

Abstract
Both proapoptotic Bax and antiapoptotic Bcl-2 are structurally homologous to the pore-forming domain of bacterial toxins. Bax proteins oligomerize in the mitochondrial outer membranes forming pores that release cytochrome c from the mitochondrial intermembrane space. Bcl-2 proteins also form pores that, however, are much smaller than the Bax pore. It is unknown whether Bcl-2 forms monomeric or oligomeric pores. Here, we characterized the Bcl-2 pore formation in liposomes using biophysical and biochemical techniques. The results show that the Bcl-2 pore enlarges as the concentration of Bcl-2 increases, suggesting that the pore is formed by Bcl-2 oligomers. As expected from oligomerization-mediated pore-formation, the small pores are formed earlier than the large ones. Bcl-2 oligomers form pores faster than the monomer, indicating that the oligomerization constitutes an intermediate step of the pore formation. A Bcl-2 mutant with higher affinity for oligomerization forms pores faster than wild type Bcl-2. Bcl-2 oligomers were detected in the liposomal membranes under conditions that Bcl-2 forms pores, and the extent of oligomerization was positively correlated with the pore-forming activity. Therefore, Bcl-2 oligomerizes in membranes forming pores, but the extent of oligomerization and the size of the resulting pores are much smaller than that of Bax, supporting the model that Bcl-2 is a defective Bax.
Keywords: Bcl-2, tBid, pore, oligomerization, mitochondria, membrane
Introduction
Apoptosis is a stereotypic cell death program that is regulated primarily by Bcl-2 family proteins functioning as either suppressors or promoters. The ratio of active anti- and pro-apoptotic Bcl-2 family proteins determines the fate of cells. Alteration of the ratio by aberrant expression of these proteins impairs the normal apoptotic program contributing to various diseases, including cancer, autoimmunity and neurodegenerative disorders(1, 2). All the Bcl-2 family members contain at least one of the four Bcl-2 homology (BH) motifs. According to the function and sequence homology, Bcl-2 family is divided to three subfamilies. The anti-apoptotic Bcl-2 subfamily proteins such as Bcl-2, Bcl-xL and Mcl-1 contain all four BH motifs. The pro-apoptotic Bax subfamily members such as Bax and Bak display homology in BH1-3 motifs, whereas the pro-apoptotic BH3-only subfamily proteins such as Bid, Bim and Bad share the sequence homology only in the BH3 motif.
Despite an overall divergence in amino acid sequence and function, the three-dimensional structures of several Bcl-2 family proteins including members from all three subfamilies are remarkably similar to each other and to that of pore-forming domains of diphtheria toxin and E. coli colicins, consisting of one or two hydrophobic helices surrounded by five to eight amphipathic helices and their connecting loops(3-7). Consistent with the structural similarity with the pore-forming domains, Bcl-2 family proteins including anti-apoptotic Bcl-2 and pro-apoptotic Bax and tBid have been shown to have pore-forming activities in membranes(8-11). After binding to tBid, Bax forms large pores in mitochondrial outer membrane (MOM) or liposomal membrane with MOM characteristic lipid composition (MOM-liposomal membrane), releasing apoptotic proteins such as cytochrome c or large fluorescent dextran, respectively(12-14). Interestingly, although Bcl-2 inhibits Bax pore formation in both membranes, Bcl-2 itself also can be activated by tBid to form pores in the liposomal membrane but the size of Bcl-2 pore is much smaller than that of a Bax pore(14). The small pore-forming activity of Bcl-2 is strongly correlated with its anti-Bax activity in liposomes and isolated mitochondria as well as its anti-apoptotic function in cells(13, 14).
Structurally similar proteins may use different mechanisms to form pores in membranes. Oligomerization of Bax was found to be required for Bax pore formation in liposomal membrane and MOM, and its deleterious effect in cells(15-17). Oligomerization of diphtheria toxin was found to facilitate their pore formation(18). However, a different model was proposed for colicin pore formation, in which the pore is formed by monomeric colicin(19). Both monomeric and oligomeric Bcl-2 have been found in cells, but their relationship with the Bcl-2 pore is elusive(17, 20, 21).
In this study we reconstituted the pore-forming process of Bcl-2 in vitro using the MOM-liposome loaded with fluorescent dyes and purified cytosolic domain of Bcl-2 (Bcl-2ΔTM) tethered to the liposomal membrane. By monitoring the size of the Bcl-2 pore formed at different Bcl-2 concentrations, the release kinetics of dyes of different sizes, the pore-forming activity of Bcl-2 monomer, oligomer and Bcl-2 mutant with a higher homo-association affinity, and the oligomerization of Bcl-2 in the membrane under pore-forming conditions, we concluded that oligomerization is involved in Bcl-2 pore formation.
Experimental Procedures
Materials
All phospholipids and lipid analogs were purchased from Avanti Polar Lipids, Cascade Blue (CB, Mr ∼ 0.5 kDa), CB-labeled dextran of 3 kDa or 10 kDa and rabbit anti-CB polyclonal antibody from Molecular Probes, bis-maleimidohexane (BMH) from Pierce, Sepharose CL-2B from Sigama, and 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) from VWR International.
