

Abstract
By sustained activation of μ-opioid receptors (MORs), chronic opioids cause analgesic tolerance, physical dependence, and opioid addiction, common clinical problems for which an effective treatment is still lacking. Chronic opioids recruit δ-opioid receptors (DORs) to plasma membrane through exocytotic trafficking, but the role of this new DOR and its interaction with existing MOR in brain functions and in these clinical problems remain largely unknown. In this study, we investigated the mechanisms underlying synaptic and behavioral actions of chronic morphine-induced DORs and their interaction with MORs in nucleus raphe magnus (NRM) neurons important for opioid analgesia. We found that the emerged DOR inhibited GABAergic IPSCs through both the phospholipase A2 (PLA2) and cAMP/protein kinase A (PKA) signaling pathways. MOR inhibition of IPSCs, normally mediated predominantly by the PLA2 pathway, was additionally mediated by the cAMP/PKA pathway, with MOR potency significantly increased after chronic morphine treatment. Isobologram analysis revealed a synergistic DOR–MOR interaction in their IPSC inhibition, which was dependent on upregulated activities of both the PLA2 and cAMP/PKA pathways. Furthermore, DOR and MOR agonists microinjected into the NRM in vivo also produced a PLA2-dependent synergism in their antinociceptive effects. These findings suggest that the cAMP/PKA pathway, upregulated by chronic opioids, becomes more important in the mechanisms of both MOR and DOR inhibition of GABA synaptic transmission after chronic opioid exposure, and DORs and MORs are synergic both synaptically and behaviorally in producing analgesic effects in a PLA2-dependent fashion, supporting the potential therapeutic use of DOR agonists in pain management under chronic opioid conditions.
Introduction
Opioids have been the mainstay in clinical pain management for several decades. However, repeated use of opioids and opioid-based analgesics, such as morphine and heroin, causes severely aversive effects including analgesic tolerance, physical dependence, and opioid addiction (Bhargava, 1994; Woolf and Hashmi, 2004; Ballantyne and LaForge, 2007). The analgesic tolerance significantly diminishes the analgesic effectiveness of opioids over time and physical dependence necessitates continued opioid administration to avoid devastating withdrawal symptoms, whereas drug addiction is a chronic psychiatric disease for which there is still a lack of effective treatment at present. These aversive effects of opioids and concerns over the risk of developing opioid addiction significantly hamper the optimal use of opioids in clinical treatment of chronic pain conditions.
Opioid effects are mediated by three major types of opioid receptors: μ-, δ-, and κ-opioid receptors (MORs, DORs, and KORs, respectively) (Pan, 1998; Waldhoer et al., 2004). Previous studies have shown that the majority of opioid effects, including analgesia, tolerance, and addiction, are primarily mediated by MORs, with the functions of the other two opioid receptors remaining illusive (Matthes et al., 1996; Contet et al., 2004). In contrast to MORs, DOR agonists have weak analgesic effects, which, as demonstrated by recent studies, was probably due to none to low levels of surface expression of functional DORs in brain and spinal neurons important in central pain-modulating circuits (Hurley and Hammond, 2000; Fields, 2004; Ma et al., 2006). Interestingly, accumulating evidence shows that prolonged exposure to morphine recruits new functional DORs to surface membrane through receptor trafficking in several brain and spinal cord sites involved in pain modulation, resulting in augmentation of a DOR-mediated antinociceptive effect (Cahill et al., 2001; Hack et al., 2005; Ma et al., 2006; Sykes et al., 2007). Repeated morphine treatment also induces new functional DORs in neurons of the central nucleus of the amygdala, a forebrain area involved in drug addiction (Bie et al., 2009).
The fact that the DOR emerges on central synaptic terminals that readily express functional MORs (Ma et al., 2006) raises an intriguing issue of DOR–MOR interaction in terms of their modulation of transmitter release, the underlying signaling transduction for synaptic transmission and systemic modification of behavioral functions. Previously, in HEK-293 cells heterologously expressing both MORs and DORs, occupancy of DORs by DOR antagonists was shown to increase MOR binding and signaling activity by physical heterodimeric association (Gomes et al., 2004), and a recent study showed that DOR and MOR agonists locally applied in the brainstem induced a synergistic behavioral effect of antinociception in naive animals in vivo (Sykes et al., 2007). However, the synaptic mechanisms and signaling pathways underlying the interaction between resident MORs and newly emerged DORs in central neurons remain unknown at present. In this study, we used the model of chronic morphine-induced DORs on central GABAergic terminals to investigate DOR–MOR interactions both synaptically in inhibition of GABA release, a crucial opioid effect for analgesia (Pan et al., 1997), and behaviorally in production of antinociception in rats in vivo.
Materials and Methods
All procedures involving the use of animals conformed to the guidelines by the University of Texas MD Anderson Cancer Center Animal Care and Use Committee.
Chronic morphine treatment.
Male Wistar rats were randomly divided into two groups: morphine group and saline/placebo group, and underwent chronic treatment with morphine or saline/placebo as described previously (Pan, 2003). For whole-cell recordings, neonatal rats (9–14 d old) in the morphine group were injected (i.p.) with increasing doses of morphine twice daily for 6 d. The morphine dose was 10 mg/kg on day 1 and was increased by 5 mg/kg each day to reach the maximum dose of 30 mg/kg on days 5 and 6. Rats in the saline group were similarly injected with saline for controls. The injection volume was 0.1–0.3 ml. Neonatal rats were used for better visualization of neurons in brainstem slices for visualized whole-cell recording. It has been shown that the physiological and pharmacological properties of neurons from these young rats are indistinguishable from those of adult rats (Pan et al., 1997). For molecular and behavioral experiments, rats (200–300 g) in the morphine group were subcutaneously implanted with one morphine pellet (75 mg) on day 1 and two more morphine pellets on day 4. In the placebo group, rats were similarly implanted with placebo pellets. Experiments were performed on day 7 for whole-cell recordings, biochemical analysis, or behavioral tests. The method of pellet implantation was primarily used in the adult rats in consideration of the fact that it required less total morphine and less work on our part. In some experiments, daily morphine injection was used to induce morphine tolerance and saline injection was used for controls in adult rats as a methodological control of their neonatal counterparts. Although developmental differences exist, our previous studies have shown that those cellular studies on neonatal rats serve well as guidelines for mechanisms underlying behavioral effects observed in adult rats (Pan et al., 1997; Bie et al., 2005).
Brian slice preparations.
The rat brain was cut in a vibratome in cold (4°C) physiological saline to obtain brainstem slices (200 μm thick) containing the nucleus raphe magnus (NRM). A single slice was submerged in a shallow recording chamber and perfused with preheated (35°C) physiological saline containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2, pH 7.2–7.4.
Whole-cell recording and synaptic currents.
Visualized whole-cell voltage-clamp recordings were obtained from identified NRM neurons with a glass pipette (resistance, 3–5 MΩ) filled with a solution containing the following (in mm): 126 KCl, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 2 ATP, and 0.25 GTP, pH adjusted to 7.3 with KOH; osmolarity, 280–290 mOsM. An AxoPatch-1D amplifier and AxoGraph software (Molecular Devices) were used for data acquisition and online/offline data analyses. A seal resistance of 2 GΩ or above and an access resistance of 15 MΩ or less were considered acceptable. Series resistance was optimally compensated and access resistance was monitored throughout the experiment. All NRM cells in this study were identified as a cell type that lacks postsynaptic MOR, as described in our previous study (Pan et al., 1990). Electrical stimuli of constant current (0.25 ms, 0.2–0.4 mA) were used to evoke GABA-mediated IPSCs with bipolar stimulating electrodes placed within the nucleus. With KCl-filled pipettes and a holding potential of −70 mV, GABA IPSCs were in an inward (downward) direction (Pan et al., 1990). GABA IPSCs were recorded in the presence of glutamate receptor antagonists d-(−)-2-amino-5-phosphonopentanoic acid (50 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (10 μm) and were completely blocked by GABAA receptor antagonist bicuculline (10 μm). A pair of IPSCs was evoked by two stimuli (100 ms interval) and a paired-pulse ratio (PPR) was calculated by dividing the amplitude of the second IPSC by the first one. The PPR has an inverse relationship with the probability of presynaptic transmitter release and has been widely used in previous studies to determine the involvement of a presynaptic action site (Zucker and Regehr, 2002; Bie and Pan, 2003). Five PPRs were averaged before (control) and 10 min after application of a drug. Some slices were preincubated for at least 1 h in vitro with the protein kinase A (PKA) inhibitor H89 (1 μm) (Bie et al., 2005) or with the phospholipase A2 (PLA2) inhibitor AACOCF3 (10 μm) (Zhu and Pan, 2005). Another structurally different PKA inhibitor, KT5720 (1 μm), and the adenylyl cyclase (AC) inhibitor SQ55236 (50 μm) were applied through the bath for at least 10 min before data collection. No morphine was added to the bath solution so that neurons in slices of the morphine group were likely under a condition of early withdrawal (Bie et al., 2005).
PLA2 activity assay.
