

Abstract
Despite a wealth of information about the structure of surface membrane immunoglobulin (smIg) on chronic lymphocytic leukemia (CLL) cells, little is known about epitopes reacting with their binding sites. Probing phage-displayed peptide libraries, we identified and characterized mimetopes for Igs of 4 patients with IGHV mutated CLL (M-CLL) and 4 with IGHV unmutated CLL (U-CLL). Six of these mAbs were representatives of stereotyped B-cell receptors characteristic of CLL. We found that mimetic epitopes for U- and M-CLL Igs differed significantly. M-CLL–derived peptides exhibited better amino acid motifs, were more similar to each other, aligned more easily, and formed tighter clusters than U-CLL–derived peptides. Mono-, oligo-, and polyreactivity of peptides correlated with structural changes within antigen-binding sites of selecting M-CLL mAbs. Although M-CLL–isolated peptides and certain U-CLL mAbs bound more effectively to the selecting mAb, others were not as specific, reacting with M-CLL and U-CLL mAbs; these data suggest that in vivo structurally diverse epitopes could bind smIgs of distinct CLL clones, thereby altering survival and growth. Finally, an M-CLL–derived peptide inhibited, in a dose-dependent manner, binding of its homologous mAb to human B lymphocytes; therefore peptides that inhibit or alter the consequences of antigen-smIg interactions may represent therapeutic modalities in CLL.
Introduction
Chronic lymphocytic leukemia (CLL), the most frequent adult leukemia among whites, follows a heterogeneous clinical course.1,2 In approximately 50% of cases, leukemic clones display somatically mutated immunoglobulin (Ig) heavy (H) chain variable (IGHV) genes (mutated CLL [M-CLL]),3 and patients with M-CLL follow a much more favorable clinical course than patients with unmutated IGHV (U-CLL).4–6 On the basis of phenotypic analyses7 and gene expression profiling,8,9 both M-CLL and U-CLL appear to derive from antigen-experienced B cells. Therefore, clonal selection by antigen is probably responsible for skewing the IGHV repertoires of both M- and U-CLL compared with normal B cells and to each other.3,10
In CLL, certain IGHVs recombine frequently with distinct IGHD and IGHJ, thereby creating highly similar H chaincomplementarity-determining region (CDR) 3 sequences11–17; these “stereotyped” receptors13 often associate with distinct Ig light (L) chain variable κ (IGKV) or λ (IGLV) genes with characteristic KCDR3 or LCDR3 sequences. For example, CLL clones with stereotyped IGHV3-21/IGHJ6 rearrangements often express IGLV3-21 with highly homologous LCDR3s11; likewise, a subset of patients with CLL whose leukemic cells produce IgG express a stereotyped IGHV4-39/IGHD6-13/IGHJ5 rearrangement associated with IGKV1-39/1D-39.12 B-cell antigen receptors (BCRs) of remarkably similar amino acid structure are not limited to these subgroups. Indeed, almost 30% of all patients fall into subsets with stereotyped HCDR3s.16,17
Collectively, these observations support antigenic selection and stimulation as playing roles in the development and evolution of CLL. Whether this selection culls susceptible cells from the entire B-cell pool or from a prebiased subset13,18 remains to be determined, although gene expression studies suggest that the susceptible pool is homogeneous.8,9
Selecting antigens for CLL Igs are largely unknown, although some Ig-reactive autoantigens have been defined.19–22 This reactivity is more promiscuous among U-CLL than M-CLL monoclonal antibodies (mAbs).22 Furthermore, many CLL Igs exhibit structural features resembling mAbs reactive with microbial antigens.18
The present study was undertaken to identify mimetic epitopes of CLL mAbs and to characterize differences in epitope reactivity between U- and M-CLL. Because little is known about the chemical nature of important antigens, that is, whether they are proteins, carbohydrates, or lipids, peptide phage display technology was used to identify ligands for CLL mAbs. This approach has successfully identified mimetic epitopes of proteins,23 carbohydrates,24–26 lipids,27 and DNA.28
Our data indicate that phage display is a feasible technique to identify and characterize ligands binding to M-CLL and U-CLL. These ligands differ in structural characteristics: those binding M-CLL mAbs exhibit clearly definable, extended amino acid motifs, whereas those interacting with BCRs of U-CLL do not. The data suggest that somatic hypermutation that shapes the antigen-binding site influences the level of epitope specificity for an individual CLL BCR.
Methods
Expression of recombinant CLL Ig molecules
The variable segments of rearranged IGH and IGL of CLL clones were expressed as human IgG1s as previously described.22,29,30 The molecular characteristics of these mAbs are listed in Table 1.
Table 1.
Molecular characteristics of recombinant CLL mAbs used in these studies
| CLL no. | Subset* | IGHV | Mut status | Mut % | IGHD | IGHJ | HCDR3 amino acid sequence | IGH GenBank no. | IGKV/IGLV | IGLJ | Mut, % | LCDR3 amino acid sequence | IGL GenBank no. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 014 | 9 | 1-69 | U-CLL | 0.0 | 3-03 | 6 | ATKNDFWSGYYEGYYYYYYMDV | AF021951 | L3-01 | LJ1 | 0.0 | QAWDSSTCYV | AY043106 |
| 068 | 6 | 1-69 | U-CLL | 0.0 | 3-16 | 3 | ARGGDYDYVWGSYRSNDAFDI | AY553640 | K3-20 | KJ4 | 0.0 | QQYGSSPT | AY574935 |
| 114 | 8 | 4-39 | U-CLL | 0.7 | 6-13 | 5 | ARRFGYSSSWYGLDWFDP | AY268372 | K1-39/1D-39 | KJ1 | 0.7 | QQSYSTPRT | AY043094 |
| 270 | 1 | 1-02 | U-CLL | 0.0 | 5-12 | 4 | ARVQWLGLRHFDY | AY055487 | K1-39/1D-39 | KJ2 | 0.0 | QQSYSTPPYT | AY574940 |
| 169 | NA | 3-33 | M-CLL | 8.8 | 3-10 | 4 | AREGGVTGQGGFDY | AY055480 | K4-01 | KJ4 | 0.0 | QQYYSTPLT | FJ039794 |
| 183 | 4 | 4-34 | M-CLL | 3.1 | 5-18 | 6 | ARGYGDTPTIRRYYYYGMDV | AF021948 | K2-30 | KJ2 | 1.7 | MQGTHWPPYT | AY043097 |
| 255 | NA | 4-59 | M-CLL | 4.2 | 3-22 | 4 | ARHRGYESSGYYSSYFDY | FJ039782 | L3-01 | LJ1 | 2.2 | QAWDSSTVV | FJ039804 |
| 412 | 2 | 3-21 | M-CLL | 2.0 | IND | 6 | ARDQNGMDV | AY553648 | L3-21 | LJ3 | 0.7 | QVWDSSSDHPWV | AY574946 |
Mut indicates mutation; U-CLL, CLL cells with unmutated IGHV; M-CLL, CLL cells with mutated IGHV; NA, not attributable to a currently defined stereotypic subset; and IND, indeterminate.
