

Abstract
Farnesyl transferase inhibitors (FTIs) have so far proved to have limited value as single agents in clinical trials. This PharmSight will focus on the use of a novel group of FTIs that are most effective in vitro when used in combination with the “statin” class of anti-hypercholesterolemic agents, which also block protein prenylation. We recently showed that these novel FTIs in combination with lovastatin reduce Ras prenylation and induce an apoptotic response in malignant peripheral nerve sheath cells. The combination of statins with these new FTIs may produce profound synergistic cytostatic and cytotoxic effects against a variety of tumors and other proliferative disorders. Since statins are well tolerated in the clinic, we suggest that this combination approach should be tested in in vivo models.
Introduction
Modification of proteins with isoprenoid groups was identified in mammalian cells in the early 1980’s (1). Approximately 0.5% of proteins are modified by isoprenoids and a fraction of these are known to regulate critical cellular processes such as growth and survival (2). We will discuss in this PharmSight a novel combination approach that can block the isoprenylation of proteins and has the potential to induce cytostatic and cytotoxic responses against hyperproliferative diseases. Numerous proteins contain a “CaaX” prenylation motif at the carboxyl terminus. This motif marks the protein to be modified by either a 15-carbon farnesyl pyrophosphate (FPP) or a 20-carbon geranylgeranyl pyrophosphate (GGPP) on the cysteine of the “CaaX” box (Fig. 1) (3). When the CaaX box ends with a serine, methionine, or glutamine, the protein is farnesylated, whereas a CaaX box ending in leucine is preferentially geranylgeranylated (4). This modification occurs on the nascent precursor protein in the cytosol and allows the protein to associate with the endoplasmic reticulum (ER). Additional steps occur at the ER where Ras converting enzyme (RCE1) proteolytically removes the three remaining amino acids (-aaX) followed by methylation of the C-terminal cysteine residue by isoprenylcysteine carboxymethyltransferase (ICMT) (5).
Figure 1.
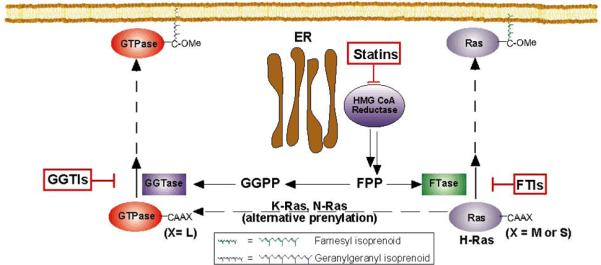
Ras small GTPases are modified at the C-terminal region by either a 15-carbon farnesyl isoprenoid or a 20-carbon geranylgeranyl isoprenoid. Ras and numerous other proteins depend on this initial posttranslational modification in order to traffic and associate with membranes where they can signal downstream to effector proteins. Since Ras has a major role in cancer development, inhibiting this prenylation step is a major focus. FTIs have been developed and were effective in preclinical studies but have not shown strong activity in the clinic. One reason may be the ability of certain proteins that are normally farnesylated, such as N-Ras and K-Ras, to be alternatively prenylated with a geranylgeranyl isoprenoid in the presence of FTIs. Thus, designing a therapy to inhibit alternative prenylation is critical. Our work has identified a therapy in which a combination of statin and novel FTIs is used to reduce Ras prenylation in a model where N-Ras is the predominant isoform expressed. This figure was adapted from Phillips M.R. and Cox A.D. (49).
Two major classes of drugs have been developed that block protein farnesylation. The first class, which includes lovastatin and various synthetic HMG-CoA reductase inhibitors, are collectively referred to as the “statins” and act to reduce production of cholesterol through inhibition of the mevalonate pathway. This pathway provides the cell, via the key branch-point intermediate farnesyl pyrophosphate (FPP), with cholesterol and the prenyl diphosphates used to modify numerous cellular proteins (6). Early studies on the potential toxicity of statins demonstrated that high levels could block cell growth (6), and there is strong evidence that this effect is due to the blockade of protein prenylation (7). Statins are known to reduce serum cholesterol, which has been shown in clinical trials to be beneficial to patients with coronary artery disease (8, 9). However, additional clinical evidence suggests that some cardiovascular benefits may not be due to reduced serum cholesterol (10). In some cases, the “non-cholesterol” effects may be due to inhibition of protein prenylation (11, 12). The potential ability of statins to block protein prenylation at clinically reasonable levels has led to significant interest in their effects on the growth of tumor cells. While retrospective analyses of clinical data from statin-treated patients have been contradictory, cellular data have clearly demonstrated antiproliferative effects of statins on tumor cells that correlated with their ability to block protein prenylation (13).