The assay protocol was adapted from the instruction of a PLA2 assay kit (Cayman Chemical) and from a previous study (Reynolds et al., 1994). NRM tissues of both morphine (n = 9 rats) and placebo (n = 6 rats) groups were homogenized in a cold buffer (50 mm HEPES, pH 7.4, with 1 mm EDTA) and centrifuged at 10,000 × g for 15 min at 4°C. The supernatant was collected for assay and stored on ice. The following assay groups were used: (1) blank (no-enzyme control), (2) bee venom PLA2 (positive control), and (3) sample. The substrate solution (200 μl) was then added to each group of wells, well mixed, and incubated for 60 min at room temperature. The reaction was stopped by adding 10 μl DTNA/EGTA. The absorbance data were read at 414 nm using a plate reader and calculated by the following formula: A414/min = [A414(sample) − A414(blank)] ÷ 60 min. PLA2 activity = (ΔA414/min ÷ 10.66 mm−1) × (0.225 ml ÷ 0.01 ml) = μmol/min/ml.
Western blotting.
Total proteins were prepared after tissue lysis and centrifugation for DSD-polyacrylamide gel electrophoresis. β-Actin was used as an internal normalizer. Protein concentration was determined before immunoblot analysis and 20 μg was mixed with SDS sample buffer, heated to 100°C for 5 min, separated under reducing conditions on a 12% SDS–polyacrylamide gel, and transferred to a nitrocellulose membrane. Nonspecific binding was blocked by incubating the membrane in 3% bovine serum albumin in TBS overnight at room temperature. The membrane was incubated with a polyclonal rabbit anti-cPLA2 or anti-phospho-cPLA2 antibody (Cell Signaling Technology) against 95 kDa cPLA2 or phospho-cPLA2 (Cayman Chemical) for 2 h, and then with horseradish peroxidase-linked secondary antibody for 1 h at room temperature. The bands were detected using enhanced chemiluminescence (GE Healthcare).
Isobologram.
The method of isobolograms has been well documented for analysis of agonist interactions at both the receptor level and the behavioral level (Hurley et al., 1999; Bolan et al., 2002; Tallarida, 2006). As both DAMGO and deltorphin are considered full agonists (Quock et al., 1999; Clark et al., 2006), their potency ratios were assumed to remain fairly constant for their effects within the effective dose range, yielding relatively parallel dose–response curves or regression lines for isobolograms. A fixed dose ratio (9:1) of DAMGO–deltorphin mixture was approximated from the ratio of their EC50 values for each agonist alone. The total dose of the mixture was the sum of DAMGO dose and deltorphin dose in weight (nanograms). In synaptic analysis, both agonist doses in weight in the mixture were converted to their corresponding nanomolar concentrations to obtain a total dose of the mixture for comparison with data of dose–response curves for each agonist alone. Synaptic effects of the mixture, at a range of total doses (3 nm to 1 μm), on the IPSC amplitude were measured as experimentally derived data. The EC50 value and the 95% confidence limits of the mixtures were obtained from a regression line of the dose-dependent effects of the mixture and compared with those of the theoretical line of additivity defined by the straight line as follows: (D/DAMGO EC50) + (d/deltorphin EC50) = 1, where D is the DAMGO dose in the total dose of the mixture and d is the deltorphin dose in the mixture. Experimental data were analyzed by isobologram and statistically compared with the theoretical line of additivity to determine the pharmacological interaction of DORs and MORs as additive, subadditive, or superadditive (synergistic) using the FlashCalc software (kindly supplied by Dr. Michael Ossipov, University of Arizona, Tucson, AZ).
Microinjection and behavioral experiments.
For NRM microinjection, a rat was anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and restrained in a stereotaxic apparatus. A 26-gauge double-guide cannula (Plastics One) was inserted into the brain and aimed at the NRM (anteroposterior, −10.0 from the bregma; lateral, 0; dorsoventral, −10.5 from the dura) (Paxinos and Watson, 1986). The guide cannula was then cemented in place to the skull and securely capped. The rat was allowed to recover for at least 5 d from the surgery before implantation of morphine or placebo pellets. Pain threshold was measured by the tail-flick test on a freely moving rat with a Hargreaves analgesic instrument (Stoelting). Tail-flick latency to a heat stimulus was measured every 5 min. The heat intensity was set to elicit stable baseline latencies with a cutoff time of 10 s. A drug was microinjected into the NRM through a 33-gauge double-injector with an infusion pump at a rate of 0.2 μl/min. The total injected volume was 0.4 μl. The antinociceptive effect of a microinjected drug was measured 20 min after the microinjection and expressed as the maximum possible effect (MPE) as follows: MPE = (test latency − baseline)/(cutoff − baseline). The confined effect within the NRM of this microinjection method has been demonstrated in our previous studies (Bie et al., 2005; Ma and Pan, 2006).
Statistical analyses and materials.
General numerical data were statistically analyzed with Students' t tests and presented as SEM. Behavioral results were statistically analyzed by an ANOVA for repeated measures and the Tukey–Kramer test of post hoc analysis. DAMGO, deltorphin, morphine sulfate, morphine, and placebo pellets were kindly supplied by the drug program of the National Institute on Drug Abuse (Bethesda, MD). All other drugs were purchased from Sigma or Tocris Cookson.
Results
DOR inhibition of IPSCs is mediated by the PLA2 and PKA signaling pathways
Consistent with our previous study (Ma et al., 2006), chronic morphine induced new functional DORs on presynaptic GABAergic terminals in brainstem neurons of the nucleus raphe magnus. Thus, the selective DOR agonist deltorphin (1 μm) was ineffective on the amplitude of GABA IPSCs in slices of the saline group from saline-treated rats (94.3 ± 5.1% of baseline control, n = 6, p > 0.05), but significantly inhibited GABA IPSCs to 53.6 ± 5.6% of control in slices of the morphine group from morphine-treated rats (n = 15, p < 0.01) (Fig. 1A). This deltorphin inhibition was dose-dependent with an estimated EC50 of 228.0 nm (Fig. 1A), and it was completely abolished by the DOR antagonist naltriben (10 μm) (91.8 ± 5.8% of control, n = 5, p > 0.05). Confirming our previous finding of DORs present on the presynaptic site (Ma et al., 2006), deltorphin increased the PPR of GABA IPSCs only in neurons from the morphine group, an effect also blocked by naltriben (saline: control, 1.35 ± 0.09, deltorphin, 1.36 ± 0.09, n = 7, p > 0.05; morphine: control, 1.28 ± 0.05, deltorphin, 1.43 ± 0.04, n = 6, p < 0.05; morphine in naltriben: control, 1.29 ± 0.04, deltorphin, 1.30 ± 0.03, n = 5, p > 0.05) (Fig. 1B).
Figure 1.
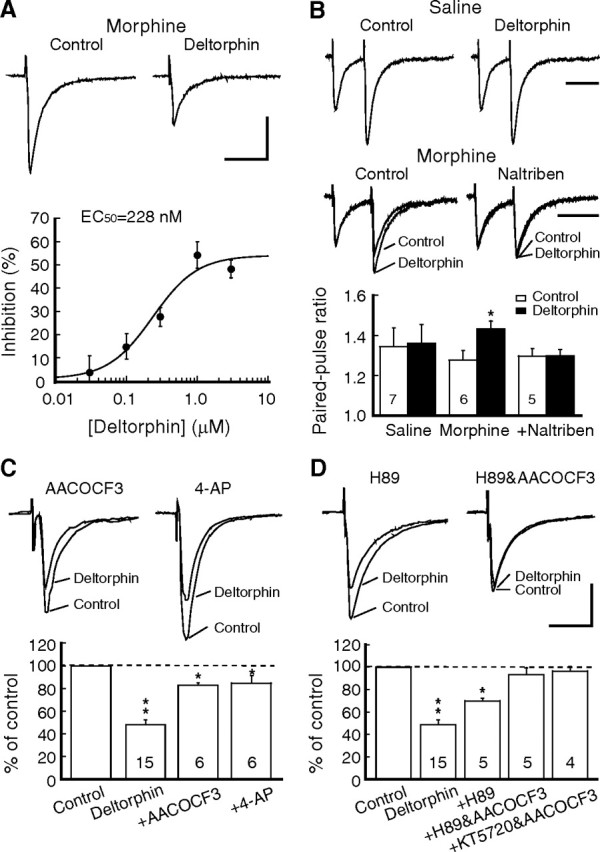
Chronic morphine-induced inhibition of GABA synaptic currents by DORs involves both the PLA2 and cAMP/PKA pathways. A, Representative GABA IPSCs (top) before (control) and during application of the DOR agonist deltorphin (1 μm) in a brainstem neuron of the NRM from a morphine-treated rat. Also shown (bottom) is a dose–response curve for the deltorphin inhibition (n = 6–7 cells for each data point). Data points were fitted by a logistic function to determine the EC50 value. B, Representative IPSC pairs (top) and summarized group data (bottom) in neurons from saline-treated rats and from morphine-treated rats without and with addition of the DOR antagonist naltriben (10 μm), showing the effects of deltorphin on the PPR. The numbers in columns denote cell numbers for that group. C, Deltorphin effects on GABA IPSCs in neurons of the morphine group in the presence of the PLA2 inhibitor AACOCF3 (10 μm) or the presynaptic potassium channel blocker 4-AP (100 μm). D, Deltorphin effects on IPSCs in the morphine group in the presence of the PKA inhibitor H89 (1 μm) or in combination of H89 and AACOCF3 or another PKA inhibitor KT5720 (1 μm) and AACOCF3. *p < 0.05, **p < 0.01. Calibration: A, C, D, 100 pA and 50 ms; B, 100 ms.