Subset according to Stamatopoulos et al.16
Probing of peptide phage display libraries with CLL mAbs
Solution-phase probing.
PhD-12 (New England Biolabs) peptide phage display library (complexities of 2.7 × 109 transformants) was used. Three rounds of isolation were performed in solution according to the manufacturer's instructions. In brief, the supplied library (10 μL) was incubated for 1 hour with CLL mAb (10 μg) in 200 μL PBS-Tween 0.1%. Phage–antibody complexes were captured on Protein G agarose beads (Pierce Biotechnology Inc), washed with PBS-Tween 0.1%, and eluted with Glycine/HCl, pH 2.2. Eluted phages were amplified and then purified with polyethylene glycol. In the second and third rounds of isolation, Tween concentration was raised to 0.5% to select for higher affinity peptide–mAb interactions. Phage–antibody complexes were alternatively captured on Protein G or Protein A agarose beads (Pierce Biotechnology Inc) in rounds 2 and 3. Negative selection on BSA-blocked Protein A and Protein G beads was carried out in rounds 2 and 3 before incubation with the CLL mAb. After isolation, randomly chosen phage clones were amplified, and DNA inserts were isolated and sequenced.
Solid-phase probing.
Purified CLL mAbs and polyclonal human IgG (Sigma) were conjugated to activated CH-Sepharose beads (Sigma). PhD-12 library (10 μL) was incubated for 30 minutes with bead slurry (25 μg) in 200 μL PBS-Tween 0.1% and then processed as for solution-phase probing. Negative selection was performed in rounds 2 and 3 by incubating the amplified eluate of the previous round with bead slurry (50 μL) of Sepharose–polyclonal human IgG.
Enzyme immunoassays
Phage ELISA.
Polystyrene plates (Nunc) were coated with polyethylene glycol–purified phages (50 μL), serially diluted in PBS. After washing with PBS-Tween 0.1%, CLL mAb (50 μL of 2 μg/mL) was incubated at room temperature for 3 hours. Plates were washed with PBS-Tween 0.1% and incubated for 1 hour at room temperature with horseradish peroxidase–conjugated goat anti–human IgG (Southern Biotechnology Associates). Plates were developed for 15 minutes with TMP Sure Blue 1-component substrate (KPL) and stopped with 1 M HCl, and absorbance was measured at 450 nm. In some instances (CLL mAbs 014, 255, and 270), direct phage enzyme-linked immunoabsorbent assay (ELISA) was performed as described.31
Peptide ELISA.
Peptides, amidated and biotinylated at the C terminus, were synthesized by Princeton Biomolecules. Polystyrene plates, coated with streptavidin (10 μg/mL; New England Biolabs) overnight at 4°C, were blocked with HSA 10%, and biotinylated peptides (2 μg/mL) were added. After washing with PBS-Tween 0.1%, wells were incubated with serially diluted CLL mAb (50 μL) at room temperature for 2 hours. ELISAs were developed and quantified as described for phage ELISA.
Computational comparisons of peptide sets isolated from phage display libraries
Pairwise alignment scores were computed for each pair of peptide sequences for each mAb by using a dynamic programming tool available on the EMBL-EBI website (www.ebi.ac.uk). Optimal scores for pairwise alignments were determined by using Gonnet matrices for matches/mismatches between amino acids with default penalties for insertions and deletions. Multiple alignment was performed for peptide sets of each mAb by using the ClustalW2 multiple sequence alignment tool32 available at http://www.ebi.ac.uk/Tools/clustalw/. Each sequence was used once, regardless of the number of clones expressing the sequence. Peptide motifs were identified by requiring 50% amino acid identity or chemical similarity for 6 or more sequences; a score of 66.7% was required when fewer than 6 sequences were available. A minimum of 4 residues at a given aligned position were necessary for an amino acid to be considered for inclusion in a motif. The following list of chemical similarities was used for these assignments: [AVLI], [NQCM], [ST], [KRH], [DE], [FWY], [P], and [G]. Subsequent to multiple alignments, edit distances were computed for every pair of sequences based on the aligned sequences. To compare data between peptide sets, pairwise alignment scores and edit distances were combined separately for sequences obtained with M- versus U-CLL mAbs. P values were computed by using Student t test.
Inhibition of CLL mAb binding to cell surfaces by synthetic peptides
CLL mAb (12.5 μg/mL) was incubated overnight at 4°C with serially diluted peptides; as controls, peptides and mAbs were incubated with PBS in a similar manner. Subsequently, RAMOS B cells (4-5 × 105 in 50 μL PBS-BSA) were added to 50 μL of the peptide-mAb mixture, the mAb-PBS mixture, the peptide-PBS mixture, or PBS alone. After incubation for 1 hour at 4°C, cells of all samples, save the unstained control, were incubated with goat anti–human IgG-FITC (5 μL; Southern Biotechnology Associates) for 30 to 45 minutes at 4°C in the dark. Cells were washed, resuspended in PBS-BSA (100 μL), and analyzed with a FACSCalibur (Becton Dickinson Immunocytometry Systems).
Results
Monoclonal antibodies used in these studies
Recombinant IgG1 mAbs from 4 patients with M-CLL and 4 patients with U-CLL were used (Table 1). Two M-CLL mAbs, 183 and 412, were members of well-recognized and relatively frequent stereotyped subsets of CLL Igs (subset 4 and 2, respectively16). Patients with mAbs falling into these 2 subsets differ in clinical outcome, with subset 4 patients having a more benign course16 and subset 2 following a more aggressive course.33 mAbs 169 and 255 have not been found to fall within stereotyped subsets; furthermore, patients with leukemic clones expressing these mAbs have not been linked to a specific clinical course that distinguishes them from patients with CLL expressing a non–stereotyped M-CLL mAb, that is, probably a more benign clinical outcome.
Each of the U-CLL mAbs (Nos. 014, 068, 114, and 270; Table 1) are representatives of distinct stereotyped subsets (9, 6, 8, and 1, respectively16). Patients with clones falling into each of these subsets usually experience aggressive disease4,5,12; initial studies suggest that those in subset 8 (mAb 114) may be at greater risk of developing a more rapidly growing lymphoma than are patients with other IGH rearrangements.34,35
Peptides reactive with soluble M-CLL and U-CLL mAbs differ in structural properties
The PhD-12 peptide phage display library was probed with each CLL mAb by using conditions that favor more specific and avid peptide–mAb interactions. After 3 rounds of selection, 15 to 30 phages were isolated for each mAb; most of these phages contained peptide inserts (supplemental Tables 1-2, available on the Blood website; see the Supplemental Materials link at the top of the online article).
