to vehicle control (DMSO) (Figure 2A) and were thus candidate inhibitors of corepressor binding to the BCL6 lateral groove. Structures and properties of the selected compounds are shown in Figure S2A and Table S2. Compounds structurally similar to the active compounds were identified based on the Tanimoto Similarity Index (Butina, 1999) and subjected to biological testing, with the exception of compound 46 analogs, which were not available. In BCL6 BTB reporter assays, several of the similar compounds retained the activity of the parental molecule, with the most potent derivatives belonging to the series 28 and series 79 families (Figure 2B). In addition, active analogs were also present for families 72, 53 and 55, suggesting that these compounds may also be suitable for further investigation. The 79 series compounds were selected for further study because this group contained the largest number of active compounds, with 11/13 compounds attenuating transcriptional repression by greater than 50%. One of the 79-series compounds (79-40) with the smallest effect on transcriptional de-repression was selected to use as a chemical control for selected subsequent experiments (Figure S2B).
Figure 2. CADD selection identifies BCL6 inhibitor compounds.

A: Reporter assays were performed to test the impact of CADD compounds on the repressor activity of a GAL4-DBD-BCL6BTB fusion construct, compared to their effect on GAL4-DBD alone. The Y-axis shows the relative percent of repression mediated by GAL-BCL6BTB in the presence of vehicle (V, which is set as 100%), compounds with activity (black bars and 79 as red bar) or selected inactive compounds (grey bars). Compounds were tested at 50 μM in DMSO. Experiments were performed in triplicate with compounds tested in quadruplicate. B: Reporter assay as in (A) was performed using families of compounds related to those in (A). The X-axis lists the compounds according to which parental compound they are similar to. The 79 series is shown in black and 79-6 is shown in red. Compounds were tested at 100 μM in DMSO. Error bars represent the SEM for replicates. See also Figure S2 and Table S2.
79-6 binds to a pocket in the lateral groove of the BCL6 BTB domain
Compound 79-6 was synthesized and purified from commercial sources. The purity and identity of compound 79-6 were verified by 1H NMR, elemental analysis and LC/MS (Figure S3). We used x-ray crystallography to determine the binding location of 79-6 on the BCL6BTB domain. Crystals of BCL6BTB were soaked with 79-6 and we observed that the crystals changed to the orange color that is characteristic of the 79 series (Figure 3A). A 2.3 Å diffraction dataset was collected on the soaked crystals, and after several rounds of refinement, difference density |Fo − Fc| maps were used to locate the positions of 79-6. Crystallographic statistics are provided in Table S3. The strong positive peak for the 79-6 bromine atom was used to anchor the molecule, but the density was not as strong at the more exposed end of the molecule (Figure 3B). As anticipated based on previous functional assays, two molecules of 79-6 bind at equivalent positions in the lateral grooves on either side of the BCL6 BTB dimer. The indolazine ring of 79-6 is positioned within a shallow preformed pocket near Tyr-58 that is empty in the apo structure of BCL6 BTB, and is occupied by SMRT residues His-1426/Ile-1428, or BCOR Trp-509/Val-511 in the two BBD corepressor structures (Ghetu et al., 2008). Thus, the presence of 79-6 in the binding pocket would prevent the interaction of BCL6 with BBD-corepressor residues that are essential for complex formation. The 79-6 indolazine ring is sandwiched between Tyr-58 from one BCL6 chain and Asn-21 and Arg-24 from the other chain (Figure 3C,D). Leu-25 lines a hydrophobic pocket that accepts the bromine from the 79-6 indolazine ring, while the main chain carbonyl oxygen of Met-51 and the guanidinium group of Arg-28 form polar interactions with the compound. The indolazine ring anchors the molecule in the lateral groove pocket of the protein, and the electron density becomes progressively weaker towards the carboxylic acid tail. The strength of the electron density correlates with the degree of burial of the compound from the fused ring system to the tail. Nevertheless, MD simulations have indicated the presence of interactions between the carboxylic acid groups and the guanidinium groups of Arg 24 and Arg 28, but these were very highly dynamic which is consistent with the weak electron density in this region (results not shown).
Figure 3. Crystallography of the BCL6BTB/79-6 complex.

A: The crystal on the right was soaked with compound 79-6 prior to transferring it to a well with no added compound. The crystal on the left was not soaked in 79-6. B: |Fo′Fc| difference electron density contoured at 4 s (red mesh) and 2 s (grey mesh) in the lateral groove site of the BCL6BTB dimer prior to the inclusion of the compound in the model. The stick model represents the final refined position of 79-6. The bromine atom of the compound is located in the region of highest electron density C: Compound 79-6 is shown in a space filling representation with green carbon atoms, and binds in the lateral groove of the BTB dimer. D: Details of the molecular interactions between 79-6 and the BCL6 BTB domain. The bromine atom of the indolazine ring of 79-6 is colored brown. Residues Asn-21, Arg-24, Leu-25 and Arg-28 are from one chain, and residues labeled with primes (Gly-55′, Tyr-58′ and Ser-59′) are from the other BCL6 chain. See also Figure S3 and Table S3.
Nuclear magnetic resonance (NMR) spectroscopy was used to characterize the binding of 79-6 to the BCL6 BTB domain in solution. BCL6BTB gave a well-dispersed 15N heteronuclear single quantum coherence (HSQC) spectrum, and assignments were obtained for 82 of the 121 observed main chain resonances using triple resonance experiments. A series of spectra were then measured with increasing concentrations of 79-6, and we observed the selective shifting of a subset of resonances that cluster near the lateral groove pocket (Figure 4 and Figure S4). To further confirm that 79-6 interacts with the lateral groove of the BTB domain, NMR HSQC measurements were performed on BCL6BTB in the presence of a two-fold molar excess of unlabelled SMRT BBD peptide (Figure S4). Notably, resonances shifted by 79-6 were also shifted by the SMRT BBD peptide indicating that 79-6 is binding to the targeted region of the protein in solution (Figure S4).
Figure 4. Nuclear magnetic resonance spectroscopy of BCL6BTB with 79-6.

A,B: Selected regions of the 15N-1H HSQC spectra of BCL6BTB with increasing amounts of 79-6. The red spectrum was obtained in the absence of the compound. The arrows indicate shifted resonances. C: Residues whose amide NMR resonances shifted by 0.05–0.09 ppm are colored light pink, 0.10–0.14 ppm are colored medium red, and >0.15 ppm are colored dark red. The surface of the BCL6 BTB dimer is colored grey and white. See also Figure S4.