Preparation of Proteins
Expression and purification of His6-tagged Bcl-2 lacking the transmembrane (TM) sequence (His6-Bcl-2ΔTM), the G154A/G155A mutant in which Gly154 and Gly155 are replaced by Ala, the Cys-null mutant (C0) in which the only Cys158 is replaced by Ala, Bax and tBid proteins were done as described before(22, 23).
Preparation of Liposomes
Liposomes were made with the MOM characteristic lipids and Ni2+-chelating lipid analog in the membrane, and CB or CB-dextran in the lumen by an extrusion method as described before(14, 24).
Assay of CB or CB-dextran Release from Liposomes
The release of fluorescent molecules from liposomes by various proteins (indicated in figure legend) was measured as described(14).
Gel filtration chromatography of Bcl-2 proteins
Soluble His6-Bcl-2ΔTM and the mutant proteins were analyzed by gel filtration chromatography using a Superdex 200 HR10/30 column as described(22). The membrane-bound Bcl-2 proteins were generated by incubating the proteins in 500 μl of buffer A (50 mM Na2HPO4, and 3 mM citric acid, pH 7.4) with tBid and liposomes at 37 °C for 3 hr, and separated from soluble proteins using a Sepharose CL-2B gel filtration column (18 cm × 0.4 cm i.d.) developed with buffer A under gravity. CHAPS (2% w/v) was added to the liposome fractions to solubilize the membrane-bound proteins. The sample was then injected into the Superdex 200 gel filtration column in an AKTA-FPLC (Amersham Biosciences), and eluted with buffer B [300 mM NaCl, 0.2 mM dithiothreitol, 2% (w/v) CHAPS, 20 mM Hepes, pH 7.4] with a flow rate of 0.4 ml/min at 25 °C. The eluted fractions (0.5-ml each) were precipitated with Cl3CCOOH and analyzed by 15% SDS-PAGE and immunoblotting using a Bcl-2-specific antibody. The membrane-bound Bax was generated and analyzed similarly, except that a Bax-specific antibody was used in the immunoblotting.
Molecular weight determination of monomeric Bcl-2 protein
His6-Bcl-2ΔTM protein (80 μg) was injected into the Superdex 200 gel filtration column in a Biocad FPLC (Perseptive Biosystems) in buffer C (200 mM NaCl, 2 mM EDTA, 0.5% (v/v) glycerol, and 50 mM Tris-HCl, pH 8.5) with a flow rate of 0.4 ml/min at 25 °C. The eluted fractions were subject to multi-angle laser light scattering analysis using a DAWN DSP laser photometer in line with an OPTILAB DSP interferometric refractometer (Wyatt Technologies). The weight average molar mass of the protein in each fraction was calculated using the Astra software, version 4.73.04.
Crosslinking of Bcl-2 proteins
To examine Bcl-2 homo-association in solution, 2 μM of His6-Bcl-2ΔTM protein or the C0 mutant were incubated in 120 μl of buffer A for 1 hr at 25 °C. Crosslinking of the Bcl-2 protein in the sample was carried out by addition of 10 μM BMH and incubation at 25 °C for 30 min. The control reaction was done in parallel by addition of the same volume of Me2SO, the solvent for BMH solution. The chemical reaction was stopped by addition of 25 mM β-mercaptoethanol. Proteins in the sample were precipitated with Cl3CCOOH and analyzed by SDS-PAGE and Coomassie Brilliant Blue staining.
To determine Bcl-2 homo-association in the membrane, 4 μM of His6-Bcl-2ΔTM protein were incubated with 1 mM of the Ni2+-chelating liposomes in 116 μl of buffer D (50 mM Na2HPO4, and 25 mM citric acid, pH 5.0) at 25 °C for 1 hour. The sample was then mixed with 184 μl of 2.2 M sucrose in buffer D to bring the final sucrose concentration to 1.35 M. The resulting sample was added to a centrifuge tube and overlaid with 500 μl of 0.8 M sucrose in buffer D and then 200 μl of 0.25 M sucrose in buffer D. The sample was centrifuged in Beckman Optima Ultracentrifuge with TLA 100.2 rotor at 436,000 × g for 6 hrs at 4 °C. The top 500 μl fraction that contains liposome-bound proteins was collected, half of which was treated with BMH and the other half was treated with Me2SO as described above, so as the following sample processing and analysis.
Fluorescence anisotropy of Bcl-2 proteins
Fluorescence anisotropy of His6-Bcl-2ΔTM and the G154A/G155A mutant was measured, and the data was fit with a model for Bcl-2 homo-association to determine the dissociation constant as described before(24).
Results
Recombinant Bcl-2 protein with the C-terminal transmembrane (TM) sequence replaced by a His6-tag (His6-Bcl-2ΔTM) was prepared. Liposome was made with the MOM characteristic lipids and a Ni2+-chelating lipid analog that binds to the His6-tag at the C-terminus of the recombinant Bcl-2, thereby generating a membrane-tethered Bcl-2 mimicking the native MOM-bound Bcl-2. The liposome was also loaded with fluorescence dye, Cascade Blue (CB, Mr ∼0.5 kDa), or CB-labled dextrans (Mr ∼3 or 10 kDa). Pore-formation by the Bcl-2 would release the fluorescent molecules from the liposome, which would be bound by the anti-CB antibody outside the liposome causing reduction (quenching) of the CB fluorescence. The extent and kinetics of fluorescence quenching indicate the extent and kinetics of dye release from the liposome, which directly correlate with the extent and kinetics of Bcl-2 pore formation in the membrane.