Peptides isolated by M-CLL mAbs differed significantly from those isolated by U-CLL mAbs. For the M-CLL mAbs, a large number of related but usually nonredundant peptides were found (Table 2; supplemental Table 1). These peptides displayed clear motifs that contained 6 to 8 identical or chemically related amino acids with shorter stretches of contiguity (Table 2). Furthermore, the occurrence rates of these key amino acids at the defined positions ranged from 50% to 100%, depending on the M-CLL peptides examined (supplemental Table 1).
Table 2.
Amino acid sequence motifs from phage clones isolated after 3 rounds of selection with either M-CLL or U-CLL mAbs
| CLL mAb no. | Mutation status | No. of sequences | Motif/occurrence cutoff, % | Amino acids comprising motifs* |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 169 | M-CLL | 8 | 50 | N | Y | — | A | AL/ST | Ali | L | — | — | R | |||||||||
| 183 | M-CLL | 8 | 50 | F | ST | Y | ST | — | — | — | L | HR | — | L | LI | |||||||
| 255 | M-CLL | 16 | 50 | FW | — | — | P | — | — | — | — | W | — | — | Ali | — | Ali | T | ||||
| 412 | M-CLL | 13 | 50 | P | — | — | — | — | Y | H | H | — | FY | — | — | — | ST | |||||
| 014 | U-CLL | 4 | 66.7 | P | ||||||||||||||||||
| 068 | U-CLL | 6 | 50 | ST | — | VL | P | |||||||||||||||
| 114 | U-CLL | 4 | 66.7 | H | ||||||||||||||||||
| 270 | U-CLL | 15 | 50 | — | ||||||||||||||||||
Multiple alignments were performed using the ClustalW2 multiple sequence alignment tool.32 Peptide motifs were identified by requiring 50% amino acid identity or chemical similarity for positions at which 6 or more residues were available; a score of 66.7% was required when fewer than 6 residues were present at a given alignment position. The following list of chemical similarities was used for these assignments: Ali(phatic): A, V, L, I; aliphatic with polar hydroxyl group: S, T; neutral with polar side chains: N, Q, C, M; cyclic imino: P; nonpolar: G; acidic: D, E; basic: H, K, R; aromatic: F, Y, W.
In contrast, U-CLL mAbs bound a series of peptides that did not contain amino acid motifs or exhibited motifs of much inferior quality (Table 2). In addition, one phage clone of those isolated with U-CLL mAbs was often numerically dominant, a feature that was far less obvious for phages isolated by M-CLL mAbs (supplemental Tables 1-2). However, difference in evaluable sequences was not the sole determinant of a lack of motifs, because for U-CLL mAb 270 we found that 16 nonredundant sequences were available for study, and a motif was not identified (Table 2).
Because U-CLL mAbs are known to be polyreactive19–22 and often bind antigens with low affinity, we tested whether altering the form and valency of the mAb would affect the selection of phages, thereby resulting in the isolation of clones with shared amino acid motifs, as for M-CLL mAbs. After chemically linking mAb 114 to Sepharose beads, selection was performed as in solution. With the use of this approach, 5 of 29 clones with inserts (supplemental Table 3) contained the identical sequence (WHDDQMLRRTVT) that had been detected when the mAb was used in soluble form (supplemental Table 2). In addition, a more dominant clone (13 of 29 phages) with the insert WNLRDTCYPYCT was also found (supplemental Table 3). Unlike with solution-phase isolation, peptides with a sequence motif were detectable by solid-phase probing, although the motif was less resolved than those found by using M-CLL mAbs and had multiple positions with disparate amino acids (supplemental Table 3). Thus, by changing the form and valency of a U-CLL mAb, peptides with greater resemblance to those found with M-CLL mAbs were isolated.
Confirmation that phages bind to selecting M- and U-CLL mAbs
To confirm the specificity of the various mAb–phage interactions, we tested binding of individual phages to the selecting Igs by enzyme immunoassay. For all 4 M-CLL mAbs, phages bearing different versions of the peptide motifs reacted with the selecting Igs, albeit with considerable and in some instances dramatic differences in binding efficiencies (Figure 1A-D). For example, mAb 169 bound much better to phage clone 169-8, with 7 of its 12 amino acids bearing the amino acid motif, than to clone 169-4, with an equal number of amino acids in the motif, and only slightly better than clones 169-2 and 169-3, which share 5 different amino acids with the motif (Figure 1A; supplemental Table 1A). For mAb 183, binding to specific phage clones followed proportionately the number of residues present in the motif, except for 183-15 that was mismatched at 4 positions and was essentially nonreactive (Figure 1B; supplemental Table 1B). Similarly, alterations in the defined motif dramatically reduced binding to clones 412-2 and 412-6, which shared only the central YHH portion of the motif (Figure 1D; supplemental Table 1D).
Figure 1.

CLL mAbs bind phage-displayed peptides in a dose-dependent manner. Individual phages isolated after 3 rounds of selection with each CLL mAb were amplified and their antibody binding properties were studied by ELISA. A random phage clone derived from the initial phage library before selection was used as a negative control. (A-D) M-CLL mAbs 169, 183, 255, and 412. (E-H) U-CLL mAbs 014, 068, 114, and 270.
As for M-CLL mAbs, each U-CLL mAb bound phages selected in the initial isolations (Figure 1E-H).
Peptide motifs identified by M-CLL mAbs are better defined
M-CLL–derived peptide motifs were more clearly defined and comprised a greater number of common amino acids in the same sequence (Table 2). To place these differences on a firm mathematical footing, we analyzed pairwise alignment scores and edit distances. Pairwise alignment scores measure the sequence similarity of any 2 peptides selected by the same mAb. After pooling these scores separately for peptides isolated with soluble M-CLL versus U-CLL mAbs, distributions were compared. Median values of distributions were separated with high statistical significance (Student t test, P < .001; Figure 2A), indicating that any 2 peptides in a pair isolated by a single M-CLL mAb were more similar to one another than those isolated by U-CLL mAbs. Thus, as a group, M-CLL–derived peptides cluster more tightly than do U-CLL–derived peptides. In a similar analysis with the use of pairwise edit distances computed on pairs of aligned sequences, the 2 distributions were significantly different (Student t test, P < .001; Figure 2B), showing that not only were peptides isolated by a single M-CLL mAb more similar to one another, they also allowed for a greater degree of alignment.