As expected, the smaller 79-6 molecule affected only a subset of the spectra shifted by the significantly larger BBD. The shifts of five strongly affected resonances with increasing concentrations of 79-6 were fit to a binding isotherm, resulting in a Kd of 138 ± 31 μM (Figure 5). We also measured the displacement of a fluorescently labeled SMRT-BBD peptide from the BCL6BTB corepressor complex by a fluorescence polarization competitive binding assay. Under these assay conditions, compound 79-6 had an IC50 value of 212 μM, which corresponds to a Ki of 147 μM while compound 79-40 had a Ki > 1 mM (Figure S5). In addition, 79-6 has a ligand efficiency of 0.21, which is comparable to other small molecule inhibitors of protein-protein interactions (Wells and McClendon, 2007).
Figure 5. Binding curves for 79-6 to the BCL6BTB.

Binding curves for five resonances that shifted (Y-axis, in ppm) upon the addition of increasing concentrations of 79-6 (X-axis). The fitting resulted in a Kd of 138 ± 31 μM. See also Figure S5.
79-6 specifically inhibits BCL6 but not other BTB-ZF proteins
Although the residues lining the lateral groove of BCL6 are not conserved in other BTB domains, it was still possible that 79-6 may nonspecifically affect the transcriptional repression of other BTB-containing proteins. To determine whether this was the case we performed reporter assays that included other BTB-zinc finger repressors in the same family as BCL6. GAL4-DBD fusions of the BTB domains of BCL6, Kaiso, HIC1 (hypermethylated in cancer 1) and PLZF (promyelocytic zinc finger) were examined for their ability to repress the (GAL4)5TK-Luc reporter in the presence of 79-6 (Figure 6A). 79-6 readily attenuated BCL6-mediated repression but had little or no effect on the other BTB domains. These assays demonstrate that 79-6 mediates specific de-repression of BCL6 transcriptional activity, but not that of other BTB containing transcriptional repressors.
Figure 6. 79-6 specifically inhibits BCL6 repressor activity.

A: Reporter assay comparing the effect of 50 μM of 79-6 on the repressor activity of GAL4-BCL6BTB, GAL4-KaisoBTB, GAL4-HIC1BTB or GAL4-PLZFBTB, compared to GAL4-DBD alone. The Y-axis shows fold repression in the presence of vehicle (black bars) compared to 79-6 (grey bars). B: Quantitative chromatin immunoprecipitation was performed in SU-DHL6 DLBCL cells to detect binding of BCL6, N-CoR and SMRT to the ATR promoter in the presence of vehicle (black bars) or 125 μM of 79-6 (grey bars). The Y-axis shows fold difference in enrichment of ATR in the various conditions. C: mRNA abundance of the BCL6 target genes ATR, TP53, CD69, p21 and CD44, and the non-target genes B2M, PCNA and HPRT was measured in SU-DLH6, SU-DHL4 (both BCL6 dependent) and Toledo (BCL6 independent) DLBCL cell lines exposed to vehicle or 50 μM of 79-6 for 8 h. The Y-axis shows 79-6 mediated fold induction of each gene normalized to RPL13A and relative to vehicle (DMSO). D: Dose-effect curves for a panel of 8 DLBCL cell lines exposed to increasing concentrations of 79-6 for 48 h. OCI-Ly7, OCI-Ly1, SU-DHL6, SU-DHL4, OCI-Ly10 and Farage are BCL6-dependent, and Toledo and OCI-Ly4 are BCL6-independent negative controls. Cell viability was determined by luminescent ATP quantization. The value between parentheses represent the drug concentration (in mM) that inhibits the growth of cell lines by 50% compared to vehicle (GI50). Assays were performed in biological triplicates, with the averages of these plotted. Error bars represent the SEM for replicates. See also Figure S6.
79-6 disrupts BCL6 transcriptional complexes and reactivates BCL6 target genes
BCL6 mediated repression of the ATR gene has been suggested to contribute to lymphomagenic actions of BCL6 and is dependent on the BCL6 lateral groove (Ranuncolo et al., 2007). To determine whether the BCL6 repression complex on the ATR promoter could be disrupted by 79-6, we exposed a BCL6-positive DLBCL cell line (SU-DHL6) to 79-6 at 125 μM or vehicle for two hours and then performed chromatin immunoprecipitations for BCL6, SMRT and N-CoR, followed by real-time PCR using primers designed to amplify the BCL6 binding site in the ATR promoter. 79-6 had no effect on BCL6, but could disrupt recruitment of N-CoR and SMRT (both of which contain a nearly identical BBD) to the ATR promoter (Figure 6B). In accordance with this result, exposure of BCL6-dependent DLBCL SU-DHL4 and SU-DHL6 cells to 79-6 at 50 μM for eight hours resulted in an increase in the abundance of ATR transcripts as well as an increase in the mRNA level of other BCL6 target genes TP53, CD69, p21 and CD44 but not the non-target genes HPRT, PCNA and B2M (Figure 6C). However, 79-6 had no effect on any of these genes in the BCL6-independent Toledo DLBCL cell line (Figure 6C). The inactive chemical control 79-40 did not relieve BCL6 mediated repression nor reactivate its target genes (Figure S6A,B). Together, these data show that 79-6 specifically disrupts BCL6 corepressor recruitment and can specifically reactivate BCL6 target genes in BCL6-dependent DLBCLs.
79-6 selectively kills BCL6-dependent DLBCL cells
DLBCL cells can be classified into BCL6-dependent and independent subtypes (Cerchietti et al., 2009; Polo et al., 2007; Ranuncolo et al., 2007). Based on these data we examined the sensitivity of eight DLBCL cell lines (six BCL6-dependent: OCI-Ly1, SU-DHL4, OCI-Ly10, Farage, SU-DHL6 and OCI-Ly7, and two BCL6-independent cell lines: Toledo and OCI-Ly4) to 79-6 in vitro. The cell lines were exposed to several concentrations of 79-6 (administered only once at time-0) and examined for cell viability at 48 hours using a metabolic luminescent assay. 79-6 induced a dose-dependent reduction of viability specifically in BCL6-dependent, but not in the BCL6-independent DLBCL cell lines (Figure 6D). In contrast, the control compound 79-40 did not specifically kill BCL6-dependent DLBCL cells (Figure S6C). The 79-6 growth inhibition 50% (GI50) values were between two to three orders of magnitude lower in the BCL6-dependent group compared to the BCL6-independent group. To determine the intracellular concentrations of 79-6, we exposed OCI-Ly7 to 0 (DMSO), 0.05, 0.1 and 0.2 mM of 79-6 for 30 minutes and measured the intracellular concentration of the drug by HPLC-MS/MS. We found that the cellular accumulation of 79-6 is between 17 and 20 times higher than the administered concentration (Figure S6D). This high intracellular-to-extracellular concentration ratio is in accordance with other anti-neoplastic and antibiotic drugs(Kuh et al., 2000; Langer et al., 2005; Lemaire et al., 2009; Meyer et al., 1993; Stamler et al., 1994). Thus, in cells, 79-6 reaches levels well above its Ki when administered at these concentrations, which is consistent with its BCL6-dependent inhibitory effects.
