

SUMMARY
PI3Kδ and PI3Kγ regulate immune cell signaling, while the related PI3Kα and PI3Kβ regulate cell survival and metabolism. Selective inhibitors of PI3Kδ/γ represent a potential class of anti-inflammatory agents lacking the anti-proliferative effects associated with PI3Kα/β inhibition. Here we report the discovery of PI3Kδ/γ inhibitors that display up to 1,000-fold selectivity over PI3Kα/β and evaluate these compounds in a high-content inflammation assay using mixtures of primary human cells. We find selective inhibition of only PI3Kδ is weakly anti-inflammatory, but PI3Kδ/γ inhibitors show superior inflammatory marker suppression through suppression of LPS-induced TNFα production and T-cell activation. Moreover, PI3Kδ/γ inhibition yields an anti-inflammatory signature distinct from pan-PI3K inhibition and known anti-inflammatory drugs, yet bears striking similarities to glucocorticoid receptor agonists. These results highlight the potential of selectively designing drugs that target kinases with shared biological function.
Introduction
Inflammatory disorders such as rheumatoid arthritis represent an important target for drug development. Therapies include naproxen, indomethacin (Backhouse et al., 1980), and corticosteroids (Gray et al., 1991). While effective, these agents have significant side effects that limit their utility (Gray et al., 1991; Rainsford, 1993). More recently, antibody therapeutics directed against tumor necrosis factor α (TNFα) have become useful for treatment of refractory chronic inflammation (Feldmann, 2002; Feldmann and Maini, 2001). These agents reduce inflammation and slow disease progression (Feldmann, 2002; Feldmann and Maini, 2001; Imperato et al., 2004), but are expensive and can generate immune-related side effects, including infection and lymphoma emergence (Imperato et al., 2004).
Recently, targeted inhibitors of the phosphoinositide-3-kinase (PI3K) pathway have been suggested as immunomodulatory agents. (Hirsch et al., 2008; Rommel et al., 2007) This interest stems from the fact that the PI3K pathway serves multiple functions in immune cell signaling, primarily through the generation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), a membrane-bound second messenger. (Cantley, 2002; Deane and Fruman, 2004; Hirsch et al., 2008; Katso et al., 2001) PIP3 recruits proteins to the cytoplasmic side of the lipid bilayer, including protein kinases and GTPases (Cantley, 2002; Hirsch et al., 2008; Katso et al., 2001), initiating a complex network of downstream signaling cascades important in the regulation of immune cell adhesion, migration, and cell-cell communication.
The four class I PI3K isoforms differ significantly in their tissue distribution. PI3Kα and PI3Kβ are ubiquitous and activated downstream of receptor tyrosine kinases (RTK) (Hirsch et al., 2008; Katso et al., 2001), while PI3Kδ and PI3Kγ are primarily limited to hematopoietic (Deane and Fruman, 2004; Rommel et al., 2007) and endothelial cells (Puri et al., 2004; Puri et al., 2005), and are activated downstream of RTKs, and G-protein coupled receptors (GPCR) respectively (Katso et al., 2001). Mouse genetic studies have revealed that PI3Kα and PI3Kβ are essential for normal development (Vanhaesebroeck et al., 2005), while loss of PI3Kδ and/or PI3Kγ yields viable offspring with selective immune deficits (Okkenhaug and Vanhaesebroeck, 2003; Swat et al., 2006; Vanhaesebroeck et al., 2005; Webb et al., 2005). The expression pattern and functions of PI3Kδ and PI3Kγ have generated much interest in developing PI3Kδ/γ inhibitors as agents for many diseases, including rheumatoid arthritis, allergies, asthma, chronic obstructive pulmonary disease and multiple sclerosis (Hirsch et al., 2008; Marone et al., 2008; Rommel et al., 2007; Ruckle et al., 2006). Studies using both pharmacologic and genetic methods have shown these two isoforms often demonstrate synergistic interactions with each other (Konrad et al., 2008; Laffargue et al., 2002). In mast cells, for example, PI3-Kδ is essential for degranulation in response to IgE crosslinking of Fc-receptors (Ali et al., 2004; Ali et al., 2008), but PI3-Kγ plays an important role in amplifying the response (Laffargue et al., 2002). Similar effects have been seen in other cellular functions, including lymphocyte homing (Reif et al., 2004) and the neutrophil respiratory burst (Condliffe et al., 2005), where PI3-Kγ plays a critical role and PI3-Kδ amplifies each process.
The non-redundant but related roles of PI3Kδ and PI3Kγ have made it difficult to determine which of the two isoforms (alone or in combination) is best targeted in a particular inflammatory disorder. Studies using mice that lack PI3Kδ and/or PI3Kγ or express kinase-dead variants of PI3Kδ and PI3Kγ have been valuable tools in understanding their roles. For example, PI3-Kδ knockout mice demonstrated diminished neutrophil chemotaxis (Puri et al., 2004), diminished antibody production (both T-cell dependent and independent) (Jou et al., 2002), and lower numbers of mature B-cells (Clayton et al., 2002; Jou et al., 2002), and a decrease in their proliferation in response to anti-IgM (Jou et al., 2002). This phenotype was replicated in the PI3Kδ kinase-dead variant (Okkenhaug et al., 2002), and with PI3Kδ selective inhibitors (Ali et al., 2004; Puri et al., 2004; Sadhu et al., 2003), along with decreased numbers of and proliferation of mast cells, and an attenuated allergic response (Ali et al., 2004). The PI3Kγ knockout contained higher numbers of, but less responsive neutrophils (Hirsch et al., 2000), lower numbers of and less responsive macrophages (Hirsch et al., 2000) and dendritic cells (Del Prete et al., 2004), displayed decreased mast cell degranulation (Laffargue et al., 2002), a higher ratio of CD4+ to CD8+ T-cells (Rodriguez-Borlado et al., 2003), increased thymocyte apoptosis (Sasaki et al., 2000), diminished induction of CXCR3 on activated T cells (Barbi et al., 2008), and decreased cardiac contractility (Crackower et al., 2002). This latter effect on cardiac tissue was a concern for chronic dosing of patients with PI3Kγ inhibitors. However, this concern was largely mitigated when the PI3Kγ kinase-dead variant (which better mimics inhibition of the kinase rather than loss of the protein) showed similar immune cell phenotypes, but importantly had no cardiac defects (Patrucco et al., 2004). The cardiac effect was later shown to be due to scaffolding effects rather than the catalytic activity of PI3Kγ. The dual PI3Kδ /PI3Kγ knockout was viable but exhibited serious defects in T-cell development (Webb et al., 2005) and thymocyte survival (Swat et al., 2006). The PI3Kγ knockout/PI3Kδ kinase-dead combination produced a similar phenotype suggesting that at least within the immune system, the role of PI3Kδ is likely only a catalytic one (Ji et al., 2007).