The second class of drugs that reduces protein farnesylation is those that directly target protein farnesyl transferase (FTase). Numerous inhibitors of FTase have been designed and include compounds that act as CaaX peptidomimetics that compete directly with the peptide substrate of FTase and those that are analogs of prenyl groups and compete with endogenous FPP for binding to FTase (14-16). Of these FTIs, two drugs derived from compound library screening efforts have progressed to phase III clinical trials – R115777/Zarnestra/tipifarnib (17) and SCH66336/Sarasar/lonafarnib (18). Both of these compounds inhibit FTase by competing with the CaaX substrate of the enzyme. They inhibit the growth of many human tumor cell lines in vitro and have resulted in either tumor growth inhibition or tumor regression in a spectrum of xenograft models (3, 19).
Statins, through their action to limit cellular prenyl substrate pools, should potentiate the action of FTIs and have a particularly synergistic effect with FTIs that are competitive with the FPP substrate of the enzyme (20, 21). In fact, our group has shown that lovastatin in combination with the FTI 3-allyfarnesol induces the relocalization of RhoB, a protein that is farnesylated or geranylgeranylated, from membrane fractions to cytosolic fractions in A10 vascular smooth muscle cells (22). The FTI 3-allyfarnesol was later modified with a pro-drug moiety that masked its modification by phosphorylation to allow improved cell penetration and efficacy. This pro-drug FTI (5b) used in combination with lovastatin also reduced RhoB prenylation and cell proliferation of STS-26T malignant peripheral nerve sheath tumor (MPNST) cells (23). We recently published work showing FTI-1 and FTI-2, which were further developed to allow improved aqueous solubility, can reduce Ras prenylation and induce apoptosis when used in combination with nanomolar doses of lovastatin in two MPNST cells lines derived from patients with Type 1 neurofibromatosis (NF1), NF90-8 and ST88-14 (24). We propose that the combination of FTIs with statins may be more efficacious towards hyperproliferative disorders such as NF1 (25).
Results
We tested the efficacy of lovastatin and FTI-1 alone or in combination against a sporadic MPNST cell line, STS-26T. The effect on protein prenylation was observed by monitoring the migration pattern of Ras by western blotting (Fig. 2). The slower mobility or upshifted band represents the precursor molecule and is consistent with an inhibition of FTase function. Single treatments of DMSO and 1 μM FTI-1 had little detectable effect on blocking Ras prenylation while 1 μM lovastatin had a minimal effect at 24 hours. However, using the compounds in combination greatly increased the presence of the upper band, with a corresponding decrease in the lower modified Ras band. Since the predominant isoform of Ras that is expressed in these cells is N-Ras (26), these results suggest that the combination of lovastatin and FTI-1 can prevent the alternative modification of N-Ras with geranylgeranyl moieties (27).
Figure 2.

Lovastatin/FTI-1 combination treatment reduced Ras prenylation in STS-26T MPNST cells. STS-26T cells were treated as indicated for 24 hours and whole cell lysates were probed for Ras prenylation status. Inhibition of prenylation is observed by the slower mobility or upshifted band via western analysis. Please refer to western blot methods from Wojtkowiak et al., (24). Single treatments of 1 μM FTI-1 or 1 μM lovastatin slightly increased the presence of the precursor Ras with continued expression of modified Ras. However, combination treatment with lovastatin plus FTI-1 greatly reduced the expression of modified Ras.
In addition to blocking Ras prenylation, Fig. 3A presents proliferation data from an MTT assay on STS-26T cells. As observed with Ras prenylation, single treatments of FTI-1 or lovastatin had little effect and did not reduce proliferation as compared to the control treatments. The combination of lovastatin plus FTI-1 significantly reduced STS-26T proliferation. Lovastatin/FTI-1 combination treatment also blocks cell cycle progression of STS-26T cells (Fig. 3B). Lovastatin/FTI-1 combination treatment significantly increased the number of cells with G1 DNA content and decreased the S phase population, which is consistent with a cytostatic effect. At this early treatment time, there is a small increase in the proportion of apparently apoptotic cells that becomes significant with more prolonged exposure to the drug combination (24).
Figure 3.