79-6 is non-toxic to animals
In order to determine whether 79-6 could serve as the basis for a clinically useful BCL6 inhibitor we first examined whether it induced toxic effects in mice. Five C57BL/6 mice were exposed to daily intraperitoneal (IP) administration of increasing doses of 79-6 ranging from 0.5 to 50 mg/kg in 10% DMSO or vehicle (10% DMSO, n=5) over the course of 12 days to a cumulative dose of 278 mg/kg (Figure S7A). In no cases were any toxic effects noted, such as lethargy, weight loss, failure to thrive or any other indicator of sickness. No evidence of tissue damage was detected by microscopic examination of mouse organs (Figure S7B). To determine if the maximal administered dose of 50 mg/kg will be tolerable in a 10-day schedule, we conducted an additional toxicity study in C57BL/6 mice. Ten mice were exposed to daily IP administration of 50 mg/kg of 79-6 in 10% DMSO or vehicle (10% DMSO, n=5) over the course of 10 days to a cumulative dose of 500 mg/kg (Figure S7A). Five mice were sacrificed immediately after the 10-day course of 79-6 administration (together with 5 control mice) and 5 mice were sacrificed after a 10-day washout period to assess delayed toxicity. No toxic effects, such as lethargy, weight loss, failure to thrive or any other indicator of sickness or tissue damage (macroscopic or microscopic), were noted with either treatment schedule (Figure S7C). Complete blood counts showed mild leucopenia in all treated mice, although bone marrow (biopsy and smear) were unaffected. Platelet and erythrocyte numbers were unaffected. All mice completely recovered after the 10-day washout period with no changes in plasma biochemistry values (Table S4).
79-6 displays favorable pharmacokinetics
In order to test whether 79-6 could perform as an anti-lymphoma therapeutic agent in vivo, we determined whether it could penetrate tumors after parenteral administration through a distal site. For this purpose 107 OCI-Ly7 cells were injected into the right flank of 10 SCID mice and allowed to form tumors. Once tumors reached ~1.5 grams, animals were injected IP with a single dose of 50 mg/kg of 79-6 in 10% DMSO or vehicle (10% DMSO) and sacrificed at 0.5, 1, 1.5, 3, 6, 12 and 24 hours after the compound administration. Blood and tumors were harvested. Quantitative HPLC/MS analysis of the serum showed that 79-6 levels peaked (to 55 μg/ml, which is equivalent to a 122 μM concentration) one hour after the IP injection (Figure 7A). 79-6 also reached its highest peak (24.5 ng/mg) at the 1-hour time point in the tumors (Figure 7A), and after a sharp decline in levels, decreased gradually over 24 hours. Therefore, 79-6 was able to reach tumors and persist within tumor tissue after the IP administration.
Figure 7. Compound 79-6 effectively distributes to lymphomas after parenteral administration and suppresses DLBCL growth in vivo.

A: The serum (blue curve) and tumor (red curve, inset) concentrations of 79-6 were determined after the intraperitoneal administration of 50 mg/kg to mice carrying OCI-Ly7 xenografts. Serum and tumors were harvested at several time points (X–axis) and the concentration of 79-6 was determined by HPLC-MS/MS and compared to control (time 0). B: Tumor growth plots in SU-DHL6, OCI-Ly7 and Toledo (as negative control) xenografted mice treated with vehicle (green circles) or 79-6 50 mg/kg/day (red circles) for 10 consecutive days. The Y-axis represents the percentage of tumor volume (in mm3) compared to day 1 of treatment and X-axis represents treatment day. C: Serum levels of human β2-microglobulin at day 10 in vehicle (green bars) and 79-6 (red bars) treated SU-DHL6, OCI-Ly7 and Toledo mice. D: Representative images from SU-DHL6, OCI-Ly7 and Toledo mice tumors after treatment with vehicle or 79-6 and assayed for apoptosis by TUNEL. Scale bars represent 125 μm and 50 μm in main images and insets respectively. Error bars represent the SEM for replicates. See also Figure S7 and Table S4.
79-6 suppresses human DLBCL xenografts in mice
In order to evaluate the anti-lymphoma activity of 79-6, a preclinical study was performed in which two BCL6-dependent DLBCL cell lines (OCI-Ly7 and SU-DHL6) were each injected into 10 SCID mice and allowed to form tumors. Once palpable tumors were detected, pairs of mice were randomized to receive either 50 mg/kg of 79-6 (n=5) per day or vehicle (10% DMSO) (n=5). In addition, one BCL6-independent cell line (Toledo) was also implanted in SCID mice (n=10) and treated in a similar way as a negative control. The volume of tumors from 79-6 treated animals was markedly reduced in both BCL6-dependent cell lines, but not in the BCL6-independent tumors (Figure 7B). The tumors in OCI-Ly7 and SU-DHL6 mice were significantly smaller than their respective vehicle control (p=0.001 and p=0.004, respectively) (Figure 7B). The levels of serum human β2-microglobulin (a surrogate marker of the xenograft burden) (Cerchietti et al., 2009) were significantly reduced by 79-6 in the xenografted mice as compared to their respective controls for the OCI-Ly7 and SU-DHL6 xenografts (p=0.0117 and p=0.0004, respectively) but not in the Toledo xenograft (p=0.7822) (Figure 7C). Histological examination of tumors revealed an increased fraction of cells undergoing apoptosis in 79-6 treated animals for the OCI-Ly7 and SU-DHL6 xenografts (p=0.0001 and p=0.00008, respectively) (Figure 7D). There was no difference in apoptosis between 79-6 and vehicle treatments in Toledo xenografts (Figure 7D). Taken together, these data indicate that therapeutic targeting of BCL6 with the small molecule 79-6 is an effective anti-lymphoma strategy in vivo.