In naive conditions, MOR inhibition of GABA and glutamate synaptic transmission is predominantly mediated by the PLA2 signaling pathway in central neurons, as the MOR effect is completely abolished by PLA2 inhibitors and by 4-aminopyridine, which blocks the presynaptic voltage-dependent potassium channels regulated by metabolites of the PLA2 pathway (Vaughan et al., 1997; Zhu and Pan, 2005). We first determined whether the PLA2 pathway was involved in inhibition of GABA IPSCs by the chronic morphine-induced new DORs on GABA terminals in the brainstem neurons. In slices of the morphine group, AACOCF3 (10 μm), a selective PLA2 inhibitor, failed to completely block deltorphin inhibition of GABA IPSCs, only partially reducing the deltorphin effect (to 83.4 ± 2.1% of control, n = 6, p < 0.05 vs 53.6 ± 5.6% of control without AACOCF3), with significant inhibition remaining (p < 0.05 vs baseline) (Fig. 1C). Increasing the dose of AACOCF3 up to 30 μm did not produce more blockade of the deltorphin inhibition (83.3 ± 6.9% of control, n = 5). Similarly, 4-AP (100 μm) also only partially blocked the deltorphin inhibition (to 84.7 ± 6.6% of control, n = 6, p < 0.05 vs normal inhibition or baseline) (Fig. 1C). This incomplete reversal of the DOR inhibition by blockade of the PLA2 pathway indicated that, unlike MOR inhibition of synaptic currents in naive conditions, DOR inhibition of GABA IPSCs likely involved an additional mechanism(s) after chronic morphine treatment.
It is known that acute opioids inhibit, but chronic opioids upregulate, the cAMP/PKA signaling pathway, whose activation facilitates GABA synaptic release (Sharma et al., 1975; Williams et al., 2001). We then examined whether the chronic morphine-upregulated cAMP/PKA pathway was involved in the remaining DOR inhibition of IPSCs. We have shown previously that the PKA inhibitor H89 is ineffective on normal GABA IPSCs but blocks chronic morphine-induced increase in GABA synaptic release, suggesting that the upregulated cAMP/PKA signaling contributes to the enhanced GABA neurotransmission by chronic opioids in these neurons (Ma and Pan, 2006). In slices of the morphine group, we found that H89 (1 μm) partially blocked deltorphin inhibition of IPSCs (to 69.9 ± 2.4% of control, n = 5, p < 0.05 vs 53.6 ± 5.6% without H89 or vs baseline) (Fig. 1D). As expected, the deltorphin inhibition was completely abolished by a combination of the PLA2 inhibitor AACOCF3 and either H89 or another structurally different PKA inhibitor KT5720 (1 μm) (AACOCF3 plus H89, 93.4 ± 6.1% of control, n = 5, p > 0.05; AACOCF3 plus KT5720, 96.5 ± 4.2% of control, n = 4, p > 0.05) (Fig. 1D). Thus, it appears that chronic morphine-induced DORs inhibit GABAergic synaptic transmission through both the PLA2 pathway and the upregulated PKA pathway in these neurons.
The potency of MOR agonist is increased after chronic morphine
In slices of the saline group, the MOR agonist DAMGO (1 μm) inhibited the IPSC amplitude to 51.8 ± 4.7% of control (n = 12, p < 0.01), which was completely blocked by the selective MOR antagonist CTAP (1 μm, 97.8 ± 6.4% of control, n = 6, p > 0.05 vs baseline). Previous studies report no significant change in MOR inhibition of GABA transmission in central neurons after chronic morphine treatment, as measured by percentage inhibition of GABA IPSC amplitude with MOR agonists (Vaughan et al., 1997; Ingram et al., 1998; Ma and Pan, 2006). In this study, we also found similar IPSC inhibition in a percentage term produced by DAMGO (1 μm) in slices of the morphine group (to 47.9 ± 4.3% of control, n = 16, p > 0.05 vs the saline group) (Fig. 2A,B). However, those previous studies also show that chronic morphine augments synaptic GABA release, resulting in an elevated baseline activity of GABA synaptic transmission. Thus, the measurement of percentage inhibition might have masked a potential change in the MOR effect due to an altered baseline. We then took efforts to compare the amplitudes of IPSCs evoked by the same stimulus intensity between saline and morphine slice groups, despite the expected difficulty of this between-group comparison due to usually large variations in IPSC amplitude among cells. We found that the averaged IPSC amplitude was significantly larger in the morphine group than that in the saline group (control, 178 ± 10 pA, n=57; morphine, 307 ± 11 pA, n = 192; p < 0.05) (Fig. 2C). This indicated that MOR agonists needed a larger net inhibition of IPSCs in the morphine group to reach the percentage inhibition similar to that in the saline group. Indeed, DAMGO produced a net inhibition of 83 ± 13 pA in IPSC amplitude in the saline group (n = 12), but 159 ± 10 pA in the morphine group (n = 16) (p < 0.01) (Fig. 2C). To reveal the underlying change, we compared the dose-dependent responses for the DAMGO inhibition of IPSCs between the two groups of slices. As shown in Figure 2D, the estimated EC50 for the saline group was 81.5 nm, but it was 29.1 nm for the morphine group, representing a 2.8-fold leftward shift in the dose– response curves without a significant change in the maximum inhibition. These results indicate that the potency, but not efficacy, of the MOR agonist is increased after chronic morphine.
Figure 2.
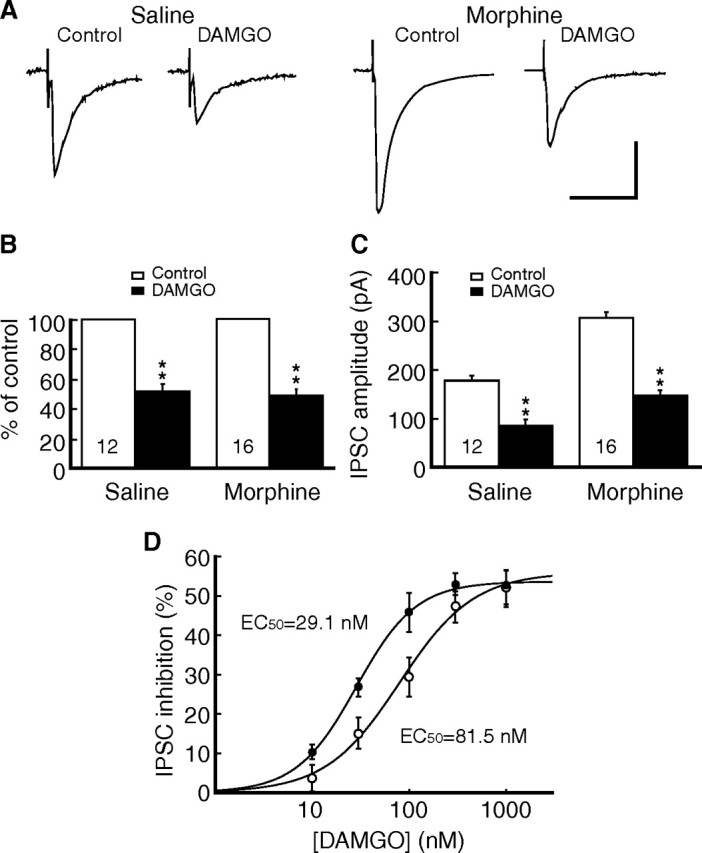
Chronic morphine increases the potency of μ-opioid receptor (MOR) agonist. A, GABA IPSCs in the absence and presence of the MOR agonist DAMGO (1 μm) in neurons of saline and morphine groups. B, C, Summarized data of the DAMGO inhibition expressed as percentage of control (B) or current amplitude (C). D, Dose–response relationship for the IPSC-inhibiting effect of DAMGO in neurons from the saline-treated rats (open circles) and from the morphine-treated rats (filled circles). n = 6–7 cells for each data point. **p < 0.01. Calibration: A, 100 pA and 50 ms.
The cAMP/PKA pathway is recruited in MOR inhibition of IPSCs after chronic morphine
Consistent with previous reports, DAMGO inhibition of IPSCs in neurons of the saline group was completely blocked by AACOCF3 (10 μm) and by 4-AP (100 μm), inhibitors of the PLA2 pathway and its targeted potassium channels, respectively (DAMGO plus AACOCF3, 96.5 ± 8.5% of control, n = 6, p > 0.05; DAMGO plus 4-AP, 99.1 ± 3.7% of control, n = 8, p > 0.05) (Fig. 3A). In contrast, in slices of the morphine group, the same inhibitors could only partially block the DAMGO effect with significant inhibition remaining (DAMGO plus AACOCF3, 84.9 ± 3.3% of control, n = 8, p < 0.05 vs baseline; DAMGO plus 4-AP, 72.3 ± 1.5% of control, n = 8, p < 0.01 vs baseline) (Fig. 3B).
Figure 3.