Interpretation of studies using knockout and kinase-dead mice can be challenging since these models provide only a steady-state picture of the immune system, lack temporal and dose control, and do not permit a full understanding of how a dynamic immune response will react to reversible inhibition. Selective inhibitors with varying profiles (PI3Kδ, PI3Kγ, and PI3Kδ/γ) are necessary for studies of leukocyte signaling in order to assess the relative contributions of each PI3K to immune cell activation.
The key challenge in developing isoform selective PI3K inhibitors is that all Class I PI3Ks share nearly identical ATP binding pockets (Knight et al., 2004). However, we and others reported that a quinazolinone-based chemical scaffold binds PI3Kδ with high selectivity (Knight et al., 2006; Puri et al., 2004). This chemotype exploits a unique, non-catalytically active conformation of PI3Kδ (and PI3Kγ) that is not easily adopted by PI3Kα and PI3Kβ (Berndt et al., 2009; Knight et al., 2006). In this work, we report the development of potent PI3Kδ and PI3Kδ/γ inhibitors. We then characterize these compounds using primary human leukocytes stimulated in co-culture with different cytokine and chemokine ligands in order to determine if targeting PI3Kγ and PI3Kδ, either alone or in combination, results in a distinct anti-inflammatory fingerprint. Finally, we compare selective PI3Kδ/γ inhibitors to other kinase inhibitors and clinical anti-inflammatory therapeutics.
Results
Design of PI3Kδ and PI3Kδ/γ Inhibitors
The ATP-binding pocket of the four class I PI3Ks (α, β, δ, γ) contains three distinct inhibitor binding elements: a hinge region with two hydrogen bonding contacts, a hydrophobic pocket deep in the protein not occupied by ATP, and a conformationally-mobile methionine residue above the adenine ring of ATP (Knight et al., 2006) (Fig. 1). The first two binding elements are common to all PI3Ks while the third appears to be significantly accessible by only PI3Kδ and PI3Kγ (Berndt et al., 2009; Knight et al., 2006).
Fig. 1. Basis of selectivity and potency for aryl quinazolinones.
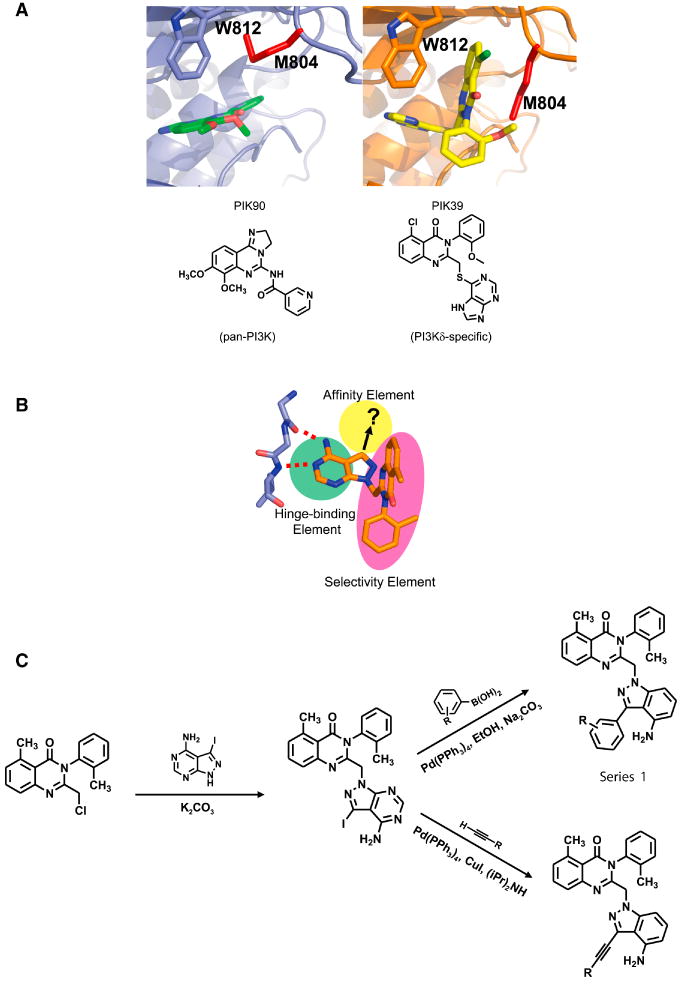
A. Crystal structure of PI3Kγ with PIK90, a pan-PI3K inhibitor (Left) and PIK39, a PI3Kδ inhibitor (Right)(Knight et al., 2006). The bi-planar binding mode of PIK39 and alternate positioning of Met804 (red) are illustrated. Chemical compound structures are shown below.
B. Compounds in this study introduce affinity elements (yellow) onto selective tolyl quinazolinone (red) scaffold maintaining selectivity for PI3Kγ and PI3Kδ.
