

Abstract
Microphthalmia-associated transcription factor, Mitf, has been shown to be necessary for regulating genes involved in osteoclast differentiation. Previously it was shown by others that Mitf translocates from the cytoplasm to the nucleus upon M-CSF/RANKL signaling in osteoclasts. Mitf’s movement is regulated by its interaction with 14-3-3 and the kinase C-TAK1. Here we demonstrate that the related family member, Tfe3, does not shuttle from the cytoplasm to the nucleus and does not interact with C-TAK1. We also demonstrate that overexpression of C-TAK1 inhibits the expression of Acp5 while a kinase dead C-TAK1 or a Mitf mutant that cannot interact with C-TAK1 increased expression of Acp5. Finally, we show that the catalytic subunit of protein phosphatase 2A is upregulated in osteoclasts with M-CSF/RANKL signaling indicating a possible mechanism for dephosphorylating Mitf on its 14-3-3 binding site and allowing Mitf to be translocated to the nucleus of osteoclasts.
Keywords: C-TAK1, 14-3-3, Mitf, Tfe3, PP2A, osteoclasts
Introduction
Bone is a dynamic tissue whose balance is maintained by the osteoblasts, which are responsible for the mineralization of bone and the osteoclasts, which are responsible for resorbing bone. One of the most informative methods for identifying genes regulating osteoclast involvement of bone development has been mouse mutants. Mutations in mice that have global effects on osteoclastogenesis typically reside in genes encoding signaling proteins or transcription factors required for osteoclast differentiation. One of these genes identified that regulates osteoclast differentiation is the transcription factor, microphthalmia-associated transcription factor (Mitf).
Mitf belongs to the MiT family of basic helix-loop-helix transcription factors that have been shown to regulate gene expression in a variety of cell types including melanocytes, macrophages and osteoclasts [1]. The MiT family includes Mitf, Tfe3, Tfeb and Tfec [2; 3]. Mitf’s ability to activate gene expression has been shown to be required for activating genes necessary for osteoclast differentiation [4; 5; 6]. Weilbaecher et al. as well as others [7; 8] have shown that Tfe3 and Mitf are expressed in osteoclasts, where they form heterodimers. It has also been shown that both Mitf and Tfe3 can activate Cathepsin K (Ctsk) [4] and that they can synergistically activate the Acp5 promoter (encoding TRACP) [5].
In hetero- or homozygous mice, Tfe3 mutations do not have a detectable phenotype that is associated with pigmentation, eye or bone development [9]. A few strong semidominant Mitf mutations induce complete or partial osteopetrosis (in particular the Mitfmi mutations); however loss of Mitf expression does not induce osteopetrosis. It has been suggested that the phenotype of the Mitfmi is caused by the dominant negative action of this allele and its interference with Tfe3 [1]. In homozygous Mitfmi mutant animals, osteoclasts express less TRACP [5] and Cathepsin K mRNA [4]. Mice null for Tfe3 and Mitf develop osteopetrosis [9]. The genetic data suggest that in osteoclasts Mitf and Tfe3 are functionally redundant and that osteoclast differentiation requires expression of either Mitf or Tfe3 unlike the biochemical data which suggested that they function as heterodimers.
Relatively little is known about proteins that interact with and potentially regulate the ability of Mitf and Tfe3 to activate osteoclast promoters during M-CSF and RANKL signaling. To identify proteins that interact with and regulate Mitf and Tfe3, we used a yeast two hybrid system designed to identify proteins that interact with transactivation domains of transcription factors such as Mitf [10]. Using this yeast two hybrid system, we had identified POH1 [11] and C-TAK1 (Cdc25C associated kinase 1) as proteins that interact with the activation domain of Mitf. C-TAK1 is a kinase that has been shown to phosphorylate various proteins including Cdc25C phosphatase [12]. C-TAK1 has been shown to regulate these proteins in vivo through the generation of 14-3-3 binding sites [13; 14]. Disruption of the 14-3-3 binding site in the proteins phosphorylated by C-TAK1 leads to aberrant protein localization [12; 13; 14]. C-TAK1 not only phosphorylates these proteins, but also has been shown to stably associate with them [12; 14]. C-TAK1 recognizes the amino acid sequence RsxS*xP, which is also a 14-3-3 binding site, and phosphorylates the S* [14; 15].