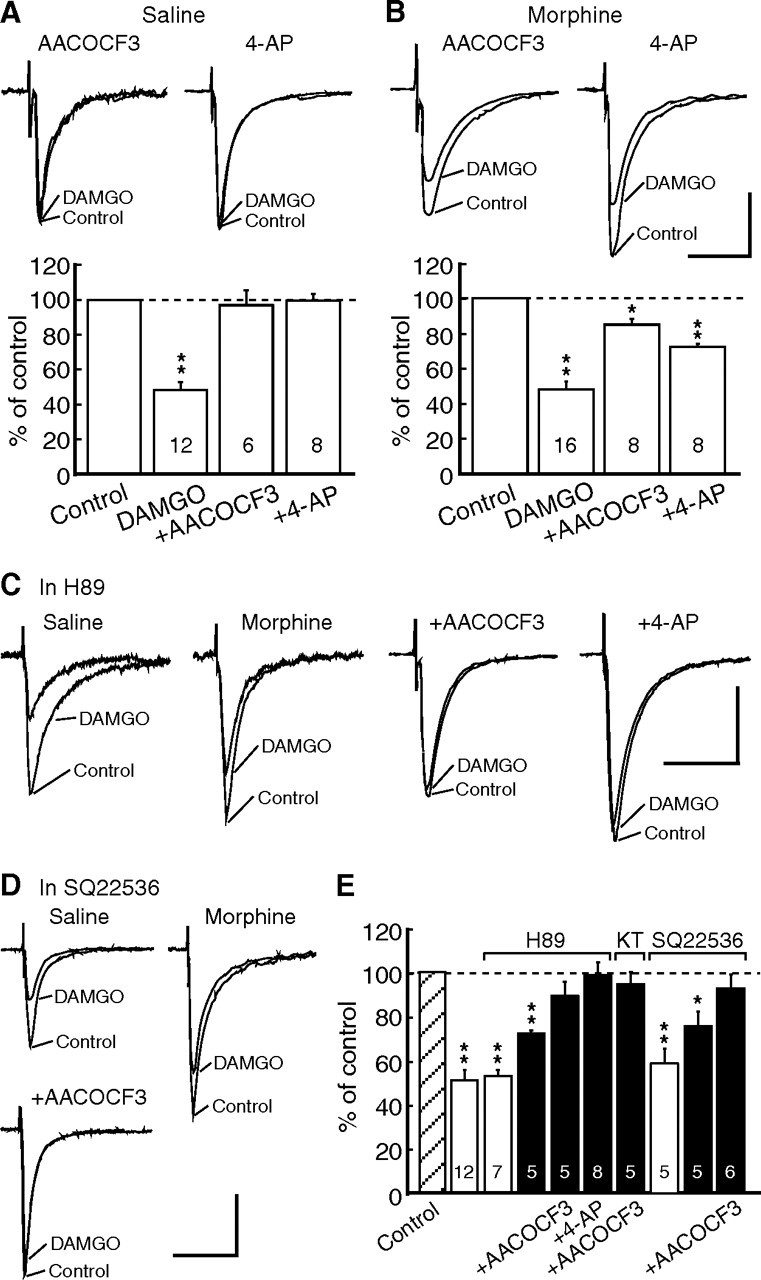
Chronic morphine recruits the cAMP/PKA pathway for MOR inhibition of GABA synaptic currents. A, B, Representative IPSCs (top) and group data (bottom) of the DAMGO inhibition in control and in the presence of AACOCF3 or 4-AP in neurons of the saline group (A) and the morphine group (B). C, DAMGO effects on IPSCs in the presence of H89 from neurons of the saline group and of the morphine group without or with addition of AACOCF3 or 4-AP. D, DAMGO effects in similar experiment groups but in the presence of the AC inhibitor SQ22536 (50 μm). E, Summarized data of the DAMGO effects on IPSCs in the saline group (open columns) and in the morphine group (filled columns). *p < 0.05, **p < 0.01. Calibration: 100 pA and 50 ms. KT, KT5720.
We then determined whether the cAMP/ PKA pathway was involved in the remaining MOR inhibition in the morphine group. In slices of the saline group, H89 (1 μm) was ineffective on DAMGO inhibition of IPSCs (to 52.6 ± 3.0% of control, n = 7, p > 0.05 vs 51.8 ± 4.7% without H89), but it significantly reduced the DAMGO inhibition in slices of the morphine group (to 72.9 ± 1.3% of control, n = 5, p < 0.01 vs 52.6 ± 3.0% for the saline slice group or vs baseline) (Fig. 3C,E). Similar to the DOR inhibition, H89 plus AACOCF3 or 4-AP completely blocked the DAMGO inhibition in the morphine group (H89 plus AACOCF3, 89.8 ± 6.5% of control, n = 5, p > 0.05 vs baseline; H89 plus 4-AP, 98.5 ± 4% of control, n = 8, p > 0.05). A combination of KT5720 and AACOCF3 also produced a complete blockade of the DAMGO inhibition (94.9 ± 5.4% of control, n = 5, p > 0.05) (Fig. 3E). Moreover, similar to H89, blockade of the upstream enzyme AC with the specific inhibitor SQ22536 (50 μm) had no effect on DAMGO inhibition of IPSCs in slices of the saline group (to 59.7 ± 6.7% of control, n = 5, p > 0.05 vs DAMGO inhibition without SQ22536), but partially reduced DAMGO inhibition in slices of the morphine group (to 75.2 ± 6.6% of control, n = 5, p < 0.05 vs the saline slice group or vs baseline) (Fig. 3D,E). The remaining inhibition was eliminated by addition of the PLA2 inhibitor AACOCF3 (Fig. 3E). Furthermore, we mimicked morphine upregulation of the cAMP/PKA pathway by using the AC activator forskolin and the cAMP analog 8-bromo-cAMP in naive slices. Basal GABAergic transmission was strongly enhanced by forskolin and 8-bromo-cAMP (IPSC amplitude in forskolin: 206.4 ± 7.5% of baseline, n = 5, p < 0.01; in 8-bromo-cAMP: 165.8 ± 8.7%, n = 5, p < 0.01) (Fig. 4A,B). Similar to the results in neurons of the morphine group from morphine-treated rats, in naive slices after application of forskolin or 8-bromo-cAMP, AACOCF3 was no longer able to completely block the effect of DAMGO, which still induced a significant inhibition of IPSCs (forskolin: to 68.9 ± 6.8% of control, n = 14, p < 0.01 vs baseline; 8-bromo-cAMP: 67.8 ± 7.6% of control, n = 9, p < 0.01) (Fig. 4A,B). The effect of the AC and cAMP activators was largely presynaptic (Bie et al., 2005), as they decreased the IPSC PPR (Fig. 4C). After blockade of the PLA2 component with AACOCF3, DAMGO significantly increased the PPR, indicating that the remaining DAMGO effect, likely mediated by the activated cAMP/PKA pathway, was probably presynaptic (forskolin, 1.07 ± 0.03, plus DAMGO, 1.41 ± 0.07, n = 14, p < 0.01; 8-bromo-cAMP, 1.08 ± 0.06, plus DAMGO, 1.29 ± 0.06, n = 9, p < 0.01) (Fig. 4D).
Figure 4.
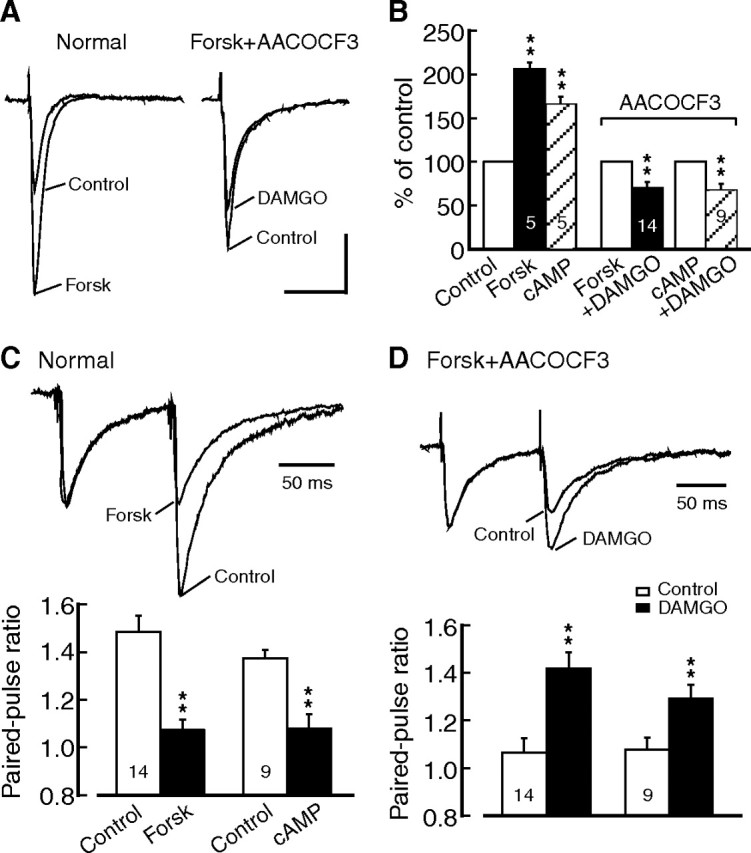
Activation of the cAMP/PKA pathway induces additional MOR inhibition of GABA synaptic currents. A, Effect of the AC activator forskolin (10 μm) on IPSCs in a naive neuron (normal) and a neuron in a naive slice treated with forskolin and AACOCF3. B, Summarized data of normalized IPSC amplitude after treatment of naive slices with forskolin, with the cAMP analog 8-bromo-cAMP (100 μm), or with AACOCF3 plus forskolin or cAMP before and after addition of DAMGO. C, D, IPSC pairs and summarized data of effects of forskolin and 8-bromo-cAMP on PPR in naive slices (normal, C), and effects of DAMGO on PPR in naive slices treated with forskolin plus AACOCF3 (D). **p < 0.01. cAMP, 8-bromo-cAMP; Forsk, Forskolin. Calibration: 100 pA and 50 ms.