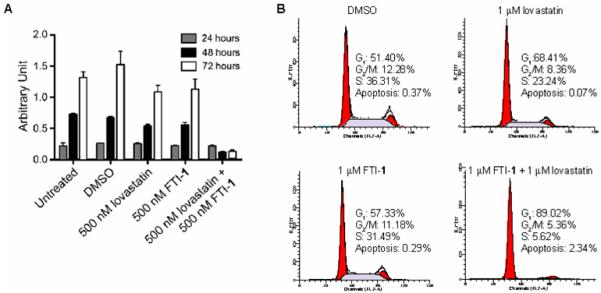
Lovastatin/FTI-1 combination treatment reduces cell proliferation and induces a G1 cell cycle arrest in STS-26T cells. A. STS-26T cells were treated as described for 24, 48, and 72 hours. Cell viability was determined based on the cells’ ability to convert MTT to formazan precipitate. Please refer to MTT assay methods from Li et al., (50). Single treatments of DMSO (vehicle), lovastatin, or FTI-1 did not reduce STS-26T proliferation. Combination treatment of lovastatin plus FTI-1 blocked cell proliferation. B. STS-26T cell cycle progression was determined using fluorescence activating cell sorting (FACS). STS-26T cultures were treated for 24 hours on the day after plating. The histograms represent 104 events. Please refer to FACS methods from Wojtkowiak et al., (24).
Discussion
FTIs were initially designed to inhibit the prenylation of Ras small GTPases as single agents. Pre-clinical studies investigating the efficacy of FTIs against cancer cell lines demonstrated reduced protein prenylation and reversal of Ras transformed phenotypes. For example, the peptidomimetic FTI L-744,832 reduced the proliferation of 70% of tumor cell lines tested and induced tumor regression in an H-Ras transgenic mouse model (28, 29).
Unfortunately, the efficacy of FTIs in cell culture and mouse studies has not yet been translated into a positive clinical response. The results of three phase II trials for tipifarnib, R115777, have been reported against pancreatic cancer, breast cancer, and non-small cell lung carcinoma (NSCLC) (30-32). No responses were observed in the pancreatic cancer and NSCLC studies (30, 31), although there were nine responses and nine stable diseases in the advanced breast cancer study (32). Phase III studies observing the efficacy of tipifarnib compared to placebo effects in colorectal cancer reported no significant effects (33). Lonafarnib, SCH66336, tested against urothelial and colorectal cancer in phase II trials had no favorable response (34, 35). Results from phase III studies involving lonafarnib have not been reported yet.
Several factors may explain why FTIs worked extremely well in preclinical studies but significantly less well as single agents against solid tumors. FTIs were designed to inhibit the prenylation of Ras proteins, with the assumption that inhibition of maturation of the driving oncogene of many human cancers would yield therapeutic benefit. It has become apparent that K-Ras and N-Ras can be alternatively geranylgeranylated in the presence of FTIs (27) [see Fig. 1], which may provide an explanation for their limited activity. Further, since K- and N-Ras are more commonly mutated in human cancer than the exclusively farnesylated H-Ras, this “escape mechanism” could allow the cancer cells to continue growing regardless of FTase inhibition. Our hypothesis is that the combination of prenylation inhibitor lovastatin with an FPP-competitive FTI (24) will provide both very effective action as a synergistic FTI approach and also, again through limitation of cellular pools of prenyl precursors, blunt the ability of proteins such as N-Ras or K-Ras to become alternatively geranylgeranylated.
Another consideration is that although Ras isoforms such as K-Ras are important during the initiation of cancer development (36, 37) additional mutations must occur for a complete transformation. Thus, Ras may not be the sole driving force in many of the cancers in which FTIs were tested. Utilizing a compound that can reduce Ras prenylation in combination with drugs that inhibit alternative cellular functions may be more beneficial in the clinic. Current chemotherapeutic agents such as doxorubicin, cisplatin, and vinblastine, and paclitaxel are now being combined with FTIs in clinical trials in an attempt to create additive and synergistic treatments (38). Results from a phase II study using lonafarnib plus paclitaxel presented a synergy that produced a clinical response in 48% percent of NSCLC patients (39). It may also be worthwhile to consider the combination of both effective inhibition of protein prenylation and another targeted therapy, such as block of driving kinase pathways (40).