C. Diversification of SW series affinity elements was achieved through Suzuki-Miyaura couplings with aryl boronic acids and Sonogashira couplings with terminal alkynes, yielding two distinct chemical series.
In order to maximize the differential binding to PI3Kδ or PI3Kγ compared to PI3Kα and PI3Kβ we exploited the rearrangement of Met804, which normally forms the roof of the ATP-binding pocket when PI3Kγ is bound to ATP and pan-PI3K inhibitors (Fig. 1A, left panel). This rearrangement reveals a new pocket, not found in any ATP bound forms of PI3Ks (Knight et al., 2006). The aryl quinazolinone inhibitor, PIK39 (Fig. 1A, right panel) exists in a conformation which projects the quinazolinone moiety into the pocket produced by the Met804 movement, affording highly selective binding to PI3Kδ and PI3Kγ (Berndt et al., 2009; Knight et al., 2006).
Synthesis and Biochemical Characterization
Both PIK39 (Fig. 1A, right panel) and the related PIK293 (Fig. 1B), form hydrogen bonds with the hinge region of PI3Ks, but do not contain elements which project into the deeper hydrophobic pocket (Knight et al., 2006) (Fig. 1B). Despite the sequence similarities in the hydrophobic binding pocket, we reasoned that it was possible to distinguish between PI3Kδ and PI3Kγ with the appropriate binding elements. PIK293 was therefore diversified into two series using palladium-catalyzed routes (Fig 1C). Series 1, synthesized with Suzuki couplings, projected substituted aryl rings into the affinity pocket, while series 2 placed sterically minimal alkyne derivatives into the affinity pocket using Sonogashira couplings (Fig. S1, S2).
Nearly fifty candidate inhibitors were synthesized and screened against all class I PI3Ks as previously described (Knight et al., 2007). As expected, all compounds were highly selective for PI3Kδ over PI3Kα and PI3Kβ (Fig. S1, S2). Addition of elements that interacted with the hydrophobic-affinity pocket increased potency against PI3Kδ and PI3Kγ without compromising selectivity against PI3Kα and PI3Kβ. SW18, one of the most potent PI3Kδ inhibitors in the series (Fig. 2A,B), was screened against 219 protein kinases at a concentration of 10μM. 218 kinases in the panel retained at least 75% activity, and only one, macrophage stimulating 1 receptor (MST1R) kinase, was inhibited to less than 60% activity (Table S1). These data demonstrate that the tolyl quinazolinone moiety responsible for selectivity within the PI3K family also confers selectivity against protein kinases.
Fig. 2. Diversification of Tolyl Quinazolinone Series.
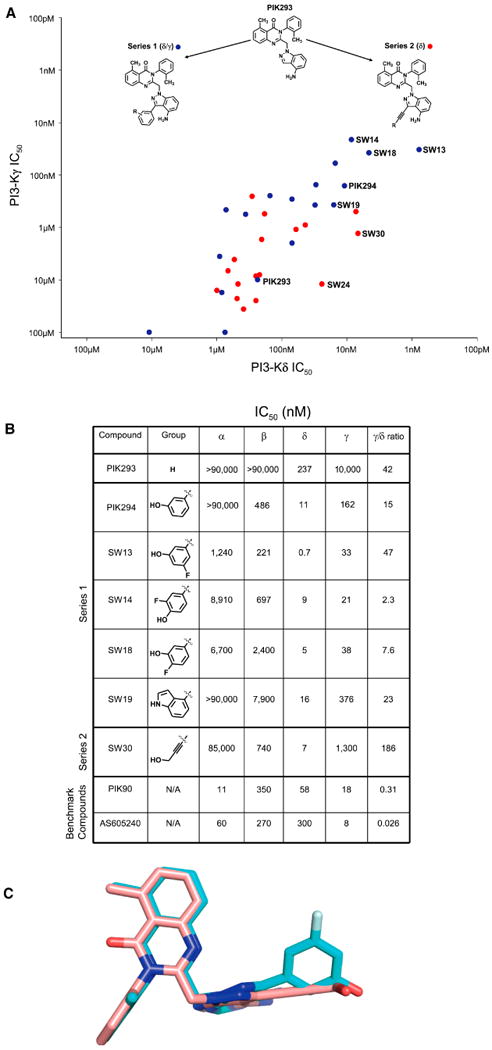
A. Introduction of affinity elements to PIK293. Series 1 (PI3Kδ/γ) introduces aryl groups and Series 2 (PI3Kδ) introduces alkynyl-linked groups. IC50s of both Series 1 (blue) and Series 2 (red) were graphed.
B. Biochemical IC50s and γ/δ ratios for Series 1 and 2 along with benchmark compounds. All compounds were tested at 10μM ATP.
C. Crystal structure of SW13(cyan), and 30(pink) as reported (Berndt et al., 2009). Hydrogen bonding elements for SW series are all aligned.
Phenol containing members of Series 1 showed the greatest potency against PI3Kδ (Fig. 2A,B), and tuning of PI3Kγ affinity was possible by substitution of the phenol ring at the 4 position (SW14, Fig. 2A,B). Movement of the fluoro-substituent to the 5 position (SW13) afforded the most potent PI3Kδ inhibitor reported to date. While there is considerable precedent for hydroxyl groups in the hydrophobic affinity pocket of PI3Ks (Palanki et al., 2007; Walker et al., 2000), phenolic substituents are often subject to first pass metabolism, limiting their oral bioavailability. We therefore explored groups that would preserve hydrogen-bonding interactions but would not contain a phenol. Of the several candidate molecules prepared, only the indole-substituted SW19 was able to retain significant PI3Kδ potency and some activity against PI3Kγ while eliminating the presence of a hydroxyl group (Fig. 2B).