Figure 2.

Comparisons of pairwise alignment scores and pairwise edit distances between M- and U-CLL mAbs. (A) Pairwise alignment scores. Pooled pairwise alignment scores for M-CLL peptides were significantly higher than those for U-CLL peptides (Student t test, P < .001). (B) Pairwise edit distances. As a group, M-CLL peptides had significantly shorter pairwise distances than did U-CLL peptides (Student t test, P < .001). The error bars extend to the extreme values of the data or 1.5× the height of the box, whichever is less. The height of the box represents the difference between the first and third quartiles. The top error bar in panel B merges with the top of the box plot for U-CLL peptides.
Binding of M- and U-CLL mAbs to synthetic peptides from selected phage clones
Because peptides displayed on the surface of phages can lose reactivity with a selecting mAb when expressed as independent molecules, we synthesized peptides corresponding to the sequences of phages selected by M- and U-CLL mAbs and determined their binding. Representative peptides isolated by M-CLL mAbs 169, 183, and 412, and by U-CLL mAbs 068 and 114 were synthesized with biotin at the C-terminus to permit binding to streptavidin. This format allowed us to perform enzyme immunoassays and immunofluorescence analyses. For each peptide, a scrambled version containing the same amino acids, but in a different order, was also synthesized to determine whether binding was due to amino acid composition alone or a particular sequence.
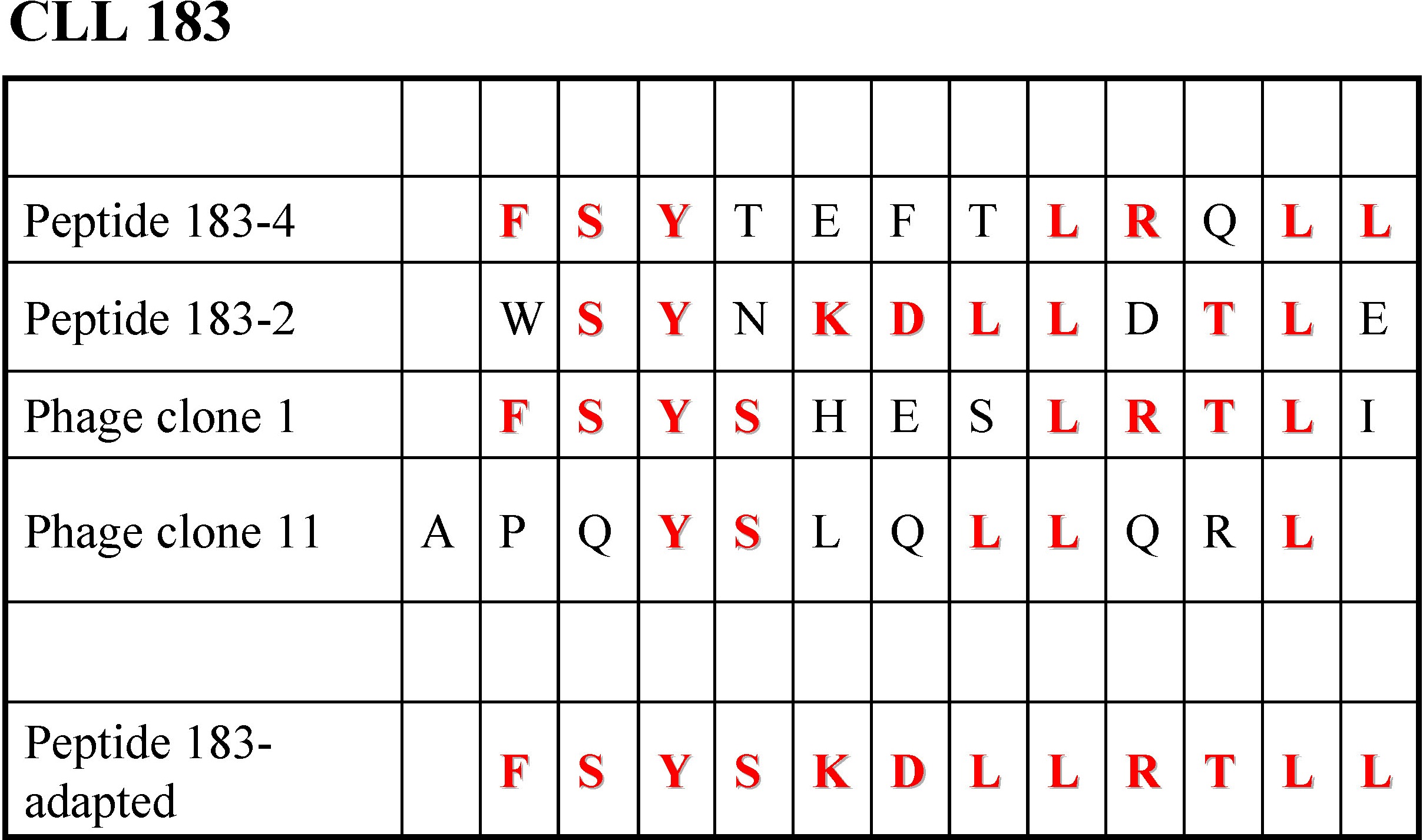
There was dose-dependent binding of mAb 169 to peptide 169-2, starting at 400 ng/mL (Figure 3A), and to peptide 169-8 (Figure 3B), beginning at 10 ng/mL; this was not observed for the corresponding scrambled peptides (Figure 3E-F). For M-CLL mAb 183, no specific binding was detected for 2 peptides, 183-2 and 183-4 (not shown). Therefore, we synthesized a peptide, “183-adapted” (FSYSKDLLRTLL), taking into account sequences of the peptide-phages that were bound best by mAb 183 in ELISA (supplemental Figure 1). This peptide showed enhanced reactivity with mAb 183 (Figure 3C).
Figure 3.

Binding of M-CLL and U-CLL mAbs to synthetic peptides derived by selection with M-CLL mAbs. Binding of M-CLL and U-CLL mAbs to 12mer peptides with sequences derived from phage selection analyses was tested by ELISA. Peptides with a scrambled amino acid order were used as negative controls. (A-D) CLL mAb reactivities with peptides 169-2, 169-8, 183-adapted, and 412-9, respectively. (E-H) Reactivities of mAbs to scrambled peptides.