Another potential problem with the interpretation of the action of FTIs is that the most critical cellular target(s) for their anti-proliferative effects has not been rigorously established. Thus, while FTIs can inhibit the prenylation of the Ras isoforms during in vitro studies, reduced prenylation of other proteins such as RhoB, CENP-E, CENP-F, and Rheb may also be responsible for or contribute to the observed effects. For example, RhoB is a small GTPase that can either be modified with an FPP or GGPP isoprenoid even in the absence of perturbation of the pathways by drug treatment. Interestingly, Prendergast and colleagues have suggested that RhoB that is geranylgeranylated in the presence of FTIs induces an apoptotic response (41, 42). On the other hand, Sebti and colleagues have suggested that both farnesylated and geranylgeranylated RhoB can be antiproliferative and induce apoptosis (43). The combination of the novel FPP-competitive FTIs and lovastatin clearly blocks the prenylation of RhoB in MPNST cells (23). Centromeric proteins (CENP-E and CENP-F) are normally farnesylated and have an important role in cell division. The ability of FTIs to induce a G2 arrest may be directly linked to inhibition of CENP farnesylation (44, 45). The combination of lovastatin and FPP-competitive FTI induces a G1 arrest (not a G2 arrest) in several cell types, however (22, 24, 26). Rheb, another exclusively farnesylated small GTPase, also appears to be a critical target of FTIs (46, 47). Inhibition of Rheb farnesylation was also shown to be antiproliferative (48).
Statins are some of the most prescribed drugs and are taken with the goal of reducing serum cholesterol levels. Statins have also been shown to have beneficial effects that are independent of reduced cholesterol and may be due to reduced protein prenylation (11, 12). Statins may sensitize tumor cells to co-administered FTIs to provide a synergistic drug combination that does not exhibit toxicity to normal cells (24). We propose that this combination approach of statins plus FPP-competitive FTIs should be tested in in vivo models of cancer and other hyperproliferative disorders.
Acknowledgments
This work was supported by grants DAMD17-03-1-0182 and W81XWH-05-1-0193 from the Department of the Army. JWW was supported by T32 ES012163 from the NIH. This project was aided by Imaging and Cytometry Core facilities that were supported by P30 ES06639 and P30 CA22453 (WSU) and CA21368 (PU) from the NIH.
References
- 1.Schmidt RA, Schneider CJ, Glomset JA. Evidence for post-translational incorporation of a product of mevalonic acid into Swiss 3T3 cell proteins. J Biol Chem. 1984;259:10175–80. [PubMed] [Google Scholar]
- 2.Tamanoi F, Kato-Stankiewicz J, Jiang C, Machado I, Thapar N. Farnesylated proteins and cell cycle progression. J Cell Biochem Suppl. 2001;(Suppl 37):64–70. doi: 10.1002/jcb.10067. [DOI] [PubMed] [Google Scholar]
- 3.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Zhang FL, Kirschmeier P, Carr D, et al. Characterization of Ha-ras, N-ras, Ki-Ras4A, and Ki-Ras4B as in vitro substrates for farnesyl protein transferase and geranylgeranyl protein transferase type I. J Biol Chem. 1997;272:10232–9. doi: 10.1074/jbc.272.15.10232. [DOI] [PubMed] [Google Scholar]
- 5.Ashby MN. CaaX converting enzymes. Curr Opin Lipidol. 1998;9:99–102. doi: 10.1097/00041433-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 7.Graaf MR, Richel DJ, van Noorden CJ, Guchelaar HJ. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat Rev. 2004;30:609–41. doi: 10.1016/j.ctrv.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 9.Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA. 2004;291:1071–80. doi: 10.1001/jama.291.9.1071. [DOI] [PubMed] [Google Scholar]
- 10.Massy ZA, Keane WF, Kasiske BL. Inhibition of the mevalonate pathway: benefits beyond cholesterol reduction? Lancet. 1996;347:102–3. doi: 10.1016/s0140-6736(96)90217-2. [DOI] [PubMed] [Google Scholar]