These results indicate that, after chronic morphine exposure, the upregulated cAMP/PKA pathway becomes involved in MOR inhibition of GABA synaptic transmission in addition to the normally involved PLA2 pathway. These presynaptic mechanisms of MOR inhibition appear similar to those for the IPSC inhibition by the newly emerged DOR described above.
Chronic morphine upregulates PLA2 activity
Next, we examined whether chronic morphine altered the PLA2 activity, which might also change the ability of opioid receptors to inhibit IPSCs. We found that the dose–response curve for AACOCF3 blockade of the DAMGO inhibition was apparently altered by chronic morphine, with the same dose of AACOCF3 producing much less antagonism of the DAMGO inhibition of IPSCs in the morphine group (Fig. 5A). Further, we isolated the PLA2-mediated inhibition by pretreatment of slices of both groups with the AC inhibitor KT5720. In slices of the saline group, AACOCF3 dose-dependently antagonized the DAMGO effect, with 10 μm DAMGO producing a nearly complete blockade (Fig. 5B). However, in slices of the morphine group, the two intermediate doses of AACOCF3 (0.3 and 1 μm) produced a much smaller amount of blockade of DAMGO inhibition (0.3 μm AACOCF3: 7.3 ± 3.7% inhibition, n = 5, p < 0.01 vs 47.2 ± 6.2% inhibition in the saline group, n = 8; 1 μm AACOCF3: 13.1 ± 2.7% inhibition, n = 5, p < 0.01 vs 56.5 ± 6.2% inhibition in the saline group, n = 11) (Fig. 5B). These findings could be indicative of an upregulated PLA2 enzymatic activity, rendering AACOCF3 relatively less effective at blocking the PLA2 activity. Indeed, our Western blot experiments showed that the expression level of phosphorylated PLA2, its active form, was significantly higher in brainstem tissues taken from morphine-treated rats (n = 9) than that from placebo-treated rats (n = 6), while there was no detectable difference in the expression of total PLA2 proteins between the two groups (Fig. 5C). Similar results were also obtained in rats treated with daily injections of morphine (Fig. 5D). Furthermore, enzymatic activity assay of absorbance (414 nm) and activity (nanomoles per minute per milliliter) showed that PLA2 enzymatic activity was consistently higher in brainstem tissues from morphine-treated rats (n = 9) than in that of placebo-treated rats (n = 6) (absorbance: placebo, 0.1443 ± 0.0069, morphine, 0.2937 ± 0.0106; activity: placebo, 5.0774 ± 0.2416, morphine, 10.3310 ± 0.3715). These pharmacological and biochemical results support the notion that the PLA2 activity in these neurons is upregulated by chronic morphine.
Figure 5.
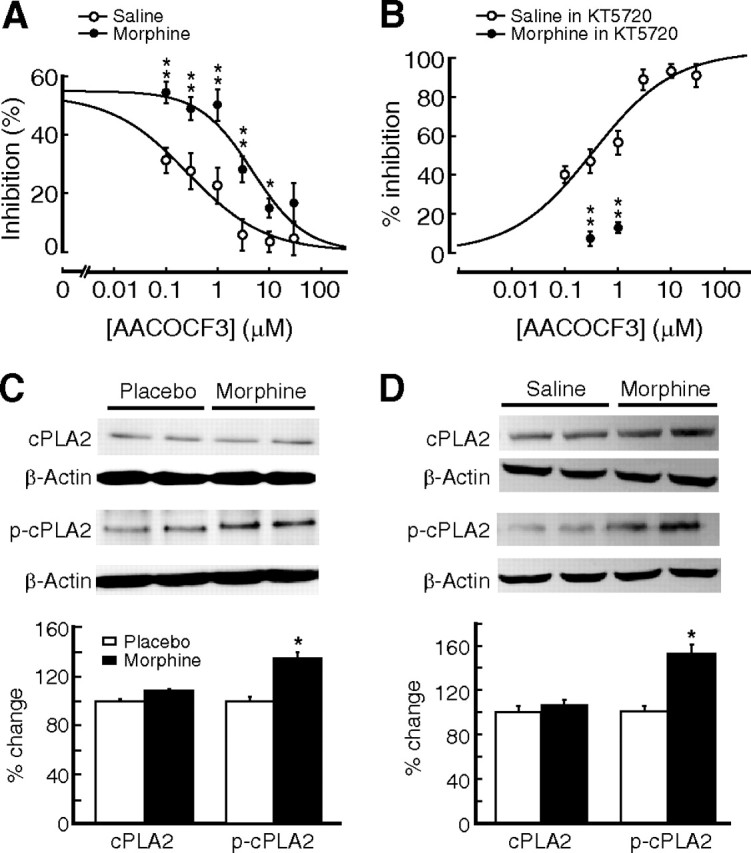
Chronic morphine upregulates PLA2 activity. A, Dose–response relationship for AACOCF3 antagonism of DAMGO-mediated inhibition of IPSCs in neurons of the saline group (open circles) and of the morphine group (filled circles). n = 6–12 cells for each data point. B, Similar experiments of AACOCF3 antagonism but in the presence of KT5720 for both the saline group (n = 8–11) and the morphine group (n = 5 for each AACOCF3 dose), showing AACOCF3 antagonism of PLA2-mediated DAMGO inhibition (normalized). C, D, Representative lanes and group data of Western blots for cPLA2 and phosphorylated cPLA2 as well as for β-actin in NRM tissues from placebo (n = 6) and morphine pellet-implanted (n = 9) rats (C) and from saline (n = 5)- and morphine (n = 5)-injected rats (D). Data of percentage changes were normalized to the expression of β-actin. The molecular mass was 95 kDa for cPLA2 and phosphorylated cPLA2. *p < 0.05, **p < 0.01.
DOR and MOR agonists produce synergistic inhibition of IPSCs
Recruitment of DORs to MOR-containing GABA terminals brought forward an intriguing issue of DOR–MOR interaction at the synaptic level. We then determined the pharmacological interaction of DORs and MORs in their IPSC inhibitions in slices of the morphine group, using their agonist mixture of fixed ratios and isobologram analysis to define the interaction as simple additive, subadditive or superadditive (synergistic) (Tallarida, 2006). At a fixed DAMGO–deltorphin ratio of 9:1 approximated from the ratio of the respective EC50 value for each agonist alone, the DAMGO–deltorphin mixture in a range of total doses (DAMGO dose plus deltorphin dose) dose-dependently inhibited IPSCs, with a maximum inhibition magnitude of ∼60% and an estimated EC50 of 8.7 nm (Fig. 6A). This was 3.3-fold and 26.2-fold smaller than the EC50 value for DAMGO alone (29.1 nm) and for deltorphin alone (228.0 nm), respectively, suggesting that the potency of this agonist mixture at equal total doses was increased when compared with that of each agonist alone. Using isobologram analysis, we found that the regression line of experimentally derived dose-dependent effects for the 9:1 mixture was shifted to the left from the theoretically calculated line of additivity (Fig. 6B). As shown in the isobolograph (Fig. 6C), the EC50 value of the 9:1 mixture was significantly smaller than those of theoretical line of additivity (p < 0.001), suggesting a synergistic interaction between deltorphin and DAMGO. Moreover, similar isobologram results were obtained with a DAMGO–deltorphin mixture at 1:1 ratio (p < 0.01), indicating that the synergism of these two agonists may be independent on dose ratios at a certain range (Fig. 6C). These results support the notion that recruitment of DORs to MOR-containing GABA terminals produces more inhibition of GABA neurotransmission than simple addition of DOR- and MOR-mediated inhibitions through a synergistic interaction on GABAergic terminals.
Figure 6.