Direct correlation between concentration and pore size of Bcl-2
Using above approach, we examined the extent of release of the three different sized fluorescent molecules by different concentrations of Bcl-2 after interaction with tBid. As shown in Figure 1A, significant release of 0.5-kDa CB started at a lower Bcl-2 concentration (25 nM) than that for 3-kDa CB-dextran (50 nM). Release of 10-kDa CB-dextran was not detected even at the highest Bcl-2 concentration (100 nM). If Bcl-2 pores are formed only by monomers, these pores should have a uniform size that will be independent of Bcl-2 concentration. Since the results show that the Bcl-2 pore size is positively dependent on Bcl-2 concentration, it suggests that Bcl-2 oligomerization is involved in the formation of the pores, at least the large one.
Figure 1. Effect of Bcl-2 concentration on release of molecules of different sizes from liposomes.

A, extent of release of 0.5-kDa CB dyes (black bar), 3 (stretched bar) or 10-kDa (white bar) CB-dextrans from 12.5 μM Ni2+-chelating liposome by various concentrations of His6-Bcl-2ΔTM protein in the presence or absence of 10 nM tBid was determined by the extent of fluorescence quenching (ΔFBcl-2ΔTM/ΔFTriton) after a 3-hr incubation at pH 7.4. Data shown are averages from three independent experiments with standard deviation (S.D., error bar). B, release kinetics of the CB dye and CB-dextrans from 12.5 μM Ni2+-liposome by 100 nM His6-Bcl-2ΔTM and 10 nM tBid at pH 7.4 was measured by continuously monitoring the extent of fluorescence quenching for 3 hr. Data shown are averages from three independent experiments with S.D. (error bars). (□) 0.5-kDa CB; (●) 3-kDa CB-dextran; (○) 10-kDa CB-dextran; (■) the negative control, 0.5-kDa CB loaded liposome incubated with protein buffer.
Inverse correlation between the kinetics of dye release by Bcl-2 and the size of the dye
To further test the model for Bcl-2 pore formation, the release kinetics of dyes of different sizes was examined. As shown in Figure 1B, even though both 0.5-kDa CB and 3-kDa CB-dextran were released from the liposome by 100 nM Bcl-2 after interaction with tBid, the small dye was released at a higher rate than the large one. The time for half of maximal release (t1/2) for CB and CB-dextran was ∼3 and 30 min, respectively. The 10-kDa CB-dextran was not released even after hours of incubation, ruling out the possibility that prolonged exposure of liposomes to Bcl-2 and tBid may cause non-specific damage to the membrane. If the Bcl-2 pores are formed only by monomers, the dyes of different sizes should be released from the liposome at the same rate that is dictated by the rate of Bcl-2 conversion from the membrane-tethered conformation to the pore conformation, a constant at a given protein concentration. In contrast, if the oligomerization is involved in the pore formation, the release rate of the small dye should be higher than that of large dye since the formation rate of small oligomeric pores should be higher than that of large oligomeric pores. Therefore, the above kinetics data provide further support to the model that Bcl-2 proteins form pores via oligomerization.
Enhancement of pore-formation by Bcl-2 oligomerization
To determine the effect of Bcl-2 oligomerization on the pore formation, we used Bcl-2 proteins in different oligomeric states or of different oligomerizing potencies. During purification of His6-Bcl-2ΔTM protein, we observed that the protein was eluted from Superdex 200 gel filtration column mainly in the monomeric and oligomeric fractions as judged by their co-elution with the protein standards of 25 and 200 kDa, respectively (Fig. 2A). The protein in both fractions is ∼90% pure according to SDS-PAGE analysis followed by Coomassie Blue staining (Fig. 2B). The identity of the protein was confirmed by immunoblotting using an antibody specific to Bcl-2 (Fig. 2C). The weight average molar mass of the monomeric protein is 24,540 ± 982 g/mol as determined by multi-angle light scattering analysis (Fig. 2D), closed to the calculated value for monomeric His6-Bcl-2ΔTM, 24,795 g/mol, confirming that the monomeric protein is the monomeric Bcl-2. Conceivably the oligomeric protein is a Bcl-2 oligomer since it eluted earlier than the monomeric protein from the gel filtration column. The purified Bcl-2 monomer and oligomer are quite stable since they were eluted in the corresponding fraction after a second gel filtration chromatography (Fig. 2E). This provided us an opportunity to test which form of the Bcl-2 protein is more efficient in pore formation. As shown in Figure 2F, both monomeric and oligomeric Bcl-2 proteins released the same amount of 3-kDa CB-dextrans from the liposomes after a 3-hr incubation. However, the kinetics of dye release by the two forms of Bcl- is different. The rate of release induced by the oligomeric Bcl-2 is much higher than that by the monomeric one since t1/2 for the former is ∼3.3 min and the latter ∼30 min. These results demonstrate that, although both Bcl-2 oligomer and monomer are competent for the pore formation, the oligomer is much more efficient in the pore formation than the monomer, indicating that the oligomerization of Bcl-2 is an intermediate step during the pore-forming reaction.