- 11.Endres M, Laufs U, Huang Z, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:8880–5. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 13.Graaf MR, Beiderbeck AB, Egberts AC, Richel DJ, Guchelaar HJ. The risk of cancer in users of statins. J Clin Oncol. 2004;22:2388–94. doi: 10.1200/JCO.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 14.Bell IM. Inhibitors of farnesyltransferase: a rational approach to cancer chemotherapy? J Med Chem. 2004;47:1869–78. doi: 10.1021/jm0305467. [DOI] [PubMed] [Google Scholar]
- 15.Brunner TB, Hahn SM, Gupta AK, Muschel RJ, McKenna WG, Bernhard EJ. Farnesyltransferase inhibitors: an overview of the results of preclinical and clinical investigations. Cancer Res. 2003;63:5656–68. [PubMed] [Google Scholar]
- 16.Gibbs RA, Zahn TJ, Sebolt-Leopold JS. Non-peptidic prenyltransferase inhibitors: diverse structural classes and surprising anti-cancer mechanisms. Curr Med Chem. 2001;8:1437–65. doi: 10.2174/0929867013372111. [DOI] [PubMed] [Google Scholar]
- 17.Zujewski J, Horak ID, Bol CJ, et al. Phase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancer. J Clin Oncol. 2000;18:927–41. doi: 10.1200/JCO.2000.18.4.927. [DOI] [PubMed] [Google Scholar]
- 18.Adjei AA, Erlichman C, Davis JN, et al. A Phase I trial of the farnesyl transferase inhibitor SCH66336: evidence for biological and clinical activity. Cancer Res. 2000;60:1871–7. [PubMed] [Google Scholar]
- 19.Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: a review. Oncologist. 2005;10:565–78. doi: 10.1634/theoncologist.10-8-565. [DOI] [PubMed] [Google Scholar]
- 20.Gibbs BS, Zahn TJ, Mu Y, Sebolt-Leopold JS, Gibbs RA. Novel farnesol and geranylgeraniol analogues: A potential new class of anticancer agents directed against protein prenylation. J Med Chem. 1999;42:3800–8. doi: 10.1021/jm9902786. [DOI] [PubMed] [Google Scholar]
- 21.Yonemoto M, Satoh T, Arakawa H, et al. J-104,871, a novel farnesyltransferase inhibitor, blocks Ras farnesylation in vivo in a farnesyl pyrophosphate-competitive manner. Mol Pharmacol. 1998;54:1–7. doi: 10.1124/mol.54.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Mattingly RR, Gibbs RA, Menard RE, Reiners JJ., Jr. Potent suppression of proliferation of a10 vascular smooth muscle cells by combined treatment with lovastatin and 3-allylfarnesol, an inhibitor of protein farnesyltransferase. J Pharmacol Exp Ther. 2002;303:74–81. doi: 10.1124/jpet.102.036061. [DOI] [PubMed] [Google Scholar]
- 23.Clark MK, Scott SA, Wojtkowiak J, et al. Synthesis, biochemical, and cellular evaluation of farnesyl monophosphate prodrugs as farnesyltransferase inhibitors. J Med Chem. 2007;50:3274–82. doi: 10.1021/jm0701829. [DOI] [PubMed] [Google Scholar]
- 24.Wojtkowiak JW, Fouad F, LaLonde DT, et al. Induction of apoptosis in neurofibromatosis type 1 malignant peripheral nerve sheath tumor cell lines by a combination of novel farnesyl transferase inhibitors and lovastatin. J Pharmacol Exp Ther. 2008;326:1–11. doi: 10.1124/jpet.107.135830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dilworth JT, Kraniak JM, Wojtkowiak JW, et al. Molecular targets for emerging anti-tumor therapies for neurofibromatosis type 1. Biochem Pharmacol. 2006;72:1485–92. doi: 10.1016/j.bcp.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 26.Mattingly RR, Kraniak JM, Dilworth JT, et al. The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J Pharmacol Exp Ther. 2006;316:456–65. doi: 10.1124/jpet.105.091454. [DOI] [PubMed] [Google Scholar]
- 27.Whyte DB, Kirschmeier P, Hockenberry TN, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–64. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 28.Kohl NE, Omer CA, Conner MW, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med. 1995;1:792–7. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 29.Sepp-Lorenzino L, Ma Z, Rands E, et al. A peptidomimetic inhibitor of farnesyl:protein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer Res. 1995;55:5302–9. [PubMed] [Google Scholar]