In this paper, we found Tfe3 expression, unlike Mitf expression, is only in the nucleus of osteoclasts. We confirm that Mitf and demonstrate that Tfe3 can interact with 14-3-3, but only Mitf’s interaction with 14-3-3 is phosphatase sensitive. We also verified the interaction between Mitf and C-TAK1 as demonstrated by our yeast two hybrid results and Bronisz et al. [15], but surprisingly we found that the related family member Tfe3 does not interact with C-TAK1. We also show that C-TAK1 overexpression affects osteoclast gene expression and formation of multinuclear TRAP-positive cells. Finally we observed that the phosphatase PP2A is expressed in osteoclasts with M-CSF signaling and could be a potential mechanism(s) that regulates Mitf subcellular location.
Materials and Methods
Cell Culture, Luciferase Assays and Transfections
293T and RAW 264.7 c4 maintenance and transfection was done as previously described [11]. Osteoclasts were isolated from bone marrow of mice as previously described [16].
Antibodies and Chemicals
Antibodies used were as follow: FLAG (M2, Sigma), Mitf (21st century Biochemicals, Malboro, Mass), Tfe3 (BD Biosciences), HA (Covance), PP2A (catalytic subunit, Cell Signaling), pan 14-3-3 (Chemicon). M-CSF and RANKL were purchased from R & D Systems (Minneapolis, MN) and used at 10 ng/mL (M-CSF) or 60 ng/mL (RANKL). Recombinant his-tagged 14-3-3 was purchased from Enzo Life Sciences (Plymouth Meeting, PA). Okadaic acid (Sigma) was used at 20 nM and incubated with the cells 5 hours before harvesting.
Protein and nucleic acid analysis
RNA was extracted and real-time RT-PCR was performed as previously described [17]. Immunoprecipitations and immunoblots were done as previously described [13]. Phosphatase assay was done as previously described [15].
Plasmids and Mutagenesis
FLAG Mitf was previously described [15]. Dr. Helen Piwnica-Worms generously provided the HA-tagged CTAK plasmid. Dr. Deborah Morrison provided the plasmid encoding the HA-tagged 14-3-3. Mitf (M105A, L178A, M105A/L178A, S100A, S173A and S100A/S173A) and kinase dead CTAK mutant (D196N) were generated by QuikChange method (Stratagene). All constructs were verified by DNA sequencing.
Statistical analysis
All experiments were run in triplicates and results are expressed as mean ± SD. Student’s t-tests were used to compare data; p < 0.05 indicates significance.
Results
Mitf and Tfe3 are localized differently during osteoclast differentiation
It has been reported previously that in osteoclasts Mitf, upon M-CSF and RANKL stimulation, translocates from the cytoplasm to the nucleus [15]. We wanted to confirm this finding and determine if Tfe3, another member of the MiT family thought to play an important role in osteoclast differentiation [9], also changes subcellular localization during osteoclast differentiation. Osteoclasts were grown in the absence of M-CSF and RANKL for 4-6 h (0 RANKL, −M-CSF), stimulated with M-CSF for 24 h (0 RANKL, +M-CSF) or M-CSF and RANKL (24 RANKL, +M-CSF) for 24 h. As shown in Figure 1, we were able to detect Mitf exclusively in the cytoplasmic fraction of osteoclasts when both M-CSF and RANKL were removed (lane 1). By 24 h of stimulation with M-CSF and RANKL, we detected Mitf exclusively in the nuclear fraction (lanes 5-6). Unlike Mitf, we could only detect Tfe3 in the nuclear fraction of osteoclasts even when both M-CSF and RANKL stimulation were removed (see Figure 1, lane 4-6). As loading controls and to ensure complete fractionation, the blots were reprobed with α-tubulin, which is a cytoplasmic protein and lamin A/C, which is a nuclear protein. These data suggest that Mitf and Tfe3 subcellular localization is regulated differently during osteoclast differentiation. These findings are intriguing since it has been suggested based on mouse models that Mitf and Tfe3 have redundant functions in osteoclasts [9].