To discriminate the key amino acid residues involved in mAb 412 recognition of peptide 412-9 (Figure 3D and Figure 4), shorter stretches of the full-length peptide were synthesized. Because peptide 412-9 (YHHAYLLPPPRM) had a highly polar N-terminus and a markedly hydrophobic C-terminus, synthetic peptides corresponding to the 2 poles of the original sequence were synthesized (412-9-1, YHHAYL, and 412-9-3, LPPPRM; Figure 4). In addition, fragments spanning the junction between the above 2 peptides were also made (412-9-2, YHHAYLLP, and 412-9-4, AYLLP). Only peptides containing YHHAYL retained binding to mAb 412, albeit less than the original full-length consensus peptide (Figure 4). Because 412-9-2 (YHHAYLLP) showed stronger binding than did 412-9-1 (YHHAYL), a combination of polar and hydrophobic residues probably contributed to the immunoreactive epitope.
Figure 4.

Binding of M-CLL mAb 412 to peptide 412-9 and its fragments. Fragments of peptide 412-9 were synthesized (peptides 412-9-1, -2, -3, -4; bottom) and tested for binding to CLL 412 mAb in ELISA. Peptide 412-9scr with a scrambled amino acid order was used as a negative control.
U-CLL mAb 068 reacted with peptide 068-4 in ELISA at a concentration of 400 ng/mL (Figure 5A). However, when U-CLL peptides 114-1 and 114-4 were synthesized and tested for binding to mAb 114, peptide 114-1 reacted (Figure 5B); peptide 114-4 did not (not shown).
Figure 5.

Binding of M- and U-CLL mAbs to synthetic peptides derived by selection with U-CLL mAbs. Binding of M- and U-CLL mAbs to 12mer peptides with sequences derived from phage selection analyses with U-CLL mAbs was tested by ELISA. (A-B) Reactivities with peptides 068-4 and 114-1, respectively. (C-D) Reactivities of mAbs with scrambled peptides used as negative controls.
Binding of U- and M-CLL mAbs to peptides selected by other mAbs
Some cases of CLL probably derive from B cells producing polyreactive, natural antibodies coded by germline IGHV and IGKV or IGLV, which either retain (U-CLL) or lose (M-CLL) this property because of somatic hypermutation.2,18,22 Consequently, we presumed that peptides selected by M-CLL mAbs would have a narrower window of antibody reactivity than those selected by U-CLL. Peptides 169-2 and 169-8, selected by the most mutated Ig of those tested, mAb 169 (∼8% difference from its germline gene; Table 1), were bound very well by their selecting mAb and were essentially nonreactive for other M- and U-CLL mAbs tested (Figure 3A-B).
Somewhat unexpectedly, M-CLL peptides 412-9 and 183-adapted, isolated by M-CLL mAbs with mid-range somatic mutations (Table 1), bound effectively to mAbs not involved in their selection. Thus, M-CLL peptide 183-adapted interacted efficiently with U-CLL mAb 114 (Figure 3C); likewise, peptide 412-9 reacted very efficiently with M-CLL mAbs 169 and 183 as well as U-CLL mAbs 068 and 114 (Figure 3D). Not surprisingly, this phenomenon also occurred with peptides isolated by polyreactive U-CLL mAbs 068 and 114 (068-4 and 114-1) that recognized both U-CLL mAbs as well as M-CLL mAb 183 (Figure 5A-B).
Viewed from the mAb side, U-CLL mAbs 068 and 114 had a broader reactivity profile, reacting with their homologous peptides (Figure 5A and B, respectively) as well as heterologous peptides isolated by either U-CLL or M-CLL mAbs. Specifically, mAb 114 bound peptides 068-4 (Figure 5A), 183-adapted (Figure 3C), and 412-9 (Figure 3D); likewise mAb 068 bound peptides 114-1 (Figure 5B) and 412-9 (Figure 3D) and had lesser reactivity to peptide 183-adapted (Figure 3C). These data were consistent with the poly-reactivity of U-CLL mAbs.22,29,30
Of interest, M-CLL mAb 169 reacted well not only with its homologous peptides (Figure 3A-B) but also with M-CLL peptide 412-9 (Figure 3D), even though the 169-2 and 169-8 peptides do not share obvious similarity in motif with peptide 412-9 (Table 2). This was surprising because mAb 169 does not react in enzyme immunoassays with a large panel of (auto)antigens,22,29 and the peptides it isolated (169-2 and 169-8) reacted solely with itself (Figure 3A-B). Thus, a significantly mutated CLL mAb that can identify a very specific ligand from the multitude contained in the phage library can still act somewhat nonspecifically by binding ligands isolated by other CLL mAbs.
Finally, M-CLL and U-CLL mAbs were analyzed for binding to various scrambled peptides. M-CLL mAbs 169 and 412 were essentially nonreactive with these variants, although M-CLL mAb 183 reacted at the highest concentration tested (50 μg/mL; Figure 3). In contrast, U-CLL mAbs 068 and 114 bound scrambled versions of peptides 068-4 and 114-1, slightly at 10 μg/mL and effectively at 50 μg/mL, respectively (Figure 5C-D).
Inhibition of CLL mAb binding to “native” antigen by a mimetope
We recently demonstrated that mAb 183, a member of stereotyped subset 4, binds the surface of viable human B (but not T) cells.36 Therefore, we tested the extent to which peptide 183-adapted and its scrambled version inhibited this natural autoreactivity. As shown in Figure 6, mAb 183 (12.5 μg/mL) bound approximately 83% of live RAMOS B cells. However, preincubation of the mAb with soluble 183 peptide (100 μg/mL) led to an almost 30% fall in mAb binding. In addition, because mean fluorescence intensities changed from 346.3 to 128.5 and the histograms indicated that virtually the entire reactive peak shifted to the left (Figure 6), competition for binding probably occurred for most of the cells. Inhibition was dose dependent as well as specific, because the scrambled version of the peptide did not alter binding (Figure 6). Thus, a soluble mimetic epitope blocked antigen binding to a CLL mAb for which a natural antigenic target is known.
Figure 6.

Inhibition of M-CLL mAb 183 binding to an autoantigen on the surface of viable RAMOS B cells by soluble peptide. mAb 183 (12.5 μg/mL) was preincubated with soluble 183-adapted peptide (pep183) or scrambled peptide (pep183scr) at indicated concentrations. Percentage and mean fluorescence intensity (in brackets) of mAb 183 staining of RAMOS cells are presented in each panel. mAb 183 binding without peptide preincubation is shown for comparison.
Discussion
The aim of this study was to characterize the binding properties of CLL BCRs by using phage-displayed 12-mer peptides. We tested 8 CLL mAbs, which differed in IGHV mutation status, amino acid structure, and associated clinical outcomes, to identify antigenic epitopes, to determine the extent to which these differed between U- and M-CLL, and to begin testing the functional activities of mimetic ligands in vitro.