79-6 can specifically kill primary human DLBCL cells
Since we intend to translate BCL6 targeted therapy to the clinical setting we next examined whether primary human DLBCL cells could also be killed by 79-6. Single cell suspensions were generated from 25 confirmed DLBCL lymph node biopsy specimens and exposed to 125 and 250 μM of 79-6 or control (DMSO) for 48 hours. 21 of 25 control specimens maintained an acceptable viability (>80%) over the 48-hour period and were analyzed together with the treated samples by metabolic luminescent-based labeling in triplicate. 19/21 DLBCLs were positive for BCL6 by western blot (Figure 8 and Figure S8). A decrease in the viability equal or greater than 25% compared to their respective controls at either dose was considered as significant. Fifteen of 19 BCL6-positive cases (79%) displayed greater than 25% loss of viability in response to 79-6 at 125 or 250 μM (Figure 8). Therefore, most BCL6 positive human primary DLBCL samples were responsive to 79-6. Although we cannot exclude that cellular pathways induced by placing primary cells in culture could potentiate the activity of the BCL6 inhibitors, the results are consistent overall with our cell line and xenograft data and support the notion that most BCL6 positive tumors are potential candidates for BCL6 inhibitor therapy.
Figure 8. Compound 79-6 inhibits the growth of primary human DLBCLs.

Single cell suspensions were obtained from lymph node biopsies of patients diagnosed with DLBCL and were treated with either vehicle, 79-6 125 μM (black bars) or 79-6 250 μM (grey bars). The Y-axis represents the percent of viable cells compared to vehicle, which is represented by the line at 100%. A significant response zone is shown as a gray shadow with 75% viability as the upper limit. Whether BCL6 was detected by western blot in these samples is indicated at the bottom of the graph by the plus and minus signs. Error bars represent the SEM for replicates. See also Figure S8.
Discussion
Transcriptional repressors usually mediate their effects on gene expression through recruitment of co-factors that either enzymatically modify the chromatin structure of target genes or serve as adaptors for such proteins(Melnick, 2005). Thus, to therapeutically target transcriptional repressors, it is therefore necessary to develop drugs that can disrupt protein-protein interactions. Such interactions have traditionally been considered difficult to target with low molecular weight compounds that have the potential to become drug candidates (Juliano et al., 2001). However, over the last decade small molecule inhibitors have been identified for an increasing number of protein-protein interactions (Wells and McClendon, 2007; Zhong et al., 2007), including ERK kinase, (Hancock et al., 2005) S100B-p53, (Markowitz et al., 2004), eIF4E-eIF4G translation initiation factors (Moerke et al., 2007) and others (Pagliaro et al., 2004). In the present study, we have now extended this list to include small molecule inhibitors of transcriptional repressor protein interactions. Experimental high-throughput screening (HTS) screening efforts for drug-like compounds typically yield hit rates in the range of 0.01 to 1% (Doman et al., 2002; Woodward et al., 2006). Here, using CADD to identify candidate compounds followed by extensive in vitro and in vivo functional testing, we achieved a hit rate of approximately 10% (10 out of 100 tested), emphasizing the utility of this combined approach in the identification of active compounds. Alternatively, experimental HTS approaches may be used to identify smaller, fragment-like molecules (Woodward et al., 2006). This approach typically yields higher hit rates, though the binding affinities are typically in the millimolar range and the compounds have low specificity (Chen and Shoichet, 2009). The advantage of fragment-based screening is the ability to chemically link different candidate fragments together, yielding higher affinity compounds with greater specificity; however, it is heavily reliant on chemical synthesis. We chose to avoid this issue in our study by searching in silico chemical databases of commercially available drug-like compounds.
Our compound screening strategy was based on the 3D structures of BCL6BTB corepressor BBD peptide complexes. Although the 17 residue BBD peptides interact over a large and mostly flat 1080 Å2 surface of the BCL6BTB lateral groove (Ahmad et al., 2003; Ghetu et al., 2008), a deeper pocket accommodates a key aromatic residue in the SMRT and BCOR complexes and we targeted this site for our rational design efforts (Ahmad et al., 2003; Ghetu et al., 2008). The 79 series was selected as a class of possible inhibitors due to their drug like characteristics and because contained several active analogs. Although the in vitro IC50 of these compounds for in vitro blockade of SMRT peptide association was in the micromolar range, and likewise, micromolar concentrations were required to elicit the observed biological effects, our extensive pharmacodynamic studies suggest that the compounds are effective and specific inhibitors of BCL6. Most importantly, the fact that 79-6 disrupts the ability of BCL6 to recruit N-CoR and SMRT to a BCL6 target gene promoter demonstrates that this drug achieves its proposed mechanism of action in living cells.
Collectively, these data support the conclusion that most of the observed activity of 79-6 is due to its inhibition of BCL6. Specifically, the following observations were made: i) 79-6 concentrates within cells to levels greater than its IC50 for BCL6 inhibition, a feature that it shares with recombinant BCL6 peptide inhibitors, ii) 79-6 does not inhibit other BTB transcriptional repressors, ii) 79-6 disrupts endogenous BCL6 repression complexes at a BCL6 target gene promoter, iv) 79-6 specifically reactivates critical BCL6 target genes only in BCL6-dependent DLBCL cells, a function which has been previously shown to be required for BCL6 blockade to kill lymphoma cells (Cerchietti et al., 2008; Cerchietti et al., 2009), v) 79-6 readily kills BCL6-dependent DLBCL cells but has almost no effect on BCL6 independent cells, a feature which implicates BCL6 as the critical target, vi) 79-6 analogs with weaker anti-BCL6 effects are also less potent in killing BCL6-dependent DLBCL cells, vii) 79-6 was non-toxic to other cell types, viii) no gain of function activities were observed with 79-6 vs. BCL6 peptide inhibitors, and ix) 79-6 only suppressed BCL6-dependent DLBCLs in vivo, but did not affect BCL6-independent tumors, indicating that its anti-tumor effect is B cell autonomous.