Figure 1. Nuclear localization of Mitf and Tfe3.
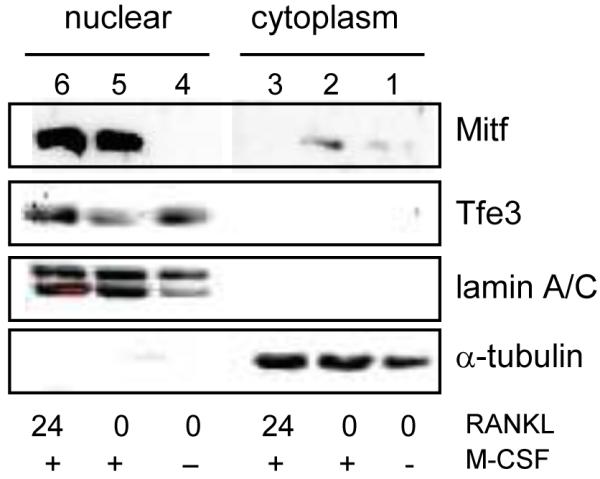
Representative immunoblot of Mitf and Tfe3 lysates from bone marrow derived-osteoclasts. M-CSF and RANKL were removed for 4-6 h, stimulated with M-CSF or M-CSF and RANKL for 24 hours, and immunoblotted.
Mitf and Tfe3 both interact with 14-3-3 protein
Overexpression of 14-3-3 in a transgenic mouse model was shown to increase the cytoplasmic localization of Mitf [15]. Since we detected a difference in localization of Mitf and Tfe3 during the initiation of osteoclast differentiation, we wanted to test if Tfe3 interacts with 14-3-3. We overexpressed a HA-tagged 14-3-3, with either a FLAG-Mitf or FLAG-Tfe3 in 293T cells. Cell lysates were immunoprecipitated with an antibody that recognized either the FLAG-tagged Mitf or Tfe3 and analyzed by immunoblot for the presence of HA-tagged 14-3-3 protein. As was previously reported, we observed that 14-3-3 is a binding partner of Mitf (Figure 2A, lane1)[15]. We also determined that Tfe3 and 14-3-3 interact (Figure 2A, lane 5), as was predicted based upon sequence similarity with other 14-3-3 interacting proteins [18]. We next wanted to determine which amino acids are necessary for Mitf to interact with 14-3-3. A loss of interaction between Mitf and 14-3-3 was obseved when both serines at amino acid residues 100 and 173 were mutated to alanine (Figure 2A, lanes 2-4). Comparable results were obtained when we used recombinant his-tagged 14-3-3 instead of HA-tagged 14-3-3 (data not shown). We were also able to detect an interaction by immunoprecipitation between endogenous 14-3-3, Mitf and Tfe3 in RAW 264.7 c4 cells stimulated with M-CSF (Figure 2B).
Figure 2. Mitf and Tfe3 bind to 14-3-3 and C-TAK1.

(A) The HA-tagged 14-3-3 construct and the FLAG-tagged Mitf constructs were expressed in 293T cells and complexes from whole-cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by immunoblot with the indicated antibodies. (B) RAW 264.7 c4 cells were stimulated with 10 ng/mL M-CSF for 24 hours. Cell lysates were immunoprecipitated with an antibody that recognizes IgG (lane 1), Mitf (lane 2) or Tfe3 (lane 3) and analyzed by immunoblot with an antibody that recognizes 14-3-3. (C) HA-tagged C-TAK1 construct and FLAG-tagged Mitf or Tfe3 were expressed in 293T cells and complexes from whole-cell lysates were immunoprecipitated with FLAG antibody and analyzed by immunoblot. (D) FLAG-Mitf or Tfe3 coexpression with HA-14-3-3. Cell lysates were untreated, treated with 20 nM okadaic acid before harvesting, or the whole-cell lysate was treated with 40 U/μl CIAP.