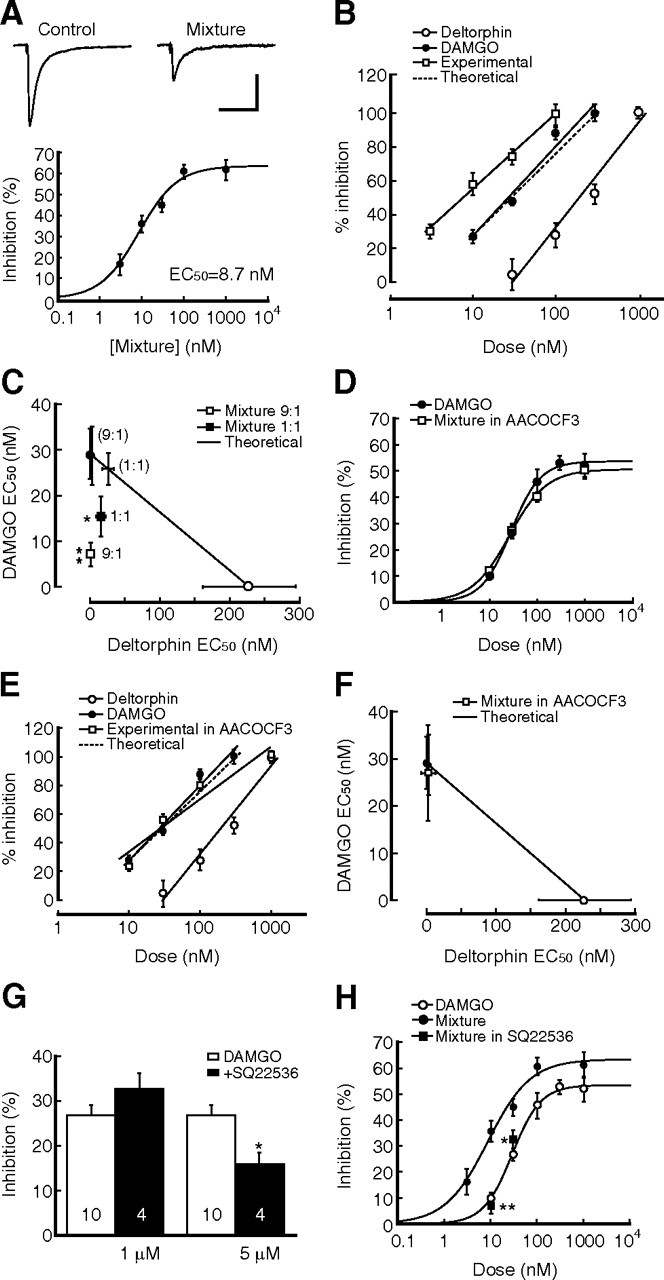
Combinations of MOR and DOR agonists produce synaptic synergism. A, Dose-dependent inhibition of IPSCs by a mixture of DAMGO and deltorphin at a fixed ratio of 9:1 in neurons from morphine-treated rats. n = 5–7 cells for each data point. B, Normalized data of dose-dependent IPSC inhibition by DAMGO, deltorphin, and the mixture (experimental), fitted by a regression line, and a calculated theoretical line of additivity in neurons from the morphine group. n = 5–16 cells for each data point. C, Isobolograph of EC50 values and their 95% confidence limits (vertical and horizontal bars) for theoretical additivity and for experimentally derived data (squares) with a DAMGO:deltorphin mixture of 9:1 and 1:1 ratios. For comparison, theoretical EC50 values for simple additivity at both 9:1 and 1:1 mixture ratios are also depicted as cross bars (95% confidence limits) without symbols on the theoretical line. D, Dose–response curves for IPSC inhibition by DAMGO and by the mixture (9:1) plus AACOCF3 (1 μm). n = 5–12 cells for each data point. E, F, Similar dose-dependent data of regression lines and a theoretical line (E) and isobolograph of EC50 values (F) except that the experimental data of the mixture (9:1) were obtained in the presence of AACOCF3 (1 μm). n = 5–16 cells for each data point. G, Effects of lower doses of the AC inhibitor SQ22536 on DAMGO inhibition of IPSCs in neurons of the morphine group. H, Antagonizing effect of 1 μm SQ22536 on the IPSC inhibition induced by the mixture at 10 nm (n = 10) and 30 nm (n = 7). n = 5–16 for other data points. *p < 0.05, **p < 0.01. Calibration: 100 pA and 50 ms.
To identify whether the upregulated PLA2 activity played a role in the DOR–MOR interaction, similar experiments with the DAMGO–deltorphin mixture (9:1 ratio) and isobologram analysis were performed in slices of the morphine group pretreated with an intermediate dose of AACOCF3 (1 μm), which had little effect on DAMGO inhibition of IPSCs in the morphine group (Fig. 5A). Our results showed that, under these conditions, the dose–response curves for the mixture of equal total doses and for DAMGO alone were similar (mixture: EC50 = 26.9 nm) (Fig. 6D). In addition, the experimentally obtained regression line of dose–response data was similar to the theoretical line of additivity, having similar EC50 values and overlapping 95% confidence limits (p > 0.05) (Fig. 6E,F). So it appears that the PLA2 activity is involved in the synaptic synergism of presynaptic DORs and MORs.
The signaling pathways involved in the mixture inhibition of GABA IPSCs remained similar, as the AC inhibitor SQ22536 (50 μm) partially blocked the IPSC inhibition by the mixture (1 μm) in the morphine group, with 12.5 ± 5.6% inhibition remaining (n = 9, p < 0.01 vs baseline), and the mixture inhibition was completely abolished by the combination of AACOCF3 and SQ22536 or AACOCF3 and KT5720 (AACOCF3 plus SQ22536: 95.3 ± 4.2% of control, n = 9, p > 0.05; AACOCF3 plus KT5720: 97.0 ± 4.3%, n = 5, p > 0.05). To determine whether the AC inhibitor blocked the DOR–MOR synergism or MOR-mediated inhibition, or both, we examined the effect of lower doses of SQ22536 on the mixture inhibition and DAMGO inhibition of IPSCs. At 1 μm, SQ22536 failed to alter the DAMGO effect, but 5 μm SQ22536 significantly reduced the DAMGO inhibition (Fig. 6G). However, 1 μm SQ22536, ineffective on the effect of DAMGO alone, significantly decreased the IPSC-inhibiting effect of the mixture at total doses of 10 nm (n = 10) and 30 nm (n = 7) (Fig. 6H), indicating that the upregulated cAMP/PKA pathway also plays a role in the DOR–MOR synergism.
DOR and MOR agonists produce synergistic antinociception
Inhibition of central GABA synaptic transmission, consequently causing disinhibition, or activation, of central pain-inhibiting neurons, is one of the major opioid actions responsible for opioid-induced analgesia (Pan et al., 1997; Ma and Pan, 2006). Thus, the synaptic synergism in augmented DOR–MOR inhibition of GABA IPSCs identified above should lead to similar behavioral synergism in producing antinociceptive effects in animals in vivo. To demonstrate that, we conducted behavioral experiments with site-specific microinjections in chronic morphine-treated rats. A single microinjection of DAMGO (10 ng) into the NRM produced a significant antinociceptive effect (n = 5 rats) (Fig. 7A,B). This DAMGO effect has been shown to be blocked by comicroinjection of the MOR antagonist CTAP, suggesting a specific MOR-mediated effect (Ma and Pan, 2006). A relatively much higher dose of deltorphin (1 μg) was needed to induce a significant antinociceptive effect (n = 5) (Fig. 7A,B), which occurred only in morphine-treated rats and was reversed by comicroinjection of the DOR antagonist naltriben (Ma et al., 2006). A subthreshold dose of DAMGO (6 ng) had no effect on baseline pain threshold (n = 5) (Fig. 7B). Therefore, we used a DAMGO–deltorphin mixture at 1:1 ratio (3 ng DAMGO plus 3 ng deltorphin) so that the total dose of the mixture was 6 ng, whose maximum effect, assuming simple additivity, would be less than the effect of 6 ng DAMGO because the potency of 3 ng deltorphin was less than that of 3 ng DAMGO (Figs. 1A, 2D). However, we found that, in contrast to the ineffectiveness of 6 ng DAMGO, microinjection of 6 ng mixture produced a significant antinociceptive effect in chronic morphine-treated rats (n = 6), but not in placebo-treated rats (n = 4) (Fig. 7C), indicating a synergistic interaction of DORs and MORs, leading to the augmented behavioral effect. Additionally, a premicroinjection of AACOCF3 (0.2 and 2 μg) 1 h before significantly reduced the mixture (6 ng)-induced antinociceptive effect (n = 6 for both AACOCF3 doses) (Fig. 7D,E). Similar behavioral synergism of DORs and MORs was also found in rats treated with daily morphine injections instead of implantation of morphine pellets (Fig. 7F). These findings further support the notion that the chronic morphine-induced new DOR interacts with resident MOR synergistically in a PLA2-dependent manner in the NRM, resulting in an enhanced antinociceptive effect of opioids.
Figure 7.
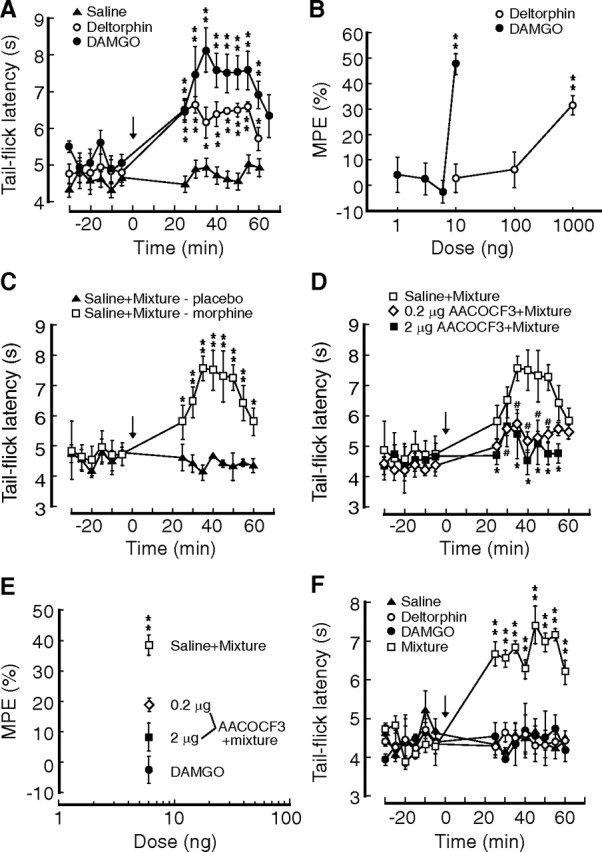
Mixture of MOR and DOR agonists produces behavioral synergism. A, Effects of a single microinjection (arrow) of saline (n = 4 rats), DAMGO (10 ng, n = 5), or deltorphin (1 μg, n = 5) into the brainstem NRM on pain threshold measured by the tail-flick test in morphine-treated rats in vivo. B, Dose-dependent effects of microinjected DAMGO and deltorphin on the pain threshold in morphine-treated rats. n = 4–6 rats. C, Behavioral effects of NRM-microinjected 1:1 mixture composed of subthreshold doses of DAMGO (3 ng) and deltorphin (3 ng) with premicroinjection (1 h before) of saline in placebo-treated (n = 4 rats) and morphine-treated rats (n = 6). D, Behavioral effects of premicroinjections of 0.2 and 2 μg AACOCF3 on the mixture-induced antinociception in morphine-treated rats. n = 6 rats for each group. E, Behavioral effects on pain threshold produced by NRM microinjections of 6 ng DAMGO and by the mixture (6 ng at 1:1 ratio) with premicroinjection of saline or AACOCF3 (0.2 μg and 0.2 μg). F, Behavioral effects of NRM microinjection of saline (n = 4 rats), deltorphin (3 ng, n = 4), DAMGO (6 ng, n = 5), and the mixture (3 ng DAMGO plus 3 ng deltorphin, n = 6) in rats treated with twice daily injections of morphine. * and # p < 0.05, **p < 0.01.