The ability of a molecule such as 79-6 to mediate its functions within cells is dependent, among other factors, on the cellular context such as the existing levels of BCL6 and corepressors and the capacity of other protein partners of SMRT and N-CoR to sequester them away from BCL6 once displaced by 79-6. It may also not be necessary for the inhibitor to totally block all BCL6-corepressor interactions to exert a biological response, but rather the inhibitor may only have to shift the equilibrium of the of BCL-6 corepressor interactions by a relatively small amount to achieve the desired biological and, possibly, therapeutic outcome. In addition, since the effect of 79-6 is to re-express silenced target genes of BCL6, the compound does not need to be present at all times for its effects to be evident. For example, we showed that 79-6 induces the expression of the critical checkpoint target genes of BCL6 ATR and TP53. Once expressed, these proteins are able to trigger their downstream cell death and anti-proliferative pathways regardless of whether 79-6 is still bound to BCL6.
It is also worth noting that BCL6 mediates several different biological effects in lymphoma cells, and these appear to be dependent on different sets of BCL6 corepressors. Specifically, the effect of BCL6 on survival is dependent on corepressor binding through the BTB lateral groove (at least in part due to BCL6 repression of ATR and TP53) (Cerchietti et al., 2008; Cerchietti et al., 2009; Polo et al., 2004; Ranuncolo et al., 2007) while its effects on B cell differentiation are dependent on MTA3/NuRD binding to the BCL6 middle domain (Fujita et al., 2004; Parekh et al., 2007), and negative autoregulation is dependent on recruitment of CtBP to the N-terminal half of BCL6 (Mendez et al., 2008). In accordance with its lateral groove-specific actions, 79-6 did not affect differentiation or BCL6 negative autoregulation (data not shown). BCL6 knockout mice develop a severe and usually fatal inflammatory syndrome due to loss of BCL6 function in T cells and macrophages (Dent et al., 1997; Ye et al., 1997). However, there was no evidence of toxicity in animals treated with either lateral groove blocking peptides (Cerchietti et al., 2009) or with 79-6. This suggests that these biological actions of BCL6 are independent of the lateral groove.
The 5-membered ring of 79-6 contains sulfur functionality, which is anticipated to be prone to oxidation, potentially leading to the loss of binding to BCL6 and enhanced metabolism and/or excretion. Current research is focused on determining the extent of oxidation, and the replacement of the sulfur-containing heterocycle in 79-6 with other heterocycles less prone to oxidation. These modifications are also anticipated to enhance oral availability, though it should be emphasized that the CADD selection criteria targeted compounds with physical properties that are predicted to have favorable bioavailability. Future efforts will be undertaken to systematically improve the therapeutic potential of the compounds identified in the present study with the goal of developing BCL6 targeted therapy for DLBCL.
Experimental Procedures
Computer aided drug design
The program DOCK 4.0.1 (Ewing et al., 2001; Kuntz, 1992; Kuntz et al., 1982) was used to screen a virtual library of approximately 1,000,000 low molecular weight compounds collected from Maybridge (80,820), Chembridge (242,869), Chemical Diversity (333,054), Specs (207,640), MDD (22,870), Tripos (78,074), Nanosyn (65,154), and TimeTec (15,875). Manipulation of the database was performed with a collection of in house programs and with the program Molecular Operating Environment (MOE); MOE was also used for chemical similarity calculations. Structural analysis and molecular dynamics (MD) simulations were performed using the program CHARMM(Brooks et al., 1983), using the CHARMM protein all-atom force field (MacKerell et al., 1998). Treatment of long-range non-bond interactions used default values supplied with the force field. Database screening targeted a region of the lateral binding groove on BCL6 occupied by residue S1424 and I1425 of the SMRT co-repressor. Site selection was based on the crystallographic structure of BCL-6 homodimer (RCSB Protein Databank(Berman et al., 2000) ID 1R2B), which also contains co-crystallized SMRT peptide. Following removal of the SMRT peptide solvent accessibilities were determined for all BCL6 residues (Chain A: F11, R13, H14, A15, D17, V18, N21; Chain B: A52, C53, S54, F89, M114, H116, V117) within 5 Å of SMRT residues S1424 and I1425; this procedure was performed for both SMRT binding grooves in the homodimer. From this analysis a putative small molecule binding pocket was identified between BCL6 residues R13A and H116B. Initial database screening referred to as first-step docking targeted the putative binding site on the crystal structure. Database screening utilizes an anchor-based ligand-docking scheme. First, each compound is divided into rigid segments containing 5 or more non-hydrogen atoms, referred to as anchors. Each anchor is then overlaid onto the sphere set in 200 different orientations and energy minimized. For each orientation, the remainder of the compound was iteratively built in a layer-by-layer fashion, with the layers separated by rotatable bonds, while minimizing new torsion angles as each layer is added. Select conformers were dynamically removed during the build-up process based on energetic and diversity considerations. For each compound the final selected conformer corresponded to that with the most favorable interaction energy, calculated as the sum of electrostatic and van der Waals interactions (vdW), with the binding site. Compounds were ranked according to their N normalized vdW attractive component (i.e.: EvdWa/N), where N is the number of heavy atoms in the ligand, in order to account for the contribution of ligand molecular size to the energy score(Pan et al., 2003). The top 50,000 compounds based on this ranking scheme were selected for use in subsequent steps. Second-step screening included simultaneous energy minimization of the anchor fragment during iterative build-up of each ligand. In addition, each anchor was placed into the sphere set in 500 different orientations and up to 5 inner layers were optimized during the build-up process. The 50,000 compounds selected from first-step screening were independently docked against three BCL6 conformations: the crystal, 8950, and 9150 conformations, corresponding to MD snapshots 0000 ps, 8950 ps, and 9150 ps, respectively. For each compound the most favorable total interaction energy from the three individual dock runs was using for compound ranking. Final ranking was based on the N2/5-normalized total interaction energy (i.e.: Etot/N2/5) from which the top 1,000 compounds were selected. Final selection of compounds for biological assays was based on their chemical diversity, solubility, and potential as drug leads. Molecular diversity was calculated by building a chemical fingerprint describing the functional groups and configurations. The 1,000 top-scoring compounds obtained in second-step screening were combined into clusters of chemically similar compounds using the clustering tool in MOE. Chemical fingerprints were generated with MACCS Structural Keys and clustered using Tanimoto coefficient metric (Butina, 1999; Godden et al., 2005). A final set of 199 diverse compounds was selected for biological assays. This similarity searching procedure was also applied to active compounds 28, 33, 72, 79, 43, 44, 49, 53 and 55 for the entire chemical database to identify chemically similar analogs of each compound.