Tfe3 does not interact with C-TAK1
Previous work has shown that Mitf interacts with the kinase C-TAK1 [15]. C-TAK1 is thought to phosphorylate Mitf, creating a 14-3-3 binding site. Since Tfe3 was detected in the nucleus fraction of osteoclasts and an interaction between Tfe3 and 14-3-3 was observed, we wanted next to determine if Tfe3 (like Mitf) interacts with C-TAK1. To do this, we overexpressed HA-CTAK1, FLAG-Mitf or FLAG-Tfe3 in 293T cells. Cell lysates were immunoprecipitated with an antibody to the FLAG-tagged Mitf or Tfe3 and analyzed by immunoblot for the presence of HA-tagged CTAK1. We found FLAG-Mitf but not FLAG-Tfe3 in the presence of the HA-tagged CTAK1 (Figure 2C, lanes 1 and 2).
Tfe3’s interaction with 14-3-3 is not phosphorylation dependent
14-3-3 binds to specific phospho-serines or threonines in their binding partner. C-TAK1 is the kinase shown to phosphorylate the serine or threonine in the 14-3-3 site. It has been shown that Mitf’s interaction with 14-3-3 is phosphorylation dependent [15], so we wanted to determine if Tfe3’s interaction with 14-3-3 is also phosphorylation dependent. Treatment of transfected cells with the phosphatase inhibitor, okadaic acid, increased the interaction between Mitf and 14-3-3 but did not change the interaction between Tfe3 and 14-3-3 (Figure 2D, lanes 1, 3, 4, and 6). Dephosphorylation of Mitf with alkaline phosphatase reduced binding to 14-3-3 but it did not affect Tfe3’s interaction with 14-3-3 (Figure 2D, compare lanes 1, 2, 4, and 5).
Amino terminus of Mitf is necessary for C-TAK1 interaction
Using site-directed mutagenesis, we mapped the Mitf amino acid residues necessary for Mitf’s interaction with C-TAK1. C-TAK1 substrates contain the following motif RxxSΦxxxΦ [14] where x is any amino acid and Φ is a hydrophobic amino acid. Mitf contains two C-TAK1 motifs. One is found from amino acid residues 97-105 and the other motif is located at amino acid residues 170-178. We determined that the methionine of Mitf at amino acid residues 105 and the lysine at amino acid residue 178 were necessary for Mitf’s interaction with C-TAK1 (Figure 3A, lane 4). It was previously found that Mitf amino acid residue 178 is necessary for Mitf’s interaction with C-TAK1 [15]. The model that both methionine at 105 and the lysine at 178 are required for the Mitf/C-TAK1 interaction may explain why Tfe3 cannot bind to C-TAK1 since the methionine at residue 105 in Mitf is not present in Tfe3 [19].
Figure 3. C-TAK1 inhibits Mitf’s ability to activate transcription.

(A) The HA-tagged C-TAK1 construct and the FLAG-tagged Mitf point mutants were transfected in 293T cells and complexes from whole-cell lysates were immunoprecipitated with FLAG antibody and analyzed by immunoblot with the indicated antibodies. Real-time RT-PCR from (B) RAW 264.7 c4 cells that were either mock transfected, transiently transfected with full length C-TAK1 **p<0.001 vs. M-CSF mock transfected, **p<0.003 vs. M-CSF C-TAK1 transfected or (C) full length C-TAK1 containing the D196N mutant or Mitf mutant M105A/L178A. Cells were stimulated with M-CSF or M-CSF and RANKL for 5 days. **p<0.0015 vs. RANKL mock transfected, ***p<0.0001 vs. RANKL mock transfected. (D) RAW 264.7c4 cells were either mock-transfected or transfected with wt full-length C-TAK1, D196N C-TAK1, wt FLAG-Mitf or FLAG-Mitf M105/L178A. Cells were cultured in M-CSF and RANKL for 7 d. The number of multinuclear cells (greater than 2 nuclei/cell) was counted and the results of two experiments performed in triplicate are shown. **p<0.007 vs. D196N and **p<0.008 vs. FLAG-Mitf.