Figure 2. Effect of Bcl-2 homo-oligomerization on pore formation.
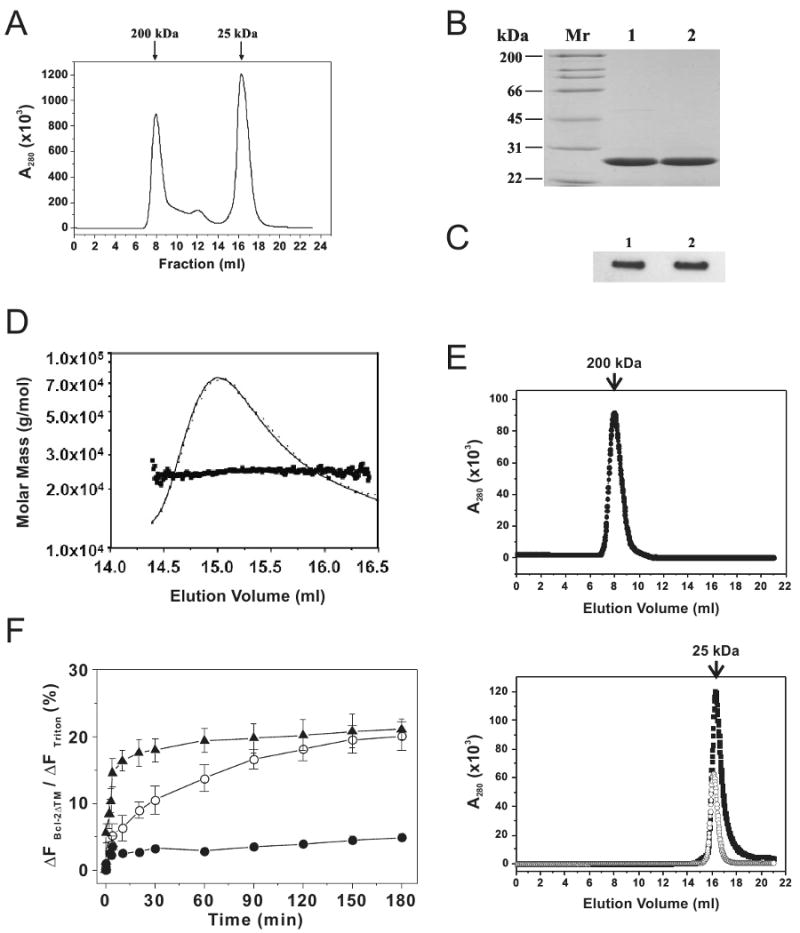
A, oligomerization of His6-Bcl-2ΔTM was examined by gel filtration chromatography. The A280 was monitored for each eluted fraction and plotted. The elution positions for protein standards are indicated on the top of the plot with corresponding Mr. B, purity of both monomeric (lane 1) and oligomeric (lane 2) His6-Bcl-2ΔTM proteins eluted from the above gel filtration column was analyzed by SDS-PAGE and Coomassie staining. Mr of protein standards is indicated on the left of the gel. C, identity of the monomeric (lane 1) and oligomeric (lane 2) His6-Bcl-2ΔTM proteins was determined by immunoblotting using a Bcl-2-specific antibody. D, weight average molar mass of His6-Bcl-2ΔTM monomer eluted from the gel filtration column was determined by multi-angle light scattering analysis. E, stability of wild-type His6-Bcl-2ΔTM oligomer (top plot, ●) and monomer (bottom plot, ■), or G154A/G155A mutant monomer (bottom plot, ○) in solution was tested by injecting the corresponding fractions from the first gel filtration column into the second one chromatography. F, release kinetics of 3-kDa CB-dextran from 12.5 μM Ni2+-liposome by 100 nM His6-Bcl-2ΔTM monomer or oligomer and 10 nM tBid at pH 7.4 was monitored as above. Data shown are averages with S.D. (error bars) from three independent experiments using the Bcl-2 oligomer (▲), monomer (○) or protein buffer (●).
In a previous study we generated a Bcl-2 mutant in which the two glycine residues at positions 154 and 155 were changed to alanine. This mutant showed a higher pore-forming activity in liposomes(14). By measuring the concentration-dependent increase of tryptophan anisotropy we found that the mutant protein has a 6.5-fold higher homo-association affinity than the wild type protein (Fig. 3A). When compared with monomeric wild-type protein, the monomeric mutant protein (Fig. 3B) had a much larger extent of formation of the pore that releases 3-kDa dye after a 3-hr incubation. In addition, the pore-forming kinetics was much faster with the mutant, with a t1/2 of ∼10 min, which is 3 times shorter than the t1/2 for the wild type (Fig. 3B). These data further support the model that oligomerization of Bcl-2 proteins facilitates the pore formation.
Figure 3. Effect of mutation on Bcl-2 homo-association and pore formation.