X-ray Crystallography
Crystals of the BCL6 BTB domain were obtained by the hanging drop vapor diffusion method by mixing 1 ml of 9.5 mg/ml BCL6BTB in 20 mM Tris pH 8.3, 450 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP) with 1 ml of reservoir solution (180 mM sodium acetate, 190 mM sodium formate, 10% Glycerol, 50 mM NaCl and 10mM N-(4-bromophenylsulfonyl)acetamide) and equilibrating with 500 ml of reservoir solution. Crystals formed in space group C2 with one BCL6BTB dimer (2 chains) per asymmetric unit, resulting in a solvent content of 54%. Crystals were soaked overnight in a 1:1 mixture of protein buffer and reservoir solution containing saturating amounts of 79-6 (2.5 mM). Crystals were next stepped through artificial mother liquor solutions containing 5, 15 and 25% ethylene glycol and flash frozen in liquid nitrogen. Diffraction images were processed and scaled with the HKL program suite (Minor et al., 2006). Molecular Replacement was performed using Phaser (Mccoy et al., 2007) with a BCL6BTB dimer from PDB ID 1R28 as the starting model. Initial model refinement was done with CNS (Brunger et al., 1998) using protein atoms only. Waters were subsequently added using Coot (Emsley and Cowtan, 2004), followed by more rounds of refinement. Once this model converged, 79-6 was built into the electron density followed by further refinement with REFMAC (Murshudov et al., 1997). Three crystallographically unique molecules of 79-6 were found in the electron density maps. 79-6 was found in both of the lateral groove pockets of the BTB dimer, as described in the main text. A third molecule was found associated with the BTB strand b3 at a crystal lattice contact site, but this binding site does not exist in the absence of the crystal lattice, and would not occur in solution. This is confirmed by the fact that we did not see any resonance shifts in this region in the 79-6 NMR titrations. Final refinement statistics are given in the Table S3. In a parallel experiment, a structure was determined for the BCL6BTB crystal form reported here without prior soaking in compound 79-6, and in this case, only water molecules were located in the lateral groove. Protein structure images were produced with PyMOL (DeLano, 2002).
Mouse xenograft studies
All procedures involving animals followed US NIH protocols and were approved by the Animal Institute Committee of the Albert Einstein College of Medicine and/or the Weill Cornell Medical College of Cornell University. Six to eight-week old male severe combined immunodeficiency (SCID) mice were purchased from the National Cancer Institute (NCI, Bethesda, MD) and housed in a clean environment. Mice were subcutaneously injected with low-passage 107 human SU-DHL6, OCI-Ly7 or Toledo cells. Tumor volume was monitored every other day using electronic digital calipers (Fisher Scientific) in two dimensions. Tumor volume was calculated using the formula: Tumor Volume (mm3) = (smallest diameter2 × largest diameter)/2. When tumors reached a palpable size (approximately 75 to 100 mm3), the mice were randomized assigned to different treatment arms. Drugs were stored in a UV light-protected vacuum dessicator at RT until used and were immediately reconstituted in DMSO and administered by intra-peritoneal injection. Mice were weighed every other day. All mice were euthanized by cervical dislocation under anesthesia when one of each pair reached the maximal tumor mass permitted by our protocol. At the moment of euthanasia, blood was collected (StatSampler, Iris, Westwood, MA) and tumors and other tissues were harvested and weighed.
Compounds
All compounds were acquired from ChemBridge (San Diego, CA) or ChemDiv (San Diego, CA). The minimum purity of all compounds is 90%, with 60% of compounds being > 95% pure. Compound identity was confirmed through mass spectrometry (ThermoFinnegan LCQ). Compound 79-6 was synthesized, purified and identity verified by ChemDiv Inc. The purity and identity were verified by mass spectrometry, 1H NMR, elemental analysis and LC/MS. LC was performed on a SpeedROD Rp-18e 50 × 4.6 mm column at a flow rate of 3.75 ml/min with 0.1% FA in AcN:Water (24.5:75.5) for 2.4 minutes, followed by 0.1% FA in AcN:Water (90:10) for 0.2 minutes and a subsequent wash with 0.1% FA in AcN:Water (24.5:75.5). MS was performed in the APCI negative ionization mode. Results show 100% purity by LC/MS in two runs, and combustion analysis that satisfied the ± 0.4% criteria.
Reporter assays
For functional screening of small molecules we transfected 293T cells in a 10 cm plate using Superfect (Qiagen, Valencia, CA) or Lipofectamine 2000 (Invitrogen) with a luciferase reporter vector containing five binding sites for the yeast GAL4 DNA binding domain and a thymidine kinase (TK) promoter, (GAL4)5TK-Luc (Polo et al., 2004) and an internal control TK-renilla reporter vector, pRL-TK (Promega) at a 10:1 ratio. Cells were also transfected with 400–500ng of a plasmid expressing the GAL4 DNA binding domain (DBD) alone (pBXG1) or GAL4-DBD fused to the BCL6BTB. Alternatively, cells were transfected with 1320 ng of plasmid containing the Kaiso-BTB domain fused to GAL4-DBD, 500ng of HIC-BTB-GAL4-DBD (Polo et al., 2004), 500ng of PLZF-BTB-GAL4-DBD (Polo et al., 2004), or 500–1320ng of GAL4-DBD alone. Twenty-four hours after transfection cells were harvested and redistributed to 24- or 96-well plates at a density of 400,000 or 20,000 cells per well, respectively, after which, cells were treated in quadruplicate with 50 μM concentrations of different compounds or DMSO for 24 or 48 h. Cell lysates were examined for the abundance of firefly luciferase relative to renilla luciferase (in counts per second) using the Dual-Luciferase Reporter Assay kit (Promega, Madison, WI) and a Synergy4 plate reader (BioTek Instruments, Winooski, VT). The repressor activity of each BTB domain was calculated as the relative fold change in repression compared to the GAL4 DBD plasmid control under the same treatment conditions.