Discussion
We have demonstrated in these pain-modulating brainstem neurons that chronic morphine-induced DORs inhibit GABA synaptic transmission through both the PLA2 and cAMP/PKA pathways, while MOR potency at inhibiting GABA synaptic transmission is increased with the cAMP/PKA pathway recruited to the mechanisms of GABA synaptic transmission after chronic morphine treatment. We have also shown that the emerged DOR and resident MOR produce functional synergism both synaptically in inhibiting presynaptic GABA release and behaviorally in producing antinociceptive effects, a synergistic interaction that is likely dependent on both the PLA2 and cAMP/PKA activities upregulated by chronic morphine.
Presynaptic mechanisms for opioid inhibition of GABA synaptic transmission
Under normal conditions, MOR agonist-induced presynaptic inhibition of central synaptic transmission in the brain, including both GABAergic and glutamatergic transmission, is primarily mediated by activation of the PLA2 pathway, of which the arachidonic acid metabolites open 4-AP- and dendrotoxin-sensitive potassium channels (Vaughan et al., 1997; Ingram et al., 1998; Zhu and Pan, 2005). We have shown recently that chronic opioids induce functional DORs, which are absent in naive conditions, on GABA and glutamate synaptic terminals in central neurons, resulting in DOR-mediated inhibition of transmitter release (Ma et al., 2006; Bie et al., 2009). The current study suggests that, in addition to the PLA2 pathway, the DOR inhibition is also mediated by the cAMP/PKA pathway, a new signaling component brought out by chronic morphine. Due to the lack of DORs in naive conditions, this new cAMP/PKA signaling is better demonstrated in MOR inhibition of GABA synaptic transmission, as it is absent in naive conditions but is partially responsible for the MOR inhibition after chronic morphine treatment. Therefore, it appears that the cAMP/PKA activity, very low in normal conditions and significantly upregulated by chronic morphine (Sharma et al., 1975; Williams et al., 2001; Nestler, 2004; Bie et al., 2005), greatly increases GABA synaptic transmission so that DOR as well as MOR is now able to produce a significant inhibition in the upregulated cAMP/PKA activity and consequently in the increased level of GABA synaptic release. A previous study in periaqueductal gray neurons indicated that the 4-AP-sensitive potassium channels were not involved in DAMGO inhibition of IPSCs in opioid withdrawal conditions, based on the observation that DAMGO still inhibited the IPSC in the presence of 4-AP (Ingram et al., 1998). We made a similar observation (Fig. 3B) and based on our current results, this observation could be accounted for by DAMGO inhibition of the newly recruited cAMP/PKA pathway. Our further analysis showed that AC and PKA inhibitors could only partially block the DAMGO effect and the complete blockade required both an AC/PKA inhibitor and a PLA2 inhibitor or 4-AP (Fig. 3C–E). These data argue that, although the cAMP/PKA pathway may be relatively more dominant, the PLA2 pathway still mediates part of opioid inhibition of IPSCs in opioid withdrawal conditions, as in naive conditions.
It is likely that the added MOR inhibition of cAMP/PKA-dependent GABA release contributes to the larger amount of net inhibition by MORs and hence the increased DAMGO potency for the inhibition. This is in contrast to a previous report of an increased efficacy, but not potency, of DAMGO during opioid withdrawal (Chieng and Williams, 1998). However, that increased efficacy was observed only in the presence of forskolin to further activate the cAMP cascade and thus could be partially related to the forskolin action itself. The enhanced potency of the MOR agonist found in this study provides further evidence that opioid tolerance in these pain-modulating brainstem neurons may be primarily not caused by a diminished action of MOR itself, but rather by its downstream signaling events and consequent adaptive alterations.
Synaptic synergism of DORs and MORs
To our knowledge, there has been no previous report of functional DORs and MORs copresent on presynaptic terminals in central neurons under naive conditions. Thus, functional interaction of DORs and MORs in regulation of neurotransmitter release has not been a research issue until recently. As we found that DORs and MORs share the same signaling pathways for their inhibition of GABA IPSCs, it was normally expected that the DOR inhibition would likely reduce the MOR inhibition through occlusion. However, we found instead a significant synergism in their inhibition of GABA neurotransmission, with more inhibition by the agonist mixture than the simple sum of each agonist-induced inhibition alone. This indicates a functionally synergistic interaction between the two types of opioid receptors, which could occur at some points along their signaling pathways, from the receptor to the molecules regulating GABA release. At the receptor level, it has been shown in heterologously expressing cell lines that occupancy of DORs by DOR antagonists increases MOR signaling and function through heterodimeric association (Gomes et al., 2004). However, we did not observe a noticeable functional interaction at the receptor level in these brainstem neurons, as the presence of a DOR antagonist did not alter the dose–response curve for DAMGO inhibition of GABA IPSCs under chronic morphine conditions (J. Ma, B. Bie, and Z. Z. Pan, unpublished observations). Interestingly, our data show that the synaptic synergism of DORs and MORs is dependent on the upregulated activity of both cAMP/PKA and PLA2 pathways that are shared by the two receptors. A presently unclear, yet interesting, issue is the potential interactions between the cAMP/PKA and PLA2 signaling pathways for the synaptic synergism after chronic opioid exposure. Chronic opioids might activate the two pathways in a parallel manner, or cross-interactions between the two signaling pathways could also occur. For example, PLA2 may be activated/upregulated by chronic opioid-induced cAMP supersensitization through the PKA-Ras/Raf-MAPK cascade (Williams et al., 2001; Murakami and Kudo, 2002; Robinson-White and Stratakis, 2002). Understanding of the underlying signaling mechanisms is important for development of DOR-based therapeutic strategies to enhance the analgesic effect of opioids under opioid-tolerant conditions.
We propose that the DOR appears on the same GABA terminals that readily express MORs, rather than on a separate population of GABA terminals distinct from MOR-containing GABA terminals, to produce the synaptic synergism in these neurons. This is based on our findings that both DORs and MORs are present on presynaptic terminals with the recorded cells lacking postsynaptic MORs (Pan et al., 1997; Ma et al., 2006), excluding postsynaptic component-mediated synergism through the response of GABAA receptors; and that blockade of the intracellularly located PLA2 inhibits the synergism, making it less likely that the synergistic interaction occurs across different GABA terminals. Direct evidence of DORs and MORs colocalizing on the same GABA terminals awaits further detailed anatomical studies.
Behavioral synergism of DORs and MORs
DOR agonists are not effective analgesics normally in clinical practice and in animal studies, as they produce weak antinociceptive effects under normal conditions (Inturrisi, 2002; Fields, 2004). As shown in recent studies, this can be largely attributed to DOR's primary localization of intracellular compartments in pain-modulating brainstem and spinal cord neurons (Fields, 2004; Hack et al., 2005; Ma et al., 2006; Zhang et al., 2006). However, a variety of chemical and behavioral stimuli, including chronic opioids and sustained pain conditions, can trigger the membrane trafficking of intracellular DORs from constitutively targeted intracellular compartments to plasma membrane, resulting in new functional DORs and DOR-mediated antinociception (Commons, 2003; Ma et al., 2006; Cahill et al., 2007; Sykes et al., 2007).
It has been long known that administration of MOR and DOR agonists together in animals produces a superadditive effect in their antinociceptive actions in vivo (Horan et al., 1992; Adams et al., 1993; Rossi et al., 1994). Recent studies also show a synergistic interaction between DOR and MOR agonists in combination of fixed ratios in their induction of antinociceptive effects (Sykes et al., 2007; Negus et al., 2009). However, the underlying cellular and molecular mechanisms for the DOR–MOR synergism are unknown. The current study provides a synaptic mechanism for this behavioral synergism of DOR and MOR agonists in antinociception, as opioid inhibition of GABA synaptic transmission is one of the main opioid actions responsible for opioid analgesia (Pan et al., 1997; Pan, 1998). Thus, augmented inhibition of GABA IPSCs by DOR and MOR synergism may account for the enhanced analgesia produced by their combined administration in vivo.