Introducing an aryl-alkynyl linkage in Series 2 reduced the bulk of the affinity pocket binding element and decreased PI3Kγ inhibition, resulting in selectivity for PI3Kδ (Fig. S2). As in Series 1, a properly positioned hydroxyl group was critical to maintain potency against PI3Kδ. For example, SW30 contained a hydroxymethyl-substituted alkyne while SW41 extended the hydroxyl group by one methylene unit and resulted in >30-fold loss of binding affinity for PI3Kδ (Fig. S2). Together, these structure-activity data suggest that potency against PI3Kδ requires the presence of a properly positioned hydrogen bond donor/acceptor and that the additional bulk provided by aryl groups in Series 1 affords activity against PI3Kγ (Fig. 2C).
The PI3Kγ/δ ratio of these compounds illustrates a wide range of biochemical activity against PI3Kγ while preserving potency against PI3Kδ. Out of nearly fifty compounds, SW13, SW14, SW19, and SW30 (Figs. 2&3) were selected for characterization in cellular assays based on potency for PI3Kδ and PI3Kγ and their range of selectivity between the two targets.
Fig. 3. Effect of selected compounds on THP-1 monocyte signaling.

A. THP-1 monocyte signaling pathway leads to phosphorylation of Akt through either RTK-linked Macrophage Colony Stimulating Factor (M-CSF) or GPCR-linked Monocyte chemoattractant protein 1 (MCP-1).
B. pAkt was measured by FACS using fluorescent pSer473 Akt antibodies. Quantitated fluorescence was used to determine an EC50 for selected compounds. Biochemical γ/δ ratios are provided for comparison.
Cellular Signaling Effects
We next assessed the inhibition of PI3Kδ and PI3Kγ in a cell model with a single readout by stimulating THP-1 monocytes with different ligands to activate either PI3Kδ or PI3Kγ. Macrophage colony stimulating factor (M-CSF/CSF-1) is a cytokine that binds to the M-CSF receptor activating RTK-linked PI3Ks (Kelley et al., 1999) while the chemokine, monocyte chemoattractant protein-1 (MCP-1) activates PI3Kγ through a GPCR (Jones et al., 2003). Both stimuli lead to phosphorylation of Akt at Ser473 (Camps et al., 2005; Jones et al., 2003; Pomel et al., 2006), which we monitored by fluorescence-activated cell sorting (FACS) (Fig. 3A). We included a literature benchmark compound, AS605240 (Camps et al., 2005; Pomel et al., 2006), and negative control (SW23) in our analysis.
M-CSF stimulated Ser473 p-Akt production was potently blocked by SW13, SW14, SW19 and SW30, but not by SW23 or AS605240 (Fig. 3B). We attribute these effects to PI3-Kδ inhibition because SW23 and AS605240, which do not significantly inhibit PI3Kδ at the doses tested, were ineffective. Only the compounds with biochemical IC50 values under 25 nM against PI3Kδ were able to inhibit Ser473 p-Akt production in this assay. Further support for the role of PI3Kδ in this assay comes from the consistent rank order (biochemical IC50 vs. cellular EC50). SW13 was most potent in the biochemical assay and was the most potent in the cellular M-CSF assay as well.
MCP-1 stimulated Ser473 p-Akt production was blocked by AS605240 and SW14, but not significantly by SW30 or SW23. SW19 blocked MCP-1 stimulated Ser473 p-Akt production but only at somewhat higher concentrations, in a manner consistent with its biochemical IC50 value (Fig. 3B). SW14, the most potent tolyl quinazolinone PI3Kγ inhibitor in the biochemical assays was of equal potency with AS605240, the most potent PI3Kγ inhibitor included in our studies. Similar to the M-CSF assay, this rank order is consistent with the biochemical potency of these compounds against PI3-Kγ. The PI3Kδ selectivity of SW13 and especially SW30 is highlighted by their low activity in this assay, while the inability of SW23 to inhibit p-Akt formation suggests that the activity in this assay is due to inhibition of PI3K rather than chemotype/scaffold-dependent effects.
Modulation of Inflammatory Signaling in Human Peripheral Blood Mononuclear Cells (PBMCs) and Umbilical Vein Endothelial Cells (HUVECs)
The above cellular data motivated us to assess the role of PI3Kδ and PI3Kγ in inflammatory signaling by use of co-cultures of primary human cells not adapted to long-term growth in vitro. There are two advantages of this approach. First, human cells rather than rodent cells are used, permitting an accurate assessment of human PI3K immune signaling. Second, the in vitro nature of the assay allows use of the compounds with highly distinct inhibitory profiles and minimizes any pharmacokinetic/pharmacodynamic issues that arise in the transition from cell-based to animal-based models. We compared the activity of selective and pan-PI3K inhibitors using the Biologically Multiplexed Activity Profiling (BioMAP) method (Berg et al., 2006; Kunkel et al., 2004). This assay utilizes primary human cells (HUVECs alone or in combination with PBMCs), stimulated with combinations of inflammatory signals (IL-1β, TNFα, IFN-γ, IL-4, Histamine, Lipopolysaccharide, and Superantigen) (Fig. 4A). The resultant expression levels of a panel of 20 receptors, cytokines, cell adhesion molecules and second messengers (Fig. 4A) – were measured following stimulation and drug treatment, and compared to the untreated (“no drug”) levels. These expression levels yield a distinctive pattern, or profile, unique to each drug that can be used to compare and contrast several types of chemical compounds and drug classes (Fig. 4B). Similar profiles are generally observed with similar mechanisms of action, but can also be observed when different chemical compounds share a functional similarity.
Fig. 4. BioMAP Analysis of PI3K Inhibitors.