Growth-inhibition determination
DLBCL cell lines were grown at respective concentrations sufficient to keep untreated cells in exponential growth over the 48h drug exposure time. Cell viability was determined using a luminescent ATP quantization method (CellTiter-Glo, Promega, Madison, Wisconsin), Trypan blue dye-exclusion (Sigma) and the EB/OG method (Easycount, Immunicon). Luminescence was determined using the Synergy4 microplate reader (BioTek). Luminescence was determined for 3 replicates per treatment condition or controls. Standard curves were obtained for each individual cell line by plotting the cell number (determined by the EB/OG method in agreement with Trypan blue) against luminescence values. The number of viable cells was calculated by using the linear least-squares regression of the standard curve and cell viability in drug-treated cells was normalized to their respective controls (fractional viability). Experiments were carried out in triplicates. For calculation purposes the drug effect was calculated as 1-fractional viability. We used the CompuSyn software (Biosoft, Cambridge, UK) to plot dose-effect curves and determine the drug concentration that inhibits the growth of cell lines by 50% compared to control (GI50). Data are presented as dose-effect curves and mean of GI50.
Primary cells
Patient de-identified tissues were obtained in accordance with the guidelines and approval of the Weill Cornell Medical College Review Board. We obtained single cells suspensions from lymph node biopsies by physical disruption of tissues followed by cell density gradient separation (Fico/Lite LymphoH, Atlanta Biologicals, Lawrenceville, Georgia). Cell number and viability were determined by an EB/AO-based method (Easycount) and cells were cultivated in medium containing 80% RPMI and 20% human serum supplemented with antibiotics, L-glutamine and HEPES for 48 h. Primary cells were exposed to 125 and 250 μM of 79-6 or control (DMSO) in triplicates. After 48 h of exposure viability was determined by using an ATP-based luminescent method (CellTiter-Glo) and EB/AO. Specimens with 20% or higher loss of viability in the controls were discarded. Lysates from these tissues were prepared using 50mM Tris pH 7.4, 150mM NaCl and 1% NP-40 lysis buffer. Protein concentrations were determined using the BCA kit (Pierce, Rockford, Illinois). Fifty μg of protein lysates were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and probed with rabbit anti-BCL6 N3 (Santa Cruz) and rabbit anti-Actin (Santa Cruz). Membranes were then incubated with a peroxidase-conjugated correspondent secondary antibody. Detection was performed using an ECL detection system (Vector, Burlingame, California) according to the manufacturer’s instructions.
Significance
BCL6 is the most commonly involved oncogene in B-cell lymphomas. Depletion or blockade of BCL6 potently kills DLBCL cells and BCL6 is thus a critical therapeutic target. Like many oncogenes and tumors suppressors, BCL6 is a transcription factor. Because such proteins usually mediate their actions through extensive protein interaction surfaces, they have been considered non-amenable to targeting with small molecules. Herein, we used an integrated biochemical and computational approach to identify an effective and specific BCL6 small molecule inhibitor. This drug displayed favorable pharmacokinetics, pharmacodynamics, toxicity and therapeutic efficacy. This work demonstrates that oncogenic transcriptional repressors can be therapeutically targeted with small molecules and presents a rationally designed transcription therapy approach for the treatment of lymphomas.
Highlights
- We identified a specific BCL6 small molecule inhibitor
- This compound targets the interaction of BCL6 with its co-repressors
- This compound displayed anti-lymphoma activity in vitro and in vivo
Supplementary Material
Acknowledgments
We are grateful to Dr. Julie White and the members of the Tri-Institutional Laboratory of Comparative Pathology for technical expertise. We also thank Dr. Nian Wu and Ms. Weige Qin from the Analytical Pharmacology Core of the Sloan Kettering Institute for technical expertise. Most of this research was made possible thanks to a program project grant from the Samuel Waxman Cancer Research Foundation to AM, GGP and ADM. AM is also supported by NCI R01 CA104348, the Chemotherapy Foundation, and is a Leukemia and Lymphoma Society Scholar. ADM is supported by HL082670, CA107331, CA120215 and the University of Maryland Computer-Aided Drug Design Center. GGP is supported by the Canadian Cancer Society.
Footnotes
Accession number. The structure raw data was deposited in the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) with the ID 3LBZ
The authors report no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad KF, Melnick A, Lax S, Bouchard D, Liu J, Kiang CL, Mayer S, Takahashi S, Licht JD, Prive GG. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol Cell. 2003;12:1551–1564. doi: 10.1016/s1097-2765(03)00454-4. [DOI] [PubMed] [Google Scholar]
- Baron BW, Anastasi J, Montag A, Huo D, Baron RM, Karrison T, Thirman MJ, Subudhi SK, Chin RK, Felsher DW, et al. The human BCL6 transgene promotes the development of lymphomas in the mouse. Proc Natl Acad Sci U S A. 2004;101:14198–14203. doi: 10.1073/pnas.0406138101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Research. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Butina D. Unsupervised Data Base Clustering on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way to Cluster Small and Large Data Sets. J Chem Inf Comput Sci. 1999;39:747–750. [Google Scholar]
- Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, Mo T, Murty VV, Dalla-Favera R. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell. 2005;7:445–455. doi: 10.1016/j.ccr.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Cerchietti LC, Polo JM, Da Silva GF, Farinha P, Shaknovich R, Gascoyne RD, Dowdy SF, Melnick A. Sequential transcription factor targeting for diffuse large B-cell lymphomas. Cancer Res. 2008;68:3361–3369. doi: 10.1158/0008-5472.CAN-07-5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchietti LC, Yang SN, Shaknovich R, Hatzi K, Polo JM, Chadburn A, Dowdy SF, Melnick A. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood. 2009;113:3397–3405. doi: 10.1182/blood-2008-07-168773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Shoichet BK. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat Chem Biol. 2009;5:358–364. doi: 10.1038/nchembio.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; 400 Oyster Point Blvd., Suite 213, San Francisco, CA 94080, USA: 2002. [Google Scholar]
- Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Geigerman C, Akyildiz A, MRM, Boss JM, Wade PA. MTA3 and Mi-2/NuRD Complex Regulate Cell Fate During B-Lymphocyte Differentiation. Cell. 2004;119:75–86. doi: 10.1016/j.cell.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Ghetu AF, Corcoran CM, Cerchietti L, Bardwell VJ, Melnick A, Prive GG. Structure of a BCOR corepressor peptide in complex with the BCL6 BTB domain dimer. Mol Cell. 2008;29:384–391. doi: 10.1016/j.molcel.2007.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godden JW, Stahura FL, Bajorath J. Anatomy of Fingerprint Search Calculations on Structurally Diverse Sets of Active Compounds. J Chem Inf Model. 2005;45:1812–1819. doi: 10.1021/ci050276w. [DOI] [PubMed] [Google Scholar]
- Hancock CN, Macias AT, Lee EK, Yu SY, MacKerell AD, Jr, Shapiro P. Identification of novel extracellular signal-regulated kinase (ERK) docking domain inhibitors. J Med Chem. 2005;48:4586–4595. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- Huang N, Nagarsekar A, Xia G, Hayashi J, MacKerell AD., Jr Identification of non-phosphate-containing small molecular weight inhibitors of the tyrosine kinase p56 Lck SH2 domain via in silico screening against the pY + 3 binding site. J Med Chem. 2004;47:3502–3511. doi: 10.1021/jm030470e. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Astriab-Fisher A, Falke D. Macromolecular Therapeutics: Emerging Strategies for Drug Discovery in the Postgenome Era. Mol Interv. 2001;1:40–53. [PubMed] [Google Scholar]
- Kuh HJ, Jang SH, Wientjes MG, Au JL. Computational model of intracellular pharmacokinetics of paclitaxel. J Pharmacol Exp Ther. 2000;293:761–770. [PubMed] [Google Scholar]
- Kuntz ID. Structure-Based Strategies of Drug Discovery and Design. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A Geometric Approach to Macromolecule-Ligand Interactions. Journal of Molecular Biology. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- Langer O, Karch R, Muller U, Dobrozemsky G, Abrahim A, Zeitlinger M, Lackner E, Joukhadar C, Dudczak R, Kletter K, et al. Combined PET and microdialysis for in vivo assessment of intracellular drug pharmacokinetics in humans. J Nucl Med. 2005;46:1835–1841. [PubMed] [Google Scholar]
- Lemaire S, Van Bambeke F, Appelbaum PC, Tulkens PM. Cellular pharmacokinetics and intracellular activity of torezolid (TR-700): studies with human macrophage (THP-1) and endothelial (HUVEC) cell lines. J Antimicrob Chemother. 2009;64:1035–1043. doi: 10.1093/jac/dkp267. [DOI] [PubMed] [Google Scholar]
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Jr, Bashford D, Bellot M, Dunbrack RL, Jr, Evanseack J, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Markowitz J, Chen I, Gitti R, Baldisseri DM, Pan Y, Udan R, Carrier F, MacKerell AD, Jr, Weber DJ. Identification and Characterization of Small Molecule Inhibitors of the Calcium-Dependent S100B-p53 Tumor Suppressor Interaction. J Med Chem. 2004;47:5085–5093. doi: 10.1021/jm0497038. [DOI] [PubMed] [Google Scholar]
- Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni L, Read RJ. Phaser crystallographic software. JApplCrystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick A. Reprogramming specific gene expression pathways in B-cell lymphomas. Cell Cycle. 2005;4:239–241. [PubMed] [Google Scholar]
- Mendez LM, Polo JM, Yu JJ, Krupski M, Ding BB, Melnick A, Ye BH. CtBP is an essential corepressor for BCL6 autoregulation. Mol Cell Biol. 2008 doi: 10.1128/MCB.01400-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AP, Bril-Bazuin C, Mattie H, van den Broek PJ. Uptake of azithromycin by human monocytes and enhanced intracellular antibacterial activity against Staphylococcus aureus. Antimicrob Agents Chemother. 1993;37:2318–2322. doi: 10.1128/aac.37.11.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Pagliaro L, Felding J, Audouze K, Nielsen SJ, Terry RB, Krog-Jensen C, Butcher S. Emerging classes of protein-protein interaction inhibitors and new tools for their development. Curr Opin Chem Biol. 2004;8:422–449. doi: 10.1016/j.cbpa.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Pan Y, Huang N, Cho S, MacKerell AD., Jr Consideration of molecular weight during compound selection in virtual target-based database screening. J Chem Inf Comput Sci. 2003;43:267–272. doi: 10.1021/ci020055f. [DOI] [PubMed] [Google Scholar]
- Parekh S, Polo JM, Shaknovich R, Juszczynski P, Lev P, Ranuncolo SM, Yin Y, Klein U, Cattoretti G, Dalla Favera R, et al. BCL6 programs lymphoma cells for survival and differentiation through distinct biochemical mechanisms. Blood. 2007;110:2067–2074. doi: 10.1182/blood-2007-01-069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054–1060. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- Polo JM, Dell’Oso T, Ranuncolo SM, Cerchietti L, Beck D, Da Silva GF, Prive GG, Licht JD, Melnick A. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat Med. 2004;10:1329–1335. doi: 10.1038/nm1134. [DOI] [PubMed] [Google Scholar]
- Polo JM, Juszczynski P, Monti S, Cerchietti L, Ye K, Greally JM, Shipp M, Melnick A. Transcriptional signature with differential expression of BCL6 target genes accurately identifies BCL6-dependent diffuse large B cell lymphomas. Proc Natl Acad Sci U S A. 2007;104:3207–3212. doi: 10.1073/pnas.0611399104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, Green R, Carroll M, Melnick A. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol. 2007;8:705–714. doi: 10.1038/ni1478. [DOI] [PubMed] [Google Scholar]
- Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol Dis. 2008;41:95–99. doi: 10.1016/j.bcmd.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler DA, Edelstein MA, Edelstein PH. Azithromycin pharmacokinetics and intracellular concentrations in Legionella pneumophila-infected and uninfected guinea pigs and their alveolar macrophages. Antimicrob Agents Chemother. 1994;38:217–222. doi: 10.1128/aac.38.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogios PJ, Chen L, Prive GG. Crystal structure of the BTB domain from the LRF/ZBTB7 transcriptional regulator. Protein Sci. 2007;16:336–342. doi: 10.1110/ps.062660907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Woodward PW, Williams C, Sewing A, Benson N. Improving the design and analysis of high-throughput screening technology comparison experiments using statistical modeling. J Biomol Screen. 2006;11:5–12. doi: 10.1177/1087057105280779. [DOI] [PubMed] [Google Scholar]
- Ye BH. BCL-6 in the pathogenesis of non-Hodgkin’s lymphoma. Cancer Invest. 2000;18:356–365. doi: 10.3109/07357900009012179. [DOI] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri-Shirazi M, Orazi A, Chaganti RS, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16:161–170. doi: 10.1038/ng0697-161. [DOI] [PubMed] [Google Scholar]
- Zhong S, Macias AT, MacKerell JAD. Computational identification of the inhibitors of protein-protein interactions. Current Topics in Medicincal Chemistry. 2007;7:63–82. doi: 10.2174/156802607779318334. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.