This DOR–MOR synergism may have significant clinical implications for improving pain management. Analgesic tolerance to repeated opioid analgesics, mainly MOR agonists, causes diminished opioid effects and undertreatment of patients with chronic pain; and long-term opioid-induced physical dependence and drug addiction limit the optimal use of opioid analgesics for fear of drug abuse (Woolf and Hashmi, 2004; Ballantyne and LaForge, 2007; Højsted and Sjøgren, 2007). In this regard, DOR agonists have much less abuse potential (Contet et al., 2004), and in addition to numerous reports of behaviorally synergistic effects of DOR and MOR agonists, we have shown that DOR agonists also reduce opioid tolerance (Ma et al., 2006). Interestingly, in contrast to their analgesia-enhancing effects, DOR agonists do not seem to enhance opioid's undesirable drug-reinforcing and sedative effects (Stevenson et al., 2005; Negus et al., 2009). Therefore, DOR agonists may have significant therapeutic potential for treatment of chronic pain conditions under a chronic opioid state.
Footnotes
This work was supported by the National Institute on Drug Abuse Grants DA023069 and DA027541.
References
- Adams JU, Tallarida RJ, Geller EB, Adler MW. Isobolographic superadditivity between delta and mu opioid agonists in the rat depends on the ratio of compounds, the mu agonist and the analgesic assay used. J Pharmacol Exp Ther. 1993;266:1261–1267. [PubMed] [Google Scholar]
- Ballantyne JC, LaForge KS. Opioid dependence and addiction during opioid treatment of chronic pain. Pain. 2007;129:235–255. doi: 10.1016/j.pain.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Bhargava HN. Diversity of agents that modify opioid tolerance, physical dependence, abstinence syndrome, and self-administrative behavior. Pharmacol Rev. 1994;46:293–324. [PubMed] [Google Scholar]
- Bie B, Pan ZZ. Presynaptic mechanism for anti-analgesic and anti-hyperalgesic actions of κ-opioid receptors. J Neurosci. 2003;23:7262–7268. doi: 10.1523/JNEUROSCI.23-19-07262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie B, Peng Y, Zhang Y, Pan ZZ. cAMP-mediated mechanisms for pain sensitization during opioid withdrawal. J Neurosci. 2005;25:3824–3832. doi: 10.1523/JNEUROSCI.5010-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie B, Zhu W, Pan ZZ. Rewarding morphine-induced synaptic function of delta-opioid receptors on central glutamate synapses. J Pharmacol Exp Ther. 2009;329:290–296. doi: 10.1124/jpet.108.148908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan EA, Tallarida RJ, Pasternak GW. Synergy between mu opioid ligands: evidence for functional interactions among mu opioid receptor subtypes. J Pharmacol Exp Ther. 2002;303:557–562. doi: 10.1124/jpet.102.035881. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A. Prolonged morphine treatment targets δ opioid receptors to neuronal plasma membranes and enhances δ-mediated antinociception. J Neurosci. 2001;21:7598–7607. doi: 10.1523/JNEUROSCI.21-19-07598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Chieng B, Williams JT. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci. 1998;18:7033–7039. doi: 10.1523/JNEUROSCI.18-17-07033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MJ, Furman CA, Gilson TD, Traynor JR. Comparison of the relative efficacy and potency of mu-opioid agonists to activate Galpha(i/o) proteins containing a pertussis toxin-insensitive mutation. J Pharmacol Exp Ther. 2006;317:858–864. doi: 10.1124/jpet.105.096818. [DOI] [PubMed] [Google Scholar]
- Commons KG. Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J Comp Neurol. 2003;464:197–207. doi: 10.1002/cne.10788. [DOI] [PubMed] [Google Scholar]
- Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5:565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A. 2004;101:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Bagley EE, Chieng BC, Christie MJ. Induction of δ-opioid receptor function in the midbrain after chronic morphine treatment. J Neurosci. 2005;25:3192–3198. doi: 10.1523/JNEUROSCI.4585-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Højsted J, Sjøgren P. Addiction to opioids in chronic pain patients: a literature review. Eur J Pain. 2007;11:490–518. doi: 10.1016/j.ejpain.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Horan P, Tallarida RJ, Haaseth RC, Matsunaga TO, Hruby VJ, Porreca F. Antinociceptive interactions of opioid delta receptor agonists with morphine in mice: supra- and sub-additivity. Life Sci. 1992;50:1535–1541. doi: 10.1016/0024-3205(92)90144-e. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. The analgesic effects of supraspinal μ and δ opioid receptor agonists are potentiated during persistent inflammation. J Neurosci. 2000;20:1249–1259. doi: 10.1523/JNEUROSCI.20-03-01249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RW, Grabow TS, Tallarida RJ, Hammond DL. Interaction between medullary and spinal delta1 and delta2 opioid receptors in the production of antinociception in the rat. J Pharmacol Exp Ther. 1999;289:993–999. [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ. Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci. 1998;18:10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inturrisi CE. Clinical pharmacology of opioids for pain. Clin J Pain. 2002;18:S3–S13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- Ma J, Pan ZZ. Contribution of brainstem GABA(A) synaptic transmission to morphine analgesic tolerance. Pain. 2006;122:163–173. doi: 10.1016/j.pain.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhang Y, Kalyuzhny AE, Pan ZZ. Emergence of functional delta-opioid receptors induced by long-term treatment with morphine. Mol Pharmacol. 2006;69:1137–1145. doi: 10.1124/mol.105.019109. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dollé P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Murakami M, Kudo I. Phospholipase A2. J Biochem. 2002;131:285–292. doi: 10.1093/oxfordjournals.jbchem.a003101. [DOI] [PubMed] [Google Scholar]
- Negus SS, Bear AE, Folk JE, Rice KC. Role of delta opioid efficacy as a determinant of mu/delta opioid interactions in rhesus monkeys. Eur J Pharmacol. 2009;602:92–100. doi: 10.1016/j.ejphar.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Historical review: Molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Pan ZZ. mu-Opposing actions of the kappa-opioid receptor. Trends Pharmacol Sci. 1998;19:94–98. doi: 10.1016/s0165-6147(98)01169-9. [DOI] [PubMed] [Google Scholar]
- Pan ZZ. Opioid tolerance in adult and neonatal rats. Methods Mol Med. 2003;84:223–232. doi: 10.1385/1-59259-379-8:223. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Williams JT, Osborne PB. Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. J Physiol. 1990;427:519–532. doi: 10.1113/jphysiol.1990.sp018185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Tershner SA, Fields HL. Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature. 1997;389:382–385. doi: 10.1038/38730. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Ed 2. Sydney: Academic; 1986. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Quock RM, Burkey TH, Varga E, Hosohata Y, Hosohata K, Cowell SM, Slate CA, Ehlert FJ, Roeske WR, Yamamura HI. The delta-opioid receptor: molecular pharmacology, signal transduction, and the determination of drug efficacy. Pharmacol Rev. 1999;51:503–532. [PubMed] [Google Scholar]
- Reynolds LJ, Hughes LL, Yu L, Dennis EA. 1-Hexadecyl-2-arachidonoylthio-2-deoxy-sn-glycero-3-phosphorylcholine as a substrate for the microtiterplate assay of human cytosolic phospholipase A2. Anal Biochem. 1994;217:25–32. doi: 10.1006/abio.1994.1079. [DOI] [PubMed] [Google Scholar]
- Robinson-White A, Stratakis CA. Protein kinase A signaling: “cross-talk” with other pathways in endocrine cells. Ann N Y Acad Sci. 2002;968:256–270. doi: 10.1111/j.1749-6632.2002.tb04340.x. [DOI] [PubMed] [Google Scholar]
- Rossi GC, Pasternak GW, Bodnar RJ. Mu and delta opioid synergy between the periaqueductal gray and the rostro-ventral medulla. Brain Res. 1994;665:85–93. doi: 10.1016/0006-8993(94)91155-x. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci U S A. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson GW, Folk JE, Rice KC, Negus SS. Interactions between delta and mu opioid agonists in assays of schedule-controlled responding, thermal nociception, drug self-administration, and drug versus food choice in rhesus monkeys: studies with SNC80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide] and heroin. J Pharmacol Exp Ther. 2005;314:221–231. doi: 10.1124/jpet.104.082685. [DOI] [PubMed] [Google Scholar]
- Sykes KT, White SR, Hurley RW, Mizoguchi H, Tseng LF, Hammond DL. Mechanisms responsible for the enhanced antinociceptive effects of micro-opioid receptor agonists in the rostral ventromedial medulla of male rats with persistent inflammatory pain. J Pharmacol Exp Ther. 2007;322:813–821. doi: 10.1124/jpet.107.121954. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. An overview of drug combination analysis with isobolograms. J Pharmacol Exp Ther. 2006;319:1–7. doi: 10.1124/jpet.106.104117. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Hashmi M. Use and abuse of opioid analgesics: potential methods to prevent and deter non-medical consumption of prescription opioids. Curr Opin Investig Drugs. 2004;5:61–66. [PubMed] [Google Scholar]
- Zhang X, Bao L, Guan JS. Role of delivery and trafficking of delta-opioid peptide receptors in opioid analgesia and tolerance. Trends Pharmacol Sci. 2006;27:324–329. doi: 10.1016/j.tips.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Zhu W, Pan ZZ. Mu-opioid-mediated inhibition of glutamate synaptic transmission in rat central amygdala neurons. Neuroscience. 2005;133:97–103. doi: 10.1016/j.neuroscience.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]