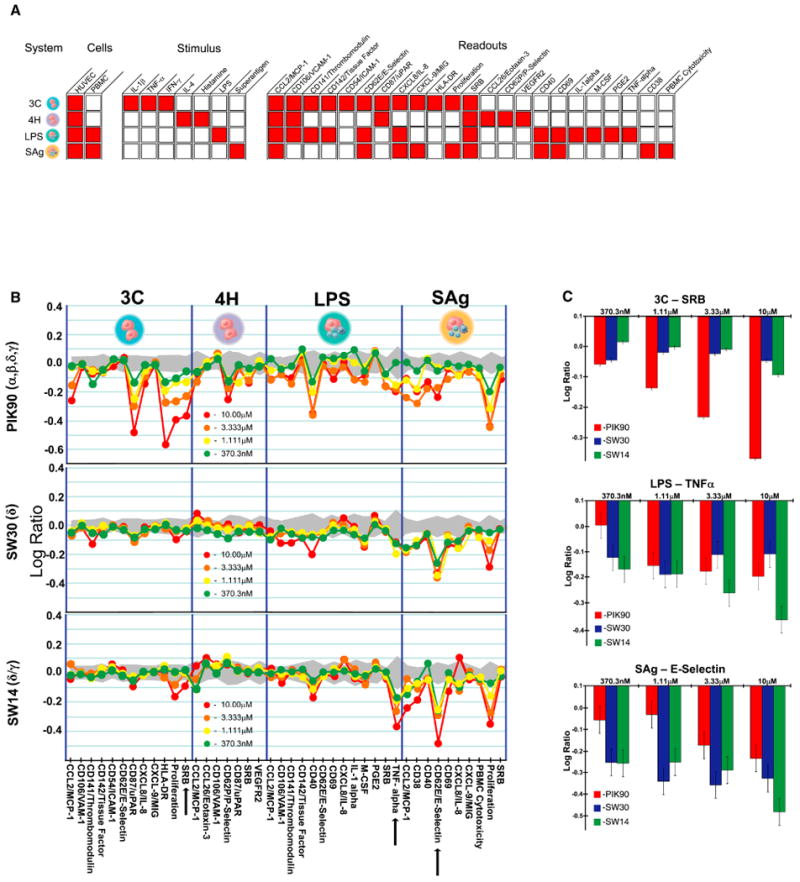
A. BioMAP Systems details.
B. BioMAP profiles for PIK90 (pan-PI3K) (top), SW30 (PI3Kδ) (middle), and SW14 (PI3Kδ/γ) (bottom). Red dots represent a 10μM dose, orange corresponds to 3.3μM, yellow represents 1.1μM and green represents 370nM. Levels of proteins were measured by ELISA and presented as log expression ratios [log10(parameter value with inhibitor/parameter value of 0.1% DMSO)]. The gray area represents the 95% prediction interval of the 0.1% DMSO data. Arrows indicate readouts that are expanded in 4C for closer inspection.
C. Log Expression Ratios of selected readouts that show divergence between pan-PI3K inhibition and selective PI3K inhibition
We evaluated pan-PI3K (α,β,δ,γ) inhibition and PI3Kγ inhibition using PIK90 and AS605240 as benchmark compounds, while PI3Kδ inhibition and dual PI3Kδ/γ inhibition were evaluated using SW30 and SW14, respectively. Each compound was tested at a range of concentrations (Fig 4B) in order to identify potential off-target activities at higher doses. For each marker analyzed, a log10 ratio for drug- treated protein levels vs. 0.1% DMSO-treated cells is indicated (Fig 4B). A log10 ratio of -1.0 indicates a ten-fold decrease in protein level upon treatment with drug as compared to the levels with 0.1% DMSO. The 0.1% DMSO measurement, compiled over several donor pools provided a benchmark for the inherent variability of the assay, and is represented by the gray shading surrounding log10 ratio = 0.0 in Figures 4 and S3.
Effects of pan-PI3K Inhibition
The pan-PI3K inhibitor PIK90 elicited both anti-inflammatory and anti-proliferative effects. In conditions designed to mimic TH1-driven endothelial cell-based inflammation, (HUVECs stimulated with IFNγ, TNFα, and IL1β) (3C; Fig. 4B), pan-PI3-K inhibition showed a moderate effect on human umbilical vein endothelial cell (HUVEC) viability (indicated by a reduction in sulforhodamine B (SRB) protein binding < -0.3 log ratio) and inhibited HUVEC proliferation (Fig. 4C). PIK90 also strongly inhibited the urokinase receptor (CD87/uPAR), important in tissue remodeling (Solberg et al., 2001) and cell motility (Kjoller and Hall, 2001), and HLA-DR, a surface receptor important in antigen presentation to T cells (Rothbard et al., 1988). In HUVECs stimulated with Histamine and IL-4 (4H; Fig 4B), the effects of PIK90 were more subtle, showing only mild inhibition of CD62/P-selectin, a cell adhesion molecule.
When introduced to a PBMC/HUVEC co-culture stimulated with lipopolysaccharide, PIK90 exhibited little effect on cell viability but significantly reduced CD40 and mildly reduced TNFα production (Fig 4B,C; LPS). In PBMCs and HUVECs stimulated with superantigen, PIK90 showed modest effects on E-selectin (CD62), CCL2/MCP-1 and CD38 production, and a decrease in PBMC (mostly monocytes and T cells) proliferation (Fig 4B; SAg). E-selectin is a cell-adhesion molecule expressed by endothelial cells that recognizes sialylated sugars on leukocytes, and is involved in the first stages of leukocyte recruitment (McEver, 1991). CCL2/MCP-1 is a cytokine that recruits monocytes to sites of injury (Daly and Rollins, 2003). Together, these results suggest that pan-PI3K inhibitors reduce some parameters associated with inflammation but exhibit general cytotoxicity and decreased HUVEC proliferation. These effects may significantly limit their utility for chronic conditions requiring frequent administration.