Interaction between C-TAK1 and Mitf regulates the activation of Mitf
To further characterize the activity of C-TAK1, RAW 264.7 c4 cells were transfected with wild type C-TAK1. Cells were then stimulated with M-CSF or M-CSF and RANKL for 5 days. Total RNA was isolated and real-time RT-PCR was performed to measure Acp5 gene expression. In mock-transfected RAW 264.7 c4 cells stimulated with M-CSF and RANKL, we detected a 6-fold increase in Acp5 gene expression, but only a 2-fold increase with the transfection of wild type C-TAK1 (Figure 3B). Transfection of the kinase dead C-TAK1 mutant D196N increased Acp5 gene expression by 4-fold compared to M-CSF-treated cells and approximately 4-fold increase in Acp5 gene expression compared to M-CSF/RANKL-treated cells (Figure 3C). When the Mitf point mutant M105A/L178A a mutant we determined did not interact with C-TAK1, we observed a similar increase (i.e., 4-fold) in Acp5 gene expression, which is comparable to what we observed when we transfected the D196N C-TAK1 mutant. However, M-CSF/RANKL-treated cells with the M105A/L178A Mitf mutant we observed a 6-fold increase in Acp5 gene expression (Figure 3D).
C-TAK1 inhibits differentiation of RAW 264.7 cells
Since C-TAK1 inhibited Mitf’s activation of osteoclast genes, we next asked whether C-TAK1 overexpression inhibited differentiation of RAW 264.7 cells. The RAW 264.7 preosteoclast cell line was transfected with full-length C-TAK1, C-TAK1 D196N mutant, FLAG-Mitf or the FLAG-MItf M105/L178A mutant (Figure 3A) and grown in presence of both M-CSF and RANKL for 7d to induce formation of multinuclear cells. As shown in Figure 3D, full length C-TAK1 significantly inhibited (2.5 fold) formation of TRAP positive cells compared to when the kinase dead C-TAK1 were overexpressed in RAW 264.7 c4 cells. Compared to FLAG-Mitf, FLAG-Mitf M105/L178A enhanced (1.5 fold) formation of TRAP positive cells when overexpressed in RAW 264.7 c4 cells.
Inhibition of PP2A activity regulates Mitf target genes
Previous work has shown that M-CSF stimulation of macrophages increases expression of the catalytic subunit of protein phosphatase 2A (PP2A, [20]). For other substrates of C-TAK1, PP2A has been shown to be necessary to dephosphorylate the protein phosphorylated by C-TAK1 and prevent the substrate from binding to 14-3-3 protein [21]. It has not been previously shown in osteoclasts that M-CSF stimulation leads to an upregulation of PP2A. To determine if M-CSF stimulation of osteoclast precursors up-regulates the catalytic subunit of PP2A, osteoclast precurors were grown in the absence of M-CSF or stimulated with M-CSF or M-CSF/RANKL for 1 to 3 days. Cytoplasmic and nuclear extracts were made from the stimulated osteoclast precursors and analyzed with an antibody that recognizes the catalytic subunit of PP2A. We detected an increase in expression of the catalytic subunit with M-CSF stimulation and when the osteoclast precursors were stimulated with M-CSF and RANKL for one day in the cytoplasmic fraction of osteoclasts. Expression in the cytoplasm would allow PP2A to interact with Mitf when osteoclasts were stimulated with M-CSF. PP2A activity could be the mechanism that releases Mitf/C-TAK1/14-3-3 and allows Mitf to translocate to the nucleus when osteoclasts are stimulated with M-CSF/RANKL.
If PP2A activity releases Mitf from 14-3-3 so that it can enter the nucleus, we would expect that expression from genes that Mitf targets during osteoclast differentiation should be affected. We inhibited PP2A activity using okadaic acid, an inhibitor with substantial preference for PP2A over PP1 [22]. Osteoclast cultures were treated with M-CSF, M-CSF/RANKL or M-CSF/RANKL/ okadaic acid and real-time RT-PCR was done to measure gene expression of the Acp5 and Ctsk. We detected a 7-fold increase of the Acp5 promoter when cells were treated with M-CSF/RANKL but only a 2-fold increase when the cells were treated with M-CSF/RANKL/OA (Figure 4B). A significant decrease in Ctsk expression was also observed when the osteoclasts were treated with okadaic acid (Figure 4B).