A, homo-association of wild-type His6-Bcl-2ΔTM or the G154A/G155A mutant was examined by measuring the fluorescence anisotropy after titrating the corresponding protein into buffer D. Data shown are averages of three independent titrations with S.D. (error bars), and (■) for wild-type and (●) for the mutant. Kd for the binding of wild-type or the mutant is 196 ± 43 or 30 ± 5 nM, respectively. B, release kinetics of 3-kDa CB-dextran from 12.5 μM Ni2+-liposome was monitored in the presence of 100 nM monomeric wild-type His6-Bcl-2ΔTM or the G154A/G155A mutant and 10 nM tBid at pH 7.4 as above. Data shown are averages with S.D. (error bars) from three independent experiments using the wild-type (○), mutant (●) or protein buffer (□).
Bcl-2 oligomerization in membrane detected by chemical cross-linking and gel filtration chromatography
To determine whether Bcl-2 oligomerization occurs in the liposomal membrane, a homobifunctional sulfhydryl-reactive cross-linker BMH was used. The His6-Bcl-2ΔTM protein contains only one cysteine at position 158. Therefore, only the dimer adduct can be generated (Fig. 4A), even though some Bcl-2 are in the oligomeric form as shown by gel filtration chromatography (Fig. 2A). The specificity of BMH reaction was demonstrated by the fact that no dimer adduct was observed even in the presence of BMH when a cysteine-null Bcl-2 mutant was used (Fig. 4A).
Figure 4. Oligomerization of Bcl-2 and Bax in liposomal membrane.
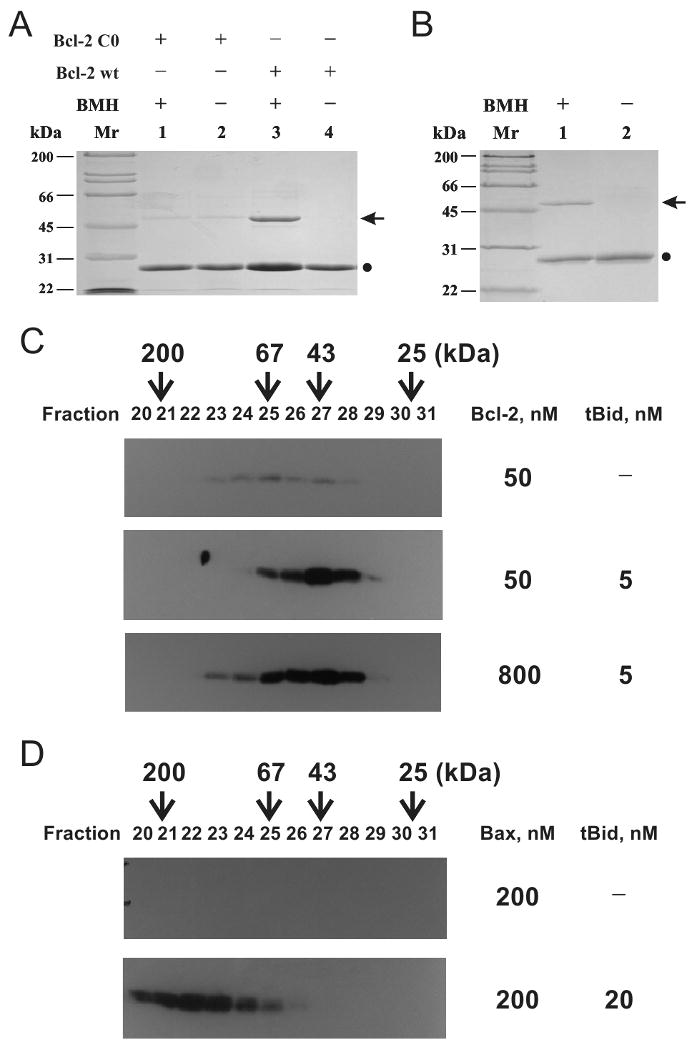
A, homo-association of His6-Bcl-2ΔTM in solution (pH 7.4) was detected by crosslinking with BMH. Data shown is a representative Coomassie-stained SDS-PAGE gel from three independent experiments. (●), Bcl-2 monomer; arrow, crosslinked Bcl-2 dimer. B, homo-association of His6-Bcl-2ΔTM in the liposomal membrane was monitored by BMH crosslinking after the protein was incubated with the liposome at pH 5.0, and the membrane-bound protein was purified using sucrose float-up centrifugation. Data shown is a representative from two independent experiments. C, oligomerization of 50 or 800 nM His6-Bcl-2ΔTM in the membrane was determined by gel filtration chromatography after incubating the protein at pH 7.4 with 12.5 μM Ni2+-liposome in the absence or presence of 5 nM tBid. The proteins in the eluted fractions were analyzed by SDS-PAGE and immunoblotting with a Bcl-2-specific antibody. The elution positions of protein standards are indicated on the top of the plot with Mr. D, oligomerization of 200 nM Bax in the membrane in the absence or presence of 20 nM tBid was determined similarly, except that a Bax-specific antibody was used in the immunoblotting.
Bcl-2 protein was incubated with the liposome at pH 5 for 3 hours, a condition that was known to promote the pore formation(14). After sucrose density float-up centrifugation, the top liposome-containing fraction was collected and subjected to BMH cross-linking. A BMH-dependent adduct was detected with a Mr ∼50 kDa, the predicted Mr of His6-Bcl-2ΔTM dimer (Fig. 4B). Since BMH contains two maleimide groups spaced by a 16 Å linker and the Bcl-2 molecule has dimensions of 50×45×35 (Å) according to the NMR structure(3), the cross-linking product is most likely formed by two Bcl-2 molecules that are bound to each other in the membrane. The data therefore demonstrate that at least some Bcl-2 proteins form dimer in the membrane under the pore-forming condition.