On the basis of current information, antigenic targets of U-CLL and M-CLL Igs/mAbs differ. Both types can be autoreactive,19–21 but U-CLL mAbs far outstrip M-CLL in this regard as well as in poly-reactivity.22 Although little is known about the precise nature of antigens recognized by CLL cells, several specific autoantigenic targets were recently identified. For example, a stereotyped subset of CLL mAbs (subset 6) binds non–muscle myosin heavy chain IIA (MYHIIA),30 a component of the cellular cytoskeleton. In addition, another stereotyped mAb30 (subset 8) as well as a nonstereotyped mAb37 react with another cytoskeletal component, vimentin. Finally, epitopes, apparently common to autoantigens but present on apoptotic cells29,36,37 and bacteria,37,38 interact with CLL mAbs. Although these epitopes are usually protein in nature, some are carbohydrate38 and others are products of oxidation29,37 or other catabolic processes.39 Because the phage display approach allows identification of mimetics for carbohydrates and other nonprotein structures as well as proteins, we used this technique to identify BCR ligands in CLL.
Characteristics of epitopes bound by M- and U-CLL mAbs
By using isolation conditions that favor selection of more specific and avid antigen–antibody interactions, we identified ligands for each CLL mAb. The characteristics of these ligands differed considerably, and this related to the structure of the selecting Ig. Whereas M-CLL mAbs recognized multiple distinct phage-displayed peptides that shared common amino acid motifs (Table 2; supplemental Table 1), U-CLL mAbs bound peptide sets that were frequently dominated by individual sequences that occurred repeatedly but did not share amino acids (Table 2; supplemental Table 2). The isolation of numerically dominant peptide-phages seems paradoxical. However, it appears that low-affinity mAb–peptide interactions often fail to isolate, especially in the initial rounds of the probing process, phages bearing unique and specific peptide sequences; rather, these low-affinity mAbs bind a broad range of disparate phages.24,25,31,40,41 This diminished “specificity” then permits those phages that grow the fastest to be amplified and enriched at each round; ultimately, this can lead to isolation of multiple phages expressing identical peptide sequences. This is not necessarily an indicator of greater binding specificity, but in fact may be the opposite, that is, low affinity, multireactivity. Finally, U-CLL mAbs did not necessarily identify the same dominant peptides when probed additional times (supplemental Table 3; data not shown).
On testing individual peptide-phage clones isolated from the 12-mer peptide-phage library, it was clear that for M-CLL mAbs slight variations in key amino acid motifs of the peptides changed or abolished binding (Figure 1A-D). Thus, peptides isolated with M-CLL mAbs can be highly specific for individual BCRs, representing defined epitopes of shared amino acids.
For the one tested U-CLL mAb, (114) the form and valency of the antibody (soluble dimeric or insoluble multimeric) appeared important, because the peptides isolated by the same mAb differed when the mAb format was altered. Specifically, changes in form and valency to insoluble multimeric altered the structure of the peptides captured from U-CLL–like to more M-CLL–like (supplemental Table 3). In CLL, in which the clonal Igs are mainly cell bound and not secreted, this might affect leukemic cell reactivity and response.
Finally, despite the fact that U-CLL mAbs are typically “polyreactive,” U-CLL mAb 068 efficiently bound its selected peptide (Figure 5A), even at a low concentration (400 ng/mL). Thus, U-CLL mAbs, which resemble natural antibodies, may have inherent, preferential specificity for certain yet-to-be-defined determinants in vivo. The reactivity of unmutated, natural antibodies with unique epitopes on human influenza viruses is consistent with this concept.42,43
Identification of key residues in peptides isolated with CLL mAbs
Common sequence motifs (Table 2) suggested the potential relative importance of conserved amino acids at distinct positions in peptides and highlighted structural distinctions between mimetopes reactive with U- versus M-CLL mAbs. These conclusions were supported by analyses of pairwise alignment scores and of edit distances based on multiple alignment of peptide set sequences (Figure 2).
Of the amino acids contained in various M-CLL motifs, 4 to 5 displayed the greatest degree of conservation and are therefore probably the most involved in mAb binding. It is intriguing that a prominent tyrosine (Y), separated by 4 amino acids from a highly resolved leucine (L), appears as important residues in peptides isolated with 2 structurally different M-CLL mAbs 169 and 183 (Table 2). M-CLL mAb 412 also had a prominent Y contained within a consistent YHH motif (Table 2).
We have searched the amino acid sequences of MYHIIA and vimentin, which we know react with CLL mAbs 068 and 114, respectively,30 to determine whether they contain residues found in the peptides isolated by these mAbs. Although there is no exact match of the peptides to any protein in the GenBank database, there is a weak match of the 068 peptide (3 of 12 residues) to MYHIIA; this degree of similarity is analogous to that observed for a phage display peptide binding a mouse antibody that recognizes MYHIIA.44 There is a somewhat better, albeit still weak match for vimentin by 114-1 peptide (4 of 12 residues). These data are provocative, although not definitive, and are being analyzed further.
The amino acid commonality between peptides isolated by distinct M-CLL mAbs suggests a restricted set of immunoreactive epitopes in the antigens that drove these B-cell clones before and possibly after leukemic transformation. These core residues may permit a definition of peptides that are even more specific for individual CLL BCRs/mAbs or for members of stereotyped subsets within which they reside. Furthermore, amino acid commonality may reflect an innate reactivity of CLL mAb binding domains with classes of antigenic epitopes for which there has been evolutionary conservation of paratope structure, for example (but not limited to) viruses. These conserved structures could be modified and lose this antigen-binding function as IGHV and IGKV or IGLV change because of somatic mutations.
Mono-, oligo-, and poly-reactivity of CLL mAbs and the peptides they select
Our studies indicate that peptides reactive with CLL mAbs can display different degrees of specificity, and this correlated with the level of structural change within the antigen-binding site of the selecting mAb. Peptides 169-2 and 169-8, isolated by M-CLL mAb 169 with 10 replacement mutations, were specific for their selecting mAb. Peptides 412-9 and 183-adapted were less specific, exhibiting a level of promiscuity consistent with the degree of somatic mutation in the M-CLL mAbs that had isolated them (3 and 4 replacement mutations, respectively). Peptides 068-4 and 114-1, isolated by U-CLL mAbs 068 and 114 with no or 1 replacement mutation, respectively, reacted with most/all of the mAbs tested (Figures 3 and 5).