Figure 4. Expression of PP2A in osteoclasts.

(A) Representative immunoblot of lysates from bone marrow-derived osteoclasts were M-CSF and RANKL was removed for 4-6 h, stimulated with M-CSF or M-CSF and RANKL for up to 72 h and immunoblotted against the catalytic subunit of PP2A, α-tubulin and lamin A/C. (B) Real-time RT-PCR analysis of osteoclasts treated with or without 20 nM oakdaic acid 6 h before harvest for expression of Acp5 ***p<0.0002 vs. M-CSF treated, ***p<0.0005 vs. RANKL treated or cathepsin K, ***p<0.0001 vs. M-CSF treated, *p<0.01 vs. RANKL treated.
Discussion
In this study, we determined that, unlike previously demonstrated for Mitf, that the related family member Tfe3 does not shuttle from the cytoplasm to the nucleus with M-CSF/RANKL signaling. We have shown that both Mitf and Tfe3 interact with 14-3-3. We also demonstrated that only the Mitf/14-3-3 interaction is phosphatase sensitive. Other proteins such as human telomerase (hTERT) and the Exoenzyme S cytotoxin have been shown to interact with 14-3-3 in a phosphorylation-independent manner and are primarily dependent on hydrophobic residues [23]. In the case of hTERT, the failure to bind to 14-3-3 prevents hTERT localization to the nucleus [24]. It was shown that 14-3-3 binding to hTERT masked the nuclear export signal enhancing nuclear localization of hTERT [24]. Based on the data with hTERT and the fact that we detect 14-3-3 protein in the nucleus of osteoclasts (data not shown), we hypothesize that Tfe3’s interaction with 14-3-3 may keep Tfe3 localized to the nucleus. It will be important to map the domain of Tfe3 that interacts with 14-3-3 to further determine the mechanism of interaction between 14-3-3 and Tfe3.
We were able to confirm the interaction between Mitf and C-TAK1 as previously reported, but where unable to detect an interaction between Tfe3 and C-TAK1. We further demonstrated that Mitf requires both the methionine at residue 105 and lysine at residue 178 to interact with C-TAK1. We hypothesize that Tfe3 cannot interact with C-TAK1 because the methionine at residue 105, which is conserved only in the MiT family member Mitf, is not present in the Tfe3 sequence [19]. Another C-TAK1 substrate, KSR1, contains two serines that are phosphorylated by C-TAK1 and are necessary for KSR1 to bind to 14-3-3 [14]. Based on our data, we conclude that both sites are necessary for Mitf’s ability to interact with C-TAK1, and since Tfe3 lacks one of the two sites may explain why Tfe3 does not interact with C-TAK1.
Other proteins that interact with 14-3-3, such as KSR1 and HDAC7, interact with protein phosphatase 2A, which dephosphorylates the phosphoserines that bind 14-3-3, and allow the protein to shuttle out of the cytoplasm [22; 25]. We showed that upon M-CSF stimulation, and the first day of stimulation with RANKL, that PP2A expression is increased in the cytoplasm of osteoclasts. We also demonstrated that treatment of osteoclasts with okadaic acid, an inhibitor of PP2A, decreased gene expression of Mitf target genes Acp5 and Ctsk. We hypothesize that PP2A is responsible for allowing Mitf to move from the cytoplasm to the nucleus of osteoclasts.
In a knockout mouse model, it was shown that Mitf and Tfe3 serve redundant roles in osteoclast differentiation [9]. Even though null mice for all the different combinations of family members were generated, only null mice for both Mitf and Tfe3 have an osteopetrotic phenotype. This data indicates that the functional MiT family unit in osteoclasts is a Mitf or Tfe3 homodimer. Why then are two related proteins that serve redundant roles in osteoclast differentiation apparently being regulated differently? Tfe3 expression, unlike Mitf expression, is up-regulated during macrophage differentiation [26]. It was further shown that Tfe3 regulates expression of MafB, a master regulator of macrophage, but not osteoclast differentiation [27]. Our data suggests that the different subcellular regulation of Mitf and Tfe3 may be due to the different roles that they play during osteoclast and macrophage differentiation.