To determine the oligomeric state of membrane-bound Bcl-2 after interaction with tBid, another condition under which Bcl-2 forms pores (Fig. 1), we first isolated the liposomes after incubation with Bcl-2 and/or tBid using CL-2B gel filtration chromatography. The membrane-bound proteins were solubilized by CHAPS, a zwitter-ionic detergent that does not change the oligomeric state of Bcl-2 family proteins(25, 26), and then subjected to Superdex 200 gel filtration chromatography. As shown in Figure 4C, membrane-bound Bcl-2 was readily detected after incubation with liposomes in the presence of tBid. The main form of the Bcl-2 is dimer and trimer as judged by their co-elution with the protein standards of 43 and 67 kDa, respectively. In the absence of tBid, only residual Bcl-2 proteins are detected in these fractions, suggesting that a small fraction of the membrane-bound Bcl-2 protein is in the dimer and trimer states even in the absence of tBid. The tBid interaction substantially increases the amount of membrane-bound, dimeric and trimeric Bcl-2 proteins. Interestingly larger Bcl-2 oligomers with apparent Mr higher than 67 kDa was detected when a higher concentration of Bcl-2 was used. Since the Bcl-2 pore size is directly correlated with its concentration (Fig. 1A), this result suggests that the larger Bcl-2 oligomers form the larger Bcl-2 pores.
In our previous study, Bax also formed pores in the liposomal membrane after interaction with tBid(14). The Bax pore is larger than Bcl-2 pore, releasing 10-kDa CB-dextran. Conceivably the Bax pore is formed by a larger oligomer. Indeed, large Bax oligomers were detected in the membrane fractions using the gel filtration assay same as that for Bcl-2. In the presence of tBid, Bax oligomers with Mr from 67 to 200 kDa and higher were detected (Fig. 4D, bottom plot). In comparison Bcl-2 oligomers are all smaller than 200 kDa even at a concentration 4-fold higher than Bax (Fig. 4C). There was no Bax detected in the membrane fraction in the absence of tBid (Fig. 4D, top plot), consistent with the fact that Bax is a soluble protein prior to interaction with tBid. Taken together, these results demonstrated that both Bcl-2 and Bax form oligomers in the membrane after interaction with tBid, but the size of Bcl-2 oligomers are smaller than Bax oligomers, which correlates directly with their pore size.
Discussion
Based on the above results, the following conclusions can be drawn about the Bcl-2 pore formation in liposomal membrane. (i) The Bcl-2 pore has variable sizes that can release either 0.5 or 3-kDa molecule, but not 10-kDa one; (ii) The larger pore is formed at a higher Bcl-2 concentration than the smaller one; (iii) The kinetics of larger pore formation is slower than the smaller one; (iv) Pre-formed Bcl-2 oligomer has a faster kinetics to form pore than the monomer; (v) Bcl-2 mutant that has a higher homo-association potency than the wild-type protein also has a faster pore-forming kinetics than the wild-type protein. All of the conclusions support the model that Bcl-2 pores, particularly the larger one, are formed by Bcl-2 oligomer (Fig. 5). Detection of Bcl-2 oligomers in the membrane under pore-forming conditions is consistent with the model.
Fig. 5. Model for Bcl-2 pore formation.

Bcl-2 pore formation in membrane may proceed with the following steps. 1. Oligomerization of Bcl-2 proteins. This step may occur when Bcl-2 is in the tail-anchoring conformation (top row) or the multi-spanning conformation (bottom row) after interaction with tBid. 2. Conformational switch from the tail-anchoring to the multi-spanning. This step may occur before or after Bcl-2 oligomerization. 3. Pore formation. This step may occur after Bcl-2 switches to the multi-spanning conformation. The size of pore increases as the size of oligomer increases. Whether the monomeric Bcl-2 forms pore is questionable. Ovals, Bcl-2 proteins; Shaded area, membrane; black circles, molecules passing through Bcl-2 pores.
In cells Bcl-2 is constitutively bound to the MOM(27-29). The membrane-bound Bcl-2 may exist in a monomer-oligomer equilibrium before changing to the pore-forming conformation(22). The conformational change can be induced by interaction with tBid or Bax in vitro and by apoptotic drugs in cells(13, 14, 30). Oligomerization of Bcl-2 may facilitate the conformational change. Whether the conformation change enhances the oligomerization is unknown, so as whether the conformationally changed Bcl-2 monomer can form pore.