Comparison of Selective PI3Kδ and Selective PI3Kδ/ PI3Kγ Inhibition
In contrast to the effects of pan-PI3K inhibition (PIK90), specific inhibitors of PI3Kδ (SW30) or PI3Kδ/γ (SW14) had effects on inflammatory markers but little anti-proliferative activity. SW30 and SW14 showed little effect on HUVECs stimulated with inflammatory cytokines (Fig. 4B; 3C,4H), but were active in suppressing markers of inflammation in co-cultures of HUVEC and PBMCs stimulated with lipopolysaccharide, the bacterial cell wall component which binds Toll-like Receptor 4 (TLR4), or superantigens, another bacterial product which universally engages the T-cell receptor (TCR) (Fig. 4B; LPS, SAg). The principal effect of the PI3Kδ selective compound SW30 occurred under superantigen stimulated conditions (reduction of E-selectin expression (Fig. 4C) and PBMC proliferation) likely because the TCR, engaged by superantigens, is coupled to PI3Kδ (Okkenhaug et al., 2002; Okkenhaug and Vanhaesebroeck, 2003). PBMC proliferation decreased under the same conditions in response to SW30 and SW14, but they did not exhibit a toxic effect as read out by Alamar Blue reduction (PBMC cytotoxicity, Fig 4B) indicating that they can inhibit PBMC growth without inducing cell death. Together, these results suggest that PI3Kδ inhibition is more selective in exerting an anti-inflammatory profile than a pan-PI3K inhibitor and may be particularly suited to treatment of disorders with a dominant T-cell component.
PI3Kδ/γ inhibition (SW14) was largely inactive in the HUVEC-only culture conditions, only slightly decreasing proliferation at the highest dose (10 μM; Fig. 4B, C). In the lipopolysaccharide-stimulated HUVEC/PBMC co-culture, PI3Kδ/γ inhibition led to decrease of TNFα production, more so than either PI3Kδ inhibition (SW30) or pan-PI3K inhibition (PIK90) (Fig. 4C). When the HUVEC/PBMC co-culture was stimulated with superantigen, SW14 decreased proliferation and CCL2/MCP-1, CD38, and E-Selectin expression. An enhanced effect of PI3Kδ/γ inhibition was observed: decreased monocyte TNFα production (under lipopolysaccharide stimulation) and more efficacious (magnitude) inhibition of T cell activation (E-selectin, PBMC proliferation; under superantigen stimulation) (Fig. 4C). Importantly, although SW14 inhibits PI3Kβ at below 1μM (Fig. 2B), related analogs (SW18, 19) with threefold and ten-fold less PI3Kβ inhibition, respectively (Fig. 2B, Fig. S1) exhibited similar enhanced activity. The profile of SW19 (Supplementary Material) also provides evidence that the effects on TNFα are truly due to additional PI3Kγ activity, and not simply more potent PI3Kδ inhibition. SW19, though less potent on PI3Kδ than SW30 (Fig 2B), displays moderate activity against PI3Kγ, which is enough to exhibit the characteristic reduction of TNFα levels and cluster it with SW14, and SW18, other PI3Kδ/γ dual inhibitors (Supplementary Material). The stronger monocyte and T-cell inhibition seen with PI3Kδ/γ inhibition may make these inhibitors particularly effective in the treatment of inflammatory disorders driven by TNFα.
Biological Activity of PI3K Inhibitors Relative to Other Agents
Having defined a potential role for PI3Kδ (SW30) in treatment of T-cell-mediated inflammatory diseases and PI3Kδ/γ (SW14) in treatment of TNFα-mediated inflammatory disease, we sought to put these agents in the broader context of approved anti-inflammatory agents. We compared PI3K inhibitors with different isoform selectivities to a panel of clinical and experimental compounds with anti-inflammatory (cyclosporin A, prednisolone) and anti-cancer activities. Included were CDK inhibitors (kenpaullone, olomoucine, roscovitine), microtubule disruptors (vincristine, colchicine), estrogens (17-β-estradiol, 2-methoxyestradiol), HSP90 inhibitors (17-AAG, geldanamycin, radicicol), taxanes (epithilone B, paclitaxel), p38 inhibitors (BIRB-796, VX-745), IKK inhibitors (Ro106-9920, SC-514), JNK inhibitors (AS602801), and mTOR inhibitors (everolimus, rapamycin) (Fig. 5). The profiles for each compound were analyzed using a Pearson correlation metric and visualized in two dimensions through a multi-dimensional scaling algorithm (Kunkel et al., 2004; Plavec et al., 2004).
Fig. 5. Function Similarity Map of PI3K inhibitors and other compounds.

A Function Similarity Map of compounds from the SW series with other anti-inflammatories, PI3K inhibitors, and kinase inhibitors. This map is generated by subjecting the pairwise correlation data of BioMAP profiles to multidimensional scaling. Significant correlations are shown by gray lines. The distance between the compounds is inversely related to the similarity of their profiles. Compounds are color-coded, grouped by target class, and the area of the circle is proportional to the dose.
Compounds targeting PI3Kγ and PI3Kδ (SW14, SW18, SW30) occupied a distinct space in this map. The more PI3Kδ selective compounds (SW30 and IC87114) clustered with lower doses of the PI3Kδ/γ compounds, confirming our observations on their specificity. Strikingly, compounds with significant activity against PI3Kγ (SW14, SW18) were functionally linked to the glucocorticoid prednisolone, the active metabolite of prednisone. This result is supported by direct comparison of the profiles of SW14, SW18, and prednisolone, which showed significant similarities, especially with regard to their effect on TNFα production in the LPS-stimulated HUVEC/PBMC co-culture (Fig. S3). Conversely, the pan-PI3K inhibitor (PIK-90), grouped with other pan-PI3K inhibitors in a different region of the similarity map (Fig. 5) with inhibitors of their downstream target mTOR, microtubule modulators, another downstream target of PI3K (Onishi et al., 2007), and estrogens which have been linked to PI3Ks (Calippe et al., 2008; Simoncini et al., 2000).