Acknowledgments
This work was supported by National Institutes of Health Grant 5R21-AR053946.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hemesath TJ, Steingrimsson E, McGill G, Hansen MJ, Vaught J, Hodgkinson CA, Arnheiter H, Copeland NG, Jenkins NA, Fisher DE. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8:2770–80. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- [2].Hershey CL, Fisher DE. Mitf and Tfe3: members of a b-HLH-ZIP transcription factor family essential for osteoclast development and function. Bone. 2004;34:689–96. doi: 10.1016/j.bone.2003.08.014. [DOI] [PubMed] [Google Scholar]
- [3].Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2004;38:365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- [4].Motyckova G, Weilbaecher KN, Horstmann M, Rieman DJ, Fisher DZ, Fisher DE. Linking osteopetrosis and pycnodysostosis: regulation of cathepsin K expression by the microphthalmia transcription factor family. Proc Natl Acad Sci U S A. 2001;98:5798–803. doi: 10.1073/pnas.091479298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Luchin A, Purdom G, Murphy K, Clark MY, Angel N, Cassady AI, Hume DA, Ostrowski MC. The microphthalmia transcription factor regulates expression of the tartrate-resistant acid phosphatase gene during terminal differentiation of osteoclasts. J Bone Miner Res. 2000;15:451–60. doi: 10.1359/jbmr.2000.15.3.451. [DOI] [PubMed] [Google Scholar]
- [6].So H, Rho J, Jeong D, Park R, Fisher DE, Ostrowski MC, Choi Y, Kim N. Microphthalmia transcription factor and PU.1 synergistically induce the leukocyte receptor osteoclast-associated receptor gene expression. J Biol Chem. 2003;278:24209–16. doi: 10.1074/jbc.M302940200. [DOI] [PubMed] [Google Scholar]
- [7].Weilbaecher KN, Motyckova G, Huber WE, Takemoto CM, Hemesath TJ, Xu Y, Hershey CL, Dowland NR, Wells AG, Fisher DE. Linkage of M-CSF signaling to Mitf, TFE3, and the osteoclast defect in Mitf(mi/mi) mice. Mol Cell. 2001;8:749–58. doi: 10.1016/s1097-2765(01)00360-4. [DOI] [PubMed] [Google Scholar]
- [8].Mansky KC, Sulzbacher S, Purdom G, Nelsen L, Hume DA, Rehli M, Ostrowski MC. The microphthalmia transcription factor and the related helix-loop-helix zipper factors TFE-3 and TFE-C collaborate to activate the tartrate-resistant acid phosphatase promoter. J Leukoc Biol. 2002;71:304–10. [PubMed] [Google Scholar]
- [9].Steingrimsson E, Tessarollo L, Pathak B, Hou L, Arnheiter H, Copeland NG, Jenkins NA. Mitf and Tfe3, two members of the Mitf-Tfe family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc Natl Acad Sci U S A. 2002;99:4477–82. doi: 10.1073/pnas.072071099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hirst M, Ho C, Sabourin L, Rudnicki M, Penn L, Sadowski I. A two-hybrid system for transactivator bait proteins. Proc Natl Acad Sci U S A. 2001;98:8726–31. doi: 10.1073/pnas.141413598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schwarz T, Sohn C, Kaiser B, Jensen ED, Mansky KC. The 19S Proteasomal Lid Subunit POH1 Enhances the Transcriptional Activation by Mitf In Osteoclasts. Journal of Cellular Biochemistry. 2009 doi: 10.1002/jcb.22475. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ogg S, Gabrielli B, Piwnica-Worms H. Purification of a serine kinase that associates with and phosphorylates human Cdc25C on serine 216. J Biol Chem. 1994;269:30461–9. [PubMed] [Google Scholar]