Similar pore-forming models have been proposed for other proteins such as diphtheria toxin. The pore-forming domain of diphtheria toxin, structurally homologous to the cytosolic domain of Bcl-2, forms pore in membrane at low pH. Similar to what we observed for Bcl-2, the size of the toxin pore increases in a concentration-dependent manner(18). In addition, self quenching of fluorescently labeled toxin and crosslinking of toxin were observed under the pore-forming condition. These data support the model that diphtheria toxin forms pore via oligomerization in membrane. The oligomerization may help insertion of the toxin into the membrane, since the insertion was detected only at high toxin concentration(31). Oligomerization should facilitate the efficient burial of hydrophilic residues that are generally more abundant in soluble proteins than transmembrane proteins(32). Since the pore-forming domain of diphtheria toxin as well as the cytosolic domain of Bcl-2 are water-soluble, they contain abundant hydrophilic residues. Oligomerization is likely the mechanism used to bury these residues in the transmembrane conformation of these proteins.
In contrast, the pore-forming domain of colicin, another structural homolog of the cytosolic domain of Bcl-2, seems to form monomeric pore. Evidence for the monomeric pore includes that the pore activity was detected at a stoichiometry of less than one colicin molecule per liposome, and that the dependence of the initial rate of ionic efflux from liposomes was linear(33). Although we cannot rule out the possibility that Bcl-2 monomer may form pore, significant release of 0.5-kDa CB dye, the smallest molecule tested, was detected at a protein:liposome ratio of ∼400:1, indicating that even the small pore is unlikely to formed by Bcl-2 monomer.
Whether Bcl-2 forms pore in vivo is unknown. However, a Bcl-2 conformational change was detected during apoptotic induction in cells and after interaction with tBid and/or Bax in isolated mitochondria(13, 30). Since a similar conformational change was detected during Bcl-2 pore formation in liposome induced by tBid and/or Bax(14), Bcl-2 may form pore in apoptotic cells. Interestingly Bcl-2 oligomers were detected in cells(17, 21). Whether the Bcl-2 oligomers form pores in these cells is elusive. Interestingly Bax oligomers and pores were formed in apoptotic cells, and oligomerization of Bax was required for its pore formation(15-17). Therefore, oligomerization is used by both pro- and anti-apoptotic proteins to form pores in the membrane. The extent of oligomerization determines the pore size and hence the activity during apoptosis induction. Since the extent of oligomerization and pore of Bcl-2 is much smaller than those of Bax, Bcl-2 can be viewed as a defective Bax(34). Moreover, Bcl-2 oligomerization and pore-formation are correlated with its anti-Bax activity(14, 22), Bcl-2 may function as a dominant-negative Bax, similar to its anti-apoptotic cousin Bcl-xL(35).
Bcl-2 can be converted to a pro-apoptotic molecule under certain condition such as interaction with nuclear receptor Nur77 or cleavage by caspase. While Nur77 interaction does not convert Bcl-2 to a Bax-like molecule that can form large pores in membranes(36, 37), caspase cleaved Bcl-2 promotes cytochrome c release from mitochondria(38), and caspase cleaved Bcl-xL forms large pore in liposomal membrane(39). Whether these caspase cleaved proteins form large oligomers in cells are unknown, however, overexpression of Bcl-2 has been shown to induce apoptosis(40, 41), consistent with the scenario that Bcl-2 at high concentrations may form large oligomers in the MOM and release cytochrome c.
Bcl-2 is overexpressed in most of human cancers and correlated with the resistance of the cancers to chemotherapeutic drugs and γ irradiation(42). One current strategy for development of anti-cancer drugs targeting Bcl-2 focuses on the BH3 mimetic that binds to Bcl-2 neutralizing its BH3-binding activity thereby its anti-apoptotic function(43, 44). The other strategy is to develop the Bcl-2 ligand that converts Bcl-2 to a pro-apoptotic protein(37). Since Bcl-2 homo-oligomerization may facilitate its pore-formation that correlates with its anti-apoptotic activity, targeting Bcl-2 oligomerization and pore-formation should be considered as an additional strategy for anti-cancer drug development. Assays established in our previous and current studies may facilitate the search for small chemicals that inhibit these Bcl-2 activities(14, 22, 24).
Acknowledgments
This work was supported in part by the grant GM062964 from the National Institutes of Health to J. L.