A comparison of compounds that target the PI3K pathway (Fig. S4) showed that agents with significant PI3Kα activity, including AS605240, clustered with inhibitors of mTOR. AS605240, often described as a PI3Kγ directed inhibitor, showed significant similarity to the multi-targeted inhibitors and clustered with them, likely due to its activity against PI3Kα (Fig S2; Supplementary Material).
Discussion
We describe the design and synthesis of highly selective PI3K inhibitors based on a quinazolinone scaffold that engages the methionine switch observed in both PI3Kγ (Knight et al., 2006) and PI3Kδ (Berndt et al., 2009). Elaboration of this scaffold with aryl and alkyne affinity elements afforded compounds with a wide range of activity against PI3Kδ and PI3Kγ but importantly did not significantly inhibit PI3Kα, PI3Kβ, or any of the 219 protein kinases evaluated. These compounds were used in conjunction with AS605240, a PI3Kγ-directed literature inhibitor, and the pan-PI3K inhibitor PIK90 in order to evaluate the effects of isoform-selective inhibition in single and multi-cellular contexts.
Several PI3K inhibitors with different isoform selectivities were tested in primary human cell models of inflammatory signaling. Pan-PI3K inhibition (PIK90) was highly anti-proliferative to both HUVECs and PMBCs whereas PI3Kδ inhibition (SW30) or PI3Kδ/γ inhibition (SW14) resulted in selective T-cell inhibition. Selective PI3Kδ inhibition (SW30) blocked T-cell cytokine production, resulting in decreased HUVEC E-selectin expression and revealing the potential to elicit a potent anti-inflammatory response without a direct effect on endothelial cells. Interestingly, SW30 and other selective PI3Kδ inhibitors such as IC87114 (Supplementary Material) did not inhibit E-selectin expression in HUVECs directly stimulated with IFNγ, TNFα, and IL1β (3C; Fig 4B) but only did so in the superantigen-stimulated HUVEC/PMBC co-culture conditions (SAg; Fig 4B, Supplementary Material). This result is consistent with these compounds indirectly blocking E-selectin expression in HUVECs as a result of lymphocyte TNFα production inhibition (TNFα is known to rapidly upregulate E-selectin expression in HUVECs (Schindler and Baichwal, 1994)), and this data clearly illustrates the ability of these assays to capture parts of the intercellular communication integral to inflammatory responses.
PI3Kδ/γ inhibition (SW14) was not cytotoxic to HUVECs and displayed more significant anti-inflammatory effects in the HUVEC/PBMC co-cultures. Dual targeting of PI3Kδ/γ resulted in enhanced inhibition of LPS-induced TNFα production (Fig. 4C) and overall T-cell activation. The inhibitory effects observed with inhibition of PI3Kδ/γ in these assays led us to wonder whether they were due to synergistic inhibition of PI3Kδ/γ or whether PI3Kγ inhibition might be sufficient. The most commonly used PI3Kγ compound in the literature and only PI3Kγ selective molecule we were able to synthesize, AS605240, inhibits all PI3K isoforms at 300nM (Camps et al., 2005), and primary human cell assays displayed a profile similar to pan-PI3K compounds (Fig. S4; Supplementary Material).
Our studies using both new and benchmark compounds show the most effective anti-inflammatory PI3K inhibitor is one that inhibits both PI3Kδ and PI3Kγ. Pan-PI3K inhibitors, on the other hand, demonstrated limited effects on anti-inflammatory markers in T-cell and monocyte-driven environments with concurrent anti-proliferative effects. Nevertheless, it remains possible that selectively inhibiting other combinations of PI3K isoforms, such as PI3Kα/β, PI3Kβ/γ, or PI3Kβ/δ, may also show synergistic anti-inflammatory activity.
Interestingly, additional inhibition of PI3Kα and β (PIK90) did not further suppress inflammatory markers (Fig. 4B; LPS, SAg). In fact, when comparing the effects on E-selectin expression (SAg), selective inhibition of PI3Kδ alone was better than pan-PI3K inhibition, but inhibition of PI3Kδ/γ was the most effective (Fig. 4C). The most effective isoform combination at inhibition of LPS-induced TNFα production was also PI3Kδ/γ (Fig. 4C). Additional PI3Kα/β inhibition (PIK90), in addition to displaying undesirable anti-proliferative properties, did not significantly decrease TNFα expression and was essentially identical to selective PI3Kδ inhibition (Fig. 4C). These results together suggest a possible role for PI3Kγ in TNFα production.
Both kinase-dependent and kinase-independent routes exist through which PI3Kγ could regulate TNFα production. PI3Kγ interacts with phosphodiesterase 3B (PDE3B), regulating cyclic AMP (cAMP) levels and activation of protein kinase A (PKA), leading to increased heart contractility (Patrucco et al., 2004) and was also linked to PDE4 (Kerfant et al., 2007). PDE4 is the main cAMP hydrolyzing enzyme in immune cells, and has been pursued as a target for generating novel anti-inflammatories (Teixeira et al., 1997) that suppress LPS-induced TNFα production in monocytes (Souness et al., 1996). Since PI3Kγ is required for PDE4 activity (Kerfant et al., 2007), PI3Kγ inhibition may lead to diminished PDE4 activity and explain the TNFα suppression. However, the first link between PDEs and PI3Kγ involved a scaffolding, non-catalytic effect (Patrucco et al., 2004) and the PDE4-PI3Kγ link was discovered in a PI3Kγ KO mouse (Kerfant et al., 2007), which has no functional PI3Kγ suggesting that this route may not involve the catalytic activity of PI3Kγ.