- [13].Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell. 2001;8:983–93. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- [14].Muller J, Ritt DA, Copeland TD, Morrison DK. Functional analysis of C-TAK1 substrate binding and identification of PKP2 as a new C-TAK1 substrate. EMBO J. 2003;22:4431–42. doi: 10.1093/emboj/cdg426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bronisz A, Sharma SM, Hu R, Godlewski J, Tzivion G, Mansky KC, Ostrowski MC. Microphthalmia-associated transcription factor interactions with 14-3-3 modulate differentiation of committed myeloid precursors. Mol Biol Cell. 2006;17:3897–906. doi: 10.1091/mbc.E06-05-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sharma SM, Bronisz A, Hu R, Patel K, Mansky KC, Sif S, Ostrowski MC. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J Biol Chem. 2007;282:15921–9. doi: 10.1074/jbc.M609723200. [DOI] [PubMed] [Google Scholar]
- [17].Rodriguez JS, Mansky KC, Jensen ED, Carlson AE, Schwarz T, Pham L, Mackenzie B, Prasad H, Rohrer MD, Petryk A, Gopalakrishnan R. Enhanced Osteoclastogenesis Causes Osteopenia in Twisted Gastrulation-Deficient Mice through Increased BMP Signaling. J Bone Miner Res. 2009 doi: 10.1359/JBMR.090507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jin J, Smith FD, Stark C, Wells CD, Fawcett JP, Kulkarni S, Metalnikov P, O’Donnell P, Taylor P, Taylor L, Zougman A, Woodgett JR, Langeberg LK, Scott JD, Pawson T. Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr Biol. 2004;14:1436–50. doi: 10.1016/j.cub.2004.07.051. [DOI] [PubMed] [Google Scholar]
- [19].Rehli M, Den Elzen N, Cassady AI, Ostrowski MC, Hume DA. Cloning and characterization of the murine genes for bHLH-ZIP transcription factors TFEC and TFEB reveal a common gene organization for all MiT subfamily members. Genomics. 1999;56:111–20. doi: 10.1006/geno.1998.5588. [DOI] [PubMed] [Google Scholar]
- [20].Wilson NJ, Moss ST, Csar XF, Ward AC, Hamilton JA. Protein phosphatase 2A is expressed in response to colony-stimulating factor 1 in macrophages and is required for cell cycle progression independently of extracellular signal-regulated protein kinase activity. Biochem J. 1999;339(Pt 3):517–24. [PMC free article] [PubMed] [Google Scholar]
- [21].Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein Phosphatase 2A Positively Regulates Ras Signaling by Dephosphorylating KSR1 and Raf-1 on Critical 14-3-3 Binding Sites. Current Biology. 2003;13:1356–1364. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- [22].Martin M, Potente M, Janssens V, Vertommen D, Twizere JC, Rider MH, Goris J, Dimmeler S, Kettmann R, Dequiedt F. Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc Natl Acad Sci U S A. 2008;105:4727–32. doi: 10.1073/pnas.0708455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ottmann C, Yasmin L, Weyand M, Veesenmeyer JL, Diaz MH, Palmer RH, Francis MS, Hauser AR, Wittinghofer A, Hallberg B. Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 2007;26:902–13. doi: 10.1038/sj.emboj.7601530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Seimiya H, Sawada H, Muramatsu Y, Shimizu M, Ohko K, Yamane K, Tsuruo T. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000;19:2652–61. doi: 10.1093/emboj/19.11.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13:1356–64. doi: 10.1016/s0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- [26].Zanocco-Marani T, Vignudelli T, Gemelli C, Pirondi S, Testa A, Montanari M, Parenti S, Tenedini E, Grande A, Ferrari S. Tfe3 expression is closely associated to macrophage terminal differentiation of human hematopoietic myeloid precursors. Exp Cell Res. 2006;312:4079–89. doi: 10.1016/j.yexcr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- [27].Zanocco-Marani T, Vignudelli T, Parenti S, Gemelli C, Condorelli F, Martello A, Selmi T, Grande A, Ferrari S. TFE3 transcription factor regulates the expression of MAFB during macrophage differentiation. Exp Cell Res. 2009;315:1798–808. doi: 10.1016/j.yexcr.2009.03.018. [DOI] [PubMed] [Google Scholar]