References
- 1.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 2.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 3.Petros AM, Medek A, Nettesheim DG, et al. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci USA. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 5.Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of Bid, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- 6.Choe S, Bennett MJ, Fujii G, et al. The crystal structure of diphtheria toxin. Nature. 1992;357:216–222. doi: 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- 7.Parker MW, Postma JP, Pattus F, Tucker AD, Tsernoglou D. Refined structure of the pore-forming domain of colicin A at 2.4 A resolution. J Mol Biol. 1992;224:639–657. doi: 10.1016/0022-2836(92)90550-4. [DOI] [PubMed] [Google Scholar]
- 8.Antonsson B, Conti F, Ciavatta A, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- 9.Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlesinger PH, Gross A, Yin XM, et al. Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic Bcl-2. Proc Nat Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schendel SL, Azimov R, Pawlowski K, Godzik A, Kagan BL, Reed JC. Ion channel activity of the BH3 only Bcl-2 family member: BID. J Biol Chem. 1999;274:21932–21936. doi: 10.1074/jbc.274.31.21932. [DOI] [PubMed] [Google Scholar]
- 12.Kuwana T, Mackey MR, Perkins G, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 13.Dlugosz PJ, Billen LP, Annis MG, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. Embo J. 2006;25:2287–2296. doi: 10.1038/sj.emboj.7601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng J, Tan C, Roberts GJ, et al. tBid elicits a conformational alteration in membrane-bound Bcl-2 such that it inhibits Bax pore formation. J Biol Chem. 2006;281:35802–35811. doi: 10.1074/jbc.M608303200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release in mitochondria. Biochem J. 2000;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- 16.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antonsson B, Montessuit S, Sanchez B, Martinou JC. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem. 2001;276:11615–11623. doi: 10.1074/jbc.M010810200. [DOI] [PubMed] [Google Scholar]
- 18.Sharpe JC, London E. Diphtheria toxin forms pores of different sizes depending on ots concentration in membranes: Probable relationship to oligomerization. J Membrane Biol. 1999;171:209–221. doi: 10.1007/s002329900572. [DOI] [PubMed] [Google Scholar]
- 19.Zakharov SD, Cramer WA. Colicin crystal structures: pathways and mechanisms for colicin insertion into membranes. Biochim Biophys Acta. 2002;1565:333–346. doi: 10.1016/s0005-2736(02)00579-5. [DOI] [PubMed] [Google Scholar]
- 20.Conus S, Kaufmann T, Fellay I, Otter I, Rosse T, Borner C. Bcl-2 is a monomeric protein: prevention of homodimerization by structural constraints. Embo J. 2000;19:1534–1544. doi: 10.1093/emboj/19.7.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Lapolla SM, Annis MG, et al. Bcl-2 homodimerization involves two distinct binding surfaces, a topographic arrangement that provides an effective mechanism for Bcl-2 to capture activated Bax. J Biol Chem. 2004;279:43920–43928. doi: 10.1074/jbc.M406412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem. 2003;278:48935–48941. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 24.Tan C, Dlugosz PJ, Peng J, et al. Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J Biol Chem. 2006;281:14764–14775. doi: 10.1074/jbc.M602374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- 26.Hsu YT, Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem. 1997;272:13829–13834. doi: 10.1074/jbc.272.21.13829. [DOI] [PubMed] [Google Scholar]
- 27.Janiak F, Leber B, Andrews DW. Assembly of Bcl-2 into microsomal and outer mitochondrial membranes. J Biol Chem. 1994;269:9842–9849. [PubMed] [Google Scholar]
- 28.Tsujimoto Y, Ikegaki N, Croce CM. Characterization of the protein product of bcl-2, the gene involved in human follicular lymphoma. Oncogene. 1987;2:3–7. [PubMed] [Google Scholar]
- 29.Chen-Levy Z, Nourse J, Cleary ML. The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol Cell Biol. 1989;9:701–710. doi: 10.1128/mcb.9.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW. During apoptosis bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol Cell. 2004;14:523–529. doi: 10.1016/s1097-2765(04)00263-1. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Malenbaum SE, Kachel K, Zhan H, Collier RJ, London E. Identification of shallow and deep membrane-penetrating forms of diphtheria toxin T domain that are regulated by protein concentration and bilayer width. J Biol Chem. 1997;272:25091–25098. doi: 10.1074/jbc.272.40.25091. [DOI] [PubMed] [Google Scholar]
- 32.Papini E, Schiavo G, Tomasi M, Colombatti M, Rappuoli R, Montecucco C. Lipid interaction of diphtheria toxin and mutants with altered fragment B. 2. Hydrophobic photolabelling and cell intoxication. Eur J Biochem. 1987;169:637–644. doi: 10.1111/j.1432-1033.1987.tb13655.x. [DOI] [PubMed] [Google Scholar]
- 33.Peterson AA, Cramer WA. Voltage-dependent, monomeric channel activity of colicin E1 in artificial membrane vesicles. J Membr Biol. 1987;99:197–204. doi: 10.1007/BF01995700. [DOI] [PubMed] [Google Scholar]
- 34.Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin B, Kolluri SK, Lin F, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 37.Kolluri SK, Zhu X, Zhou X, et al. A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell. 2008;14:285–298. doi: 10.1016/j.ccr.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirsch DG, Doseff A, Chau BN, et al. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J Biol Chem. 1999;274:21155–21161. doi: 10.1074/jbc.274.30.21155. [DOI] [PubMed] [Google Scholar]
- 39.Basanez G, Zhang J, Chau BN, et al. Pro-apoptotic cleavage products of Bcl-xL form cytochrome c-conducting pores in pure lipid membranes. J Biol Chem. 2001;276:31083–31091. doi: 10.1074/jbc.M103879200. [DOI] [PubMed] [Google Scholar]
- 40.Shinoura N, Yoshida Y, Nishimura M, et al. Expression level of Bcl-2 determines anti- or proapoptotic function. Cancer Res. 1999;59:4119–4128. [PubMed] [Google Scholar]
- 41.Wang NS, Unkila MT, Reineks EZ, Distelhorst CW. Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J Biol Chem. 2001;276:44117–44128. doi: 10.1074/jbc.M101958200. [DOI] [PubMed] [Google Scholar]
- 42.Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Neill J, Manion M, Schwartz P, Hockenbery DM. Promises and challenges of targeting Bcl-2 anti-apoptotic proteins for cancer therapy. Biochim Biophys Acta. 2004;1705:43–51. doi: 10.1016/j.bbcan.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 44.Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ. 2008;15:977–987. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]