The latter findings are consistent with several different peptides binding the paratope of a single CLL mAb. This phenomenon has been documented for germline antibodies,40,41 in agreement with our data for U-CLL. Surprisingly, we found the same for M-CLL mAbs. Although the ability of more than one peptide to bind to a single mAb binding site related to the level of IGHV somatic mutations present in the mAb (U-CLL > M-CLL), this was not absolute. The antigen-binding site of the highly mutated mAb 169 could be occupied by peptides selected by another M-CLL mAb (183; Figure 3C), and the paratope of the less-mutated M-CLL mAb 183 could accommodate U-CLL peptide 068-4 (Figure 5A). There is no a priori reason that any ligand will not be bound by a given receptor; however, because in most settings IGHV mutated mAbs tend to bind more specifically to epitope targets, one might expect that such bound epitopes would be more specific for the selecting mutated mAb than others. Thus, although somatically mutated antigen-binding sites such as in M-CLL can be shaped to bind more defined determinants, this remodeling may not completely exclude “nonspecific” binding regions or regions that serve as superantigen binding sites. A key unresolved question is whether this remodeling is less efficient in CLL Igs compared to Igs from normal human B cells or B-cell subsets. The latter may be relevant for mAb 183, which as a member of subset 4 expresses IGHV4-34 that can bind N-acetyllactosamine through key residues in the FR1 region.45 Of note, these residues are conserved in mAb 183 and in most IGHV4-34 using CLL mAbs in subset 4.17
Finally, in vivo our data suggest that multiple distinct structural epitopes could bind M-CLL as well as U-CLL BCRs. Such interactions could lead to transmission of BCR-mediated signals, thereby affecting survival and growth of leukemic cells.
Targeting CLL BCRs with mimetic antigens
Although the unique structural features of BCRs from patients with U-CLL and patients with M-CLL may provide leukemic cells with an opportunity to receive survival signals, their antigen-binding properties may also provide an avenue to selectively target leukemic cells. Using a relatively specific, although not apparently especially avid peptide, we specifically inhibited, in a dose-dependent manner, the interaction of a selecting mAb with a natural ligand on viable human B cells (Figure 6). Because this peptide could bind the soluble mAb, it would very likely bind immobilized surface membrane Ig of the patient's B cells from which the selecting mAb was derived, were they available for testing. Thus, this peptide or others with similar properties might be used to target and eliminate CLL cells via the BCR, either by inducing apoptosis via BCR crosslinking or by delivering cytotoxic agents. Indeed, preliminary data indicate that peptide 169-8 binds to the surface of CLL 169 cells and induces cell death.46 However, should in vivo BCR signaling be involved in CLL cell survival and expansion, nonagonistic peptides might block such antigen–Ig interactions and indirectly lead to clonal exhaustion of the cells. Further studies along this line are under way.
In conclusion, phage display is a feasible technology to identify specific ligands to BCRs of both M- and U-CLL clones. By analyzing epitopes recognized by 4 M-CLL and 4 U-CLL mAbs it appears that these ligands differ substantially, with the former binding defined structures and the latter recognizing multiple different epitopes; additional studies will be necessary to confirm these findings. The level of somatic mutations shaping the antibody's binding site seems to influence both the degree of paratope specificity of the selected peptides as well as the ability of more than one epitope to bind a single mAb paratope. These differences could affect the clinical behavior of CLL clones after BCR binding; therefore, mimetic ligands to CLL BCRs might serve as valuable therapeutics by specifically targeting leukemic cells.
Supplementary Material
Acknowledgments
We thank Des Higgins for valuable communications about aspects of the bioinformatics analysis.
This work was supported in part by grant (R01 CA81554) and by General Clinical Research Center grant (M01 RR018535) and by The Karches Foundation, The Prince Family Foundation, The Marks Foundation, The Jerome Levy Foundation, The Leon Levy Foundation, The Tebil Foundation, Inc, and the Joseph Eletto Leukemia Research Fund.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: T.S., M.W., S.Y., R.C., W.L., K.H., M.T., S.P., H.D., S.S., C.C.C., and N.C. designed the research and analyzed results; T.S., M.W., R.C., C.M., and K.H. performed experiments; T.S., M.W., S.Y., C.C.C., and N.C. wrote the paper and made the figures; and M.S.K., S.L.A., J.E.K., and K.R.R. contributed samples and clinical information and correlation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current address for T.S. is Department of Internal Medicine III, University of Munich, Campus Grosshadern, Munich, Germany.
Correspondence: Nicholas Chiorazzi, The Feinstein Institute for Medical Research, 350 Community Dr, Manhasset, NY 11030; e-mail: nchizzi@nshs.edu.
References
- 1.Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med. 1995;333(16):1052–1057. doi: 10.1056/NEJM199510193331606. [DOI] [PubMed] [Google Scholar]
- 2.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 3.Fais F, Ghiotto F, Hashimoto S, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J Clin Invest. 1998;102(8):1515–1525. doi: 10.1172/JCI3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. [PubMed] [Google Scholar]
- 5.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig VH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. [PubMed] [Google Scholar]
- 6.Krober A, Seiler T, Benner A, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood. 2002;100(4):1410–1416. [PubMed] [Google Scholar]
- 7.Damle RN, Ghiotto F, Valetto A, et al. B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood. 2002;99(11):4087–4093. doi: 10.1182/blood.v99.11.4087. [DOI] [PubMed] [Google Scholar]
- 8.Klein U, Tu Y, Stolovitzky GA, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):1639–1647. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schroeder HW, Jr, Dighiero G. The pathogenesis of chronic lymphocytic leukemia: analysis of the antibody repertoire. Immunol Today. 1994;15(6):288–294. doi: 10.1016/0167-5699(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 11.Tobin G, Thunberg U, Johnson A, et al. Chronic lymphocytic leukemias utilizing the VH3-21 gene display highly restricted V{lambda}2-14 gene use and homologous CDR3s: implicating recognition of a common antigen epitope. Blood. 2003;101(12):4952–4957. doi: 10.1182/blood-2002-11-3485. [DOI] [PubMed] [Google Scholar]