- 30.Adjei AA, Mauer A, Bruzek L, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21:1760–6. doi: 10.1200/JCO.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 31.Cohen SJ, Ho L, Ranganathan S, et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J Clin Oncol. 2003;21:1301–6. doi: 10.1200/JCO.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 32.Johnston SR, Hickish T, Ellis P, et al. Phase II study of the efficacy and tolerability of two dosing regimens of the farnesyl transferase inhibitor, R115777, in advanced breast cancer. J Clin Oncol. 2003;21:2492–9. doi: 10.1200/JCO.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 33.Rao S, Cunningham D, de Gramont A, et al. Phase III double-blind placebocontrolled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–7. doi: 10.1200/JCO.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 34.Sharma S, Kemeny N, Kelsen DP, et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann Oncol. 2002;13:1067–71. doi: 10.1093/annonc/mdf173. [DOI] [PubMed] [Google Scholar]
- 35.Winquist E, Moore MJ, Chi KN, et al. A multinomial Phase II study of lonafarnib (SCH 66336) in patients with refractory urothelial cancer. Urol Oncol. 2005;23:143–9. doi: 10.1016/j.urolonc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 36.Ho CL, Kurman RJ, Dehari R, Wang TL, Shih Ie M. Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer Res. 2004;64:6915–8. doi: 10.1158/0008-5472.CAN-04-2067. [DOI] [PubMed] [Google Scholar]
- 37.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 38.Moasser MM, Sepp-Lorenzino L, Kohl NE, et al. Farnesyl transferase inhibitors cause enhanced mitotic sensitivity to taxol and epothilones. Proc Natl Acad Sci USA. 1998;95:1369–74. doi: 10.1073/pnas.95.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim ES, Kies MS, Fossella FV, et al. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer. 2005;104:561–9. doi: 10.1002/cncr.21188. [DOI] [PubMed] [Google Scholar]
- 40.Dilworth JT, Wojtkowiak JW, Mathieu P, et al. Suppression of Proliferation of two Independent NF1 Malignant Peripheral Nerve Sheath Tumor Cell Lines by the pan-ErbB Inhibitor CI-1033. Cancer Biol Ther. 2008;7:1938–46. doi: 10.4161/cbt.7.12.6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du W, Lebowitz PF, Prendergast GC. Cell growth inhibition by farnesyltransferase inhibitors is mediated by gain of geranylgeranylated RhoB. Mol Cell Biol. 1999;19:1831–40. doi: 10.1128/mcb.19.3.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du W, Prendergast GC. Geranylgeranylated RhoB mediates suppression of human tumor cell growth by farnesyltransferase inhibitors. Cancer Res. 1999;59:5492–6. [PubMed] [Google Scholar]
- 43.Mazieres J, Tillement V, Allal C, et al. Geranylgeranylated, but not farnesylated, RhoB suppresses Ras transformation of NIH-3T3 cells. Exp Cell Res. 2005;304:354–64. doi: 10.1016/j.yexcr.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Ashar HR, James L, Gray K, et al. Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J Biol Chem. 2000;275:30451–7. doi: 10.1074/jbc.M003469200. [DOI] [PubMed] [Google Scholar]
- 45.Schafer-Hales K, Iaconelli J, Snyder JP, et al. Farnesyl transferase inhibitors impair chromosomal maintenance in cell lines and human tumors by compromising CENP-E and CENP-F function. Mol Cancer Ther. 2007;6:1317–28. doi: 10.1158/1535-7163.MCT-06-0703. [DOI] [PubMed] [Google Scholar]
- 46.Basso AD, Mirza A, Liu G, Long BJ, Bishop WR, Kirschmeier P. The farnesyl transferase inhibitor (FTI) SCH66336 (lonafarnib) inhibits Rheb farnesylation and mTOR signaling. Role in FTI enhancement of taxane and tamoxifen anti-tumor activity. J Biol Chem. 2005;280:31101–8. doi: 10.1074/jbc.M503763200. [DOI] [PubMed] [Google Scholar]
- 47.Clark GJ, Kinch MS, Rogers-Graham K, Sebti SM, Hamilton AD, Der CJ. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J Biol Chem. 1997;272:10608–15. doi: 10.1074/jbc.272.16.10608. [DOI] [PubMed] [Google Scholar]
- 48.Gau CL, Kato-Stankiewicz J, Jiang C, Miyamoto S, Guo L, Tamanoi F. Farnesyltransferase inhibitors reverse altered growth and distribution of actin filaments in Tsc-deficient cells via inhibition of both rapamycin-sensitive and -insensitive pathways. Mol Cancer Ther. 2005;4:918–26. doi: 10.1158/1535-7163.MCT-04-0347. [DOI] [PubMed] [Google Scholar]
- 49.Philips MR, Cox AD. Geranylgeranyltransferase I as a target for anti-cancer drugs. J Clin Invest. 2007;117:1223–5. doi: 10.1172/JCI32108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–29. doi: 10.1593/neo.07970. [DOI] [PMC free article] [PubMed] [Google Scholar]