- 12.Ghiotto F, Fais F, Valetto A, et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113(7):1008–1016. doi: 10.1172/JCI19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messmer BT, Albesiano E, Efremov DG, et al. Multiple distinct sets of stereotyped antigen receptors indicate a key role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–525. doi: 10.1084/jem.20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tobin G, Thunberg U, Karlsson K, et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood. 2004;104(9):2879–2885. doi: 10.1182/blood-2004-01-0132. [DOI] [PubMed] [Google Scholar]
- 15.Widhopf GF, II, Rassenti LZ, Toy TL, Gribben JG, Wierda WG, Kipps TJ. Chronic lymphocytic leukemia B cells of more than 1% of patients express virtually identical immunoglobulins. Blood. 2004;104(8):2499–2504. doi: 10.1182/blood-2004-03-0818. [DOI] [PubMed] [Google Scholar]
- 16.Stamatopoulos K, Belessi C, Moreno C, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. 2007;109(1):259–270. doi: 10.1182/blood-2006-03-012948. [DOI] [PubMed] [Google Scholar]
- 17.Murray F, Darzentas N, Hadzidimitriou A, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111(3):1524–1533. doi: 10.1182/blood-2007-07-099564. [DOI] [PubMed] [Google Scholar]
- 18.Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. 2003;21:841–894. doi: 10.1146/annurev.immunol.21.120601.141018. [DOI] [PubMed] [Google Scholar]
- 19.Broker BM, Klajman A, Youinou P, et al. Chronic lymphocytic leukemic cells secrete multispecific autoantibodies. J Autoimmun. 1988;1(5):469–481. doi: 10.1016/0896-8411(88)90068-6. [DOI] [PubMed] [Google Scholar]
- 20.Sthoeger ZM, Wakai M, Tse DB, et al. Production of autoantibodies by CD5-expressing B lymphocytes from patients with chronic lymphocytic leukemia. J Exp Med. 1989;169(1):255–268. doi: 10.1084/jem.169.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borche L, Lim A, Binet JL, Dighiero G. Evidence that chronic lymphocytic leukemia B lymphocytes are frequently committed to production of natural autoantibodies. Blood. 1990;76(3):562–569. [PubMed] [Google Scholar]
- 22.Herve M, Xu K, Ng YS, et al. Unmutated and mutated chronic lymphocytic leukemias derive from self-reactive B cell precursors despite expressing different antibody reactivity. J Clin Invest. 2005;115(6):1636–1643. doi: 10.1172/JCI24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97(2):391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 24.Valadon P, Nussbaum G, Boyd LF, Margulies DH, Scharff MD. Peptide libraries define the fine specificity of anti-polysaccharide antibodies to Cryptococcus neoformans. J Mol Biol. 1996;261(1):11–22. doi: 10.1006/jmbi.1996.0438. [DOI] [PubMed] [Google Scholar]
- 25.Valadon P, Nussbaum G, Oh J, Scharff MD. Aspects of antigen mimicry revealed by immunization with a peptide mimetic of Cryptococcus neoformans polysaccharide. J Immunol. 1998;161(4):1829–1836. [PubMed] [Google Scholar]
- 26.Krishnan L, Lomash S, Raj BP, Kaur KJ, Salunke DM. Paratope plasticity in diverse modes facilitates molecular mimicry in antibody response. J Immunol. 2007;178(12):7923–7931. doi: 10.4049/jimmunol.178.12.7923. [DOI] [PubMed] [Google Scholar]
- 27.Mertens P, Walgraffe D, Laurent T, Deschrevel N, Letesson JJ, De Bolle X. Selection of phage-displayed peptides recognised by monoclonal antibodies directed against the lipopolysaccharide of Brucella. Int Rev Immunol. 2001;20(2):181–199. doi: 10.3109/08830180109043033. [DOI] [PubMed] [Google Scholar]
- 28.Putterman C, Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med. 1998;188(1):29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Catera R, Silverman GJ, Hatzi K, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol Med. 2008;14(11–12):665–674. doi: 10.2119/2008-00102.Catera. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu CC, Catera R, Hatzi K, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood. 2008;112(13):5122–5129. doi: 10.1182/blood-2008-06-162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valadon P, Scharff MD. Enhancement of ELISAs for screening peptides in epitope phage display libraries. J Immunol Methods. 1996;197(1–2):171–179. doi: 10.1016/0022-1759(96)00133-0. [DOI] [PubMed] [Google Scholar]
- 32.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tobin G, Thunberg U, Johnson A, et al. Somatically mutated Ig V(H)3-21 genes characterize a new subset of chronic lymphocytic leukemia. Blood. 2002;99(6):2262–2264. doi: 10.1182/blood.v99.6.2262. [DOI] [PubMed] [Google Scholar]
- 34.Rossi D, Cerri M, Capello D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2009;142(2):202–215. doi: 10.1111/j.1365-2141.2008.07166.x. [DOI] [PubMed] [Google Scholar]
- 35.Rossi D, Spina V, Cerri M, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15(13):4415–4422. doi: 10.1158/1078-0432.CCR-08-3266. [DOI] [PubMed] [Google Scholar]
- 36.Catera R, Hatzi K, Chu CC, et al. Polyreactive monoclonal antibodies synthesized by some B-CLL cells recognize specific antigens on viable and apoptotic T cells [abstract]. Blood. 2006;108(11):796a. Abstract 2813. [Google Scholar]
- 37.Lanemo Myhrinder A, Hellqvist E, Sidorova E, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008;111(7):3838–3848. doi: 10.1182/blood-2007-11-125450. [DOI] [PubMed] [Google Scholar]
- 38.Hatzi K, Catera R, Ferrarini M, et al. B-cell chronic lymphocytic leukemia (B-CLL) cells express antibodies reactive with antigenic epitopes expressed on the surface of common bacteria [abstract]. Blood. 2006;108(11):12a. Abstract 25. [Google Scholar]
- 39.Catera R, Silverman GJ, Hatzi K, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and catabolic chemical modifications[abstract]. Blood. 2008;112(11):1080. doi: 10.2119/2008-00102.Catera. Abstract 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manivel V, Bayiroglu F, Siddiqui Z, Salunke DM, Rao KV. The primary antibody repertoire represents a linked network of degenerate antigen specificities. J Immunol. 2002;169(2):888–897. doi: 10.4049/jimmunol.169.2.888. [DOI] [PubMed] [Google Scholar]
- 41.Sethi DK, Agarwal A, Manivel V, Rao KV, Salunke DM. Differential epitope positioning within the germline antibody paratope enhances promiscuity in the primary immune response. Immunity. 2006;24(4):429–438. doi: 10.1016/j.immuni.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Sui J, Hwang WC, Perez S, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16(3):265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khurana S, Suguitan AL, Jr, Rivera Y, et al. Antigenic fingerprinting of H5N1 avian influenza using convalescent sera and monoclonal antibodies reveals potential vaccine and diagnostic targets. PLoS Med. 2009;6(4):e1000049. doi: 10.1371/journal.pmed.1000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M, Alicot EM, Chiu I, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203(1):141–152. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Potter KN, Hobby P, Klijn S, Stevenson FK, Sutton BJ. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J Immunol. 2002;169(7):3777–3782. doi: 10.4049/jimmunol.169.7.3777. [DOI] [PubMed] [Google Scholar]
- 46.Woelfle M, Seiler T, Catera R, et al. Induction of apoptosis in CLL by peptides binding the B-cell antigen receptor in vitro [abstract]. Blood. 2008;112(11):1080. Abstract 3151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}