TNFα is involved in several autocrine signaling loops (Lisby et al., 2007) and it is possible that PI3Kγ is involved in amplifying such signals. TNFα activates PI3Kγ in endothelial cells leading to oxidant generation and NF-κB activation (Frey et al., 2006). NF-κB regulates production of TNFα (Li and Stark, 2002) so TNFα may activate its own production by NF-κB through PI3Kγ. TNFα can upregulate its own mRNA synthesis in keratinocytes (Lisby et al., 2007), and if this happens in PBMCs, it provides an attractive model for PI3Kγ involvement in TNFα production and may explain why PI3Kγ inhibition lowers TNFα levels.
Our data suggest that inhibition of PI3Kγ's catalytic activity is sufficient to regulate TNFα levels, but they do not necessarily rule out other mechanisms. It is possible that application of BioMAP analysis to this question may resolve the conflict. If the combination of a PDE4 inhibitor and a PI3Kγ inhibitor did not alter TNFα levels more than either inhibitor alone, this would suggest that PI3Kγ is signaling through PDE4, if however, combined inhibition of PDE4 and PI3Kγ further decreased TNFα levels, this might suggest that they work through independent pathways (as mentioned above) Results obtained by treating with more than one compound may be unreliable because of potential off-target effects, thus a combination of a specific inhibitor with siRNA in a BioMAP system might better resolve the dilemma.
In broad terms, PI3K signaling can be viewed into two divisions, a survival pathway (α/β) and inflammatory PI3K (δ/γ) pathway, yet inhibitors that target both units can lead to functional antagonism. Essentially, inhibition of Class I PI3-Ks is not necessarily additive, and inhibition of PI3-Kα and PI3-Kβ can actually antagonize the effects of more isoform selective inhibition. For example, inhibition of PI3Kδ/γ by SW14 results in suppression of E-selectin or TNFα but additional inhibition of PI3Kα/β using PIK90 actually results in a smaller degree of suppression (Fig. 4B).
The actual mechanisms by which PI3Kα/β inhibition may antagonize PI3Kδ/γ inhibition are not perfectly clear. In the case of TNFα suppression, pan-PI3K inhibition with wortmannin and LY294002 has been shown to reverse amlodipine-induced inhibition of TNFα production in LPS-stimulated rat cardiomyocytes (Li et al., 2009), and of more relevance, treatment of PBMCs and THP-1 monocytes with the same compounds increases LPS induced TNFα expression (Guha and Mackman, 2002). Both wortmannin and LY294002 have more activity outside of the Class PI3Ks than PIK90, (Knight et al., 2006; Knight and Shokat, 2005) which may explain why PIK90 did not increase TNFα levels, but our results with pan-PI3K inhibition are not necessarily surprising. Studies with pan-PI3K inhibitors cannot identify the actual isoforms responsible for the increase in TNFα levels, but a recent study showed that p110α deficient THP-1 monocytes display increased TNFα production when stimulated by LPS, suggesting that PI3Kα inhibition is responsible for the increase in TNFα production (Lee et al., 2007). Together this evidence suggests that PI3Kα inhibition increases TNFα production, while PI3Kδ/γ inhibition decreases TNFα production, and when all isoforms are inhibited, both activities are balanced, leaving TNFα levels unchanged, but if PI3Kα is not inhibited (in the case of our PI3Kδ/γ inhibitors) then TNFα levels will decrease.
This work illustrates how targeted chemical inhibition can access information not available from genetic inactivation of one or more PI3K isoforms, which is difficult to obtain because these animals often suffer significant defects during development. Furthermore, results obtained using targeted inhibitors can be different from those obtained in animals with sustained genetic inactivation (Knight and Shokat, 2007). PI3Kδ/PI3Kγ knockout mice exhibited a more severe immune phenotype than mice lacking either isoform alone, (Swat et al., 2006; Webb et al., 2005) but the severity of that phenotype is likely due to sustained absence of both PI3Kδ and PI3Kγ and may not be duplicated with pharmacological treatment.
Many clinical anti-inflammatory agents function through different targets yet all have the property of inhibiting immune cell function while leaving non-immune cells relatively unaffected. We asked if the PI3Kδ/γ inhibitors exhibited this property and which current anti-inflammatory agents they might resemble. The most closely related profile was that of the glucocorticoid receptor (GR) agonist, prednisolone. It is interesting that a particular multi-cellular profile can be achieved through two distinct mechanisms (kinase inhibition vs. nuclear hormone receptor activation). Although prednisolone is an effective anti-inflammatory agent, there have been efforts to identify “dissociating GR agonists” that separate anti-inflammatory from other GR effects (bone loss, cardiovascular disease) that limit their long term use (Schaecke et al., 2007). The discovery that PI3Kδ/γ inhibition can functionally mimic several anti-inflammatory features of prednisolone opens a new way to improve upon a proven class of anti-inflammatories (GR agonists) while targeting completely different enzymes, and could only have been realized through analysis of this compound series on primary human cells.
Despite the related responses of prednisolone and PI3Kδ/γ inhibitors, the similarities may be limited to the cell types we analyzed. Cell types which have documented GR agonist responses (macrophages, coronary artery cells) are not included in our assays. The exceptionally broad cellular effects of GR agonists would likely be distinguished from PI3Kδ/γ inhibitors if more cell types were analyzed. Despite these caveats, the use of primary human cells provides a powerful early assessment of differential inhibition of important signaling nodes (PI3Ks, nuclear receptors, JNKs, calcineurin, IKK).
With the new availability of small molecules capable of inhibiting the inflammatory PI3Ks (δ/γ) without inhibition of the ubiquitous growth-linked PI3Ks we are poised to begin to resolve the opposing effects of pan-PI3K inhibition and selective inflammatory PI3K inhibition and to begin further validation of PI3Kδ/γ as a target for the treatment of inflammatory disorders.
Significance
The work describes the discovery of small molecule inhibitors of unusual potency and selectivity for immune cell expressed PI3Ks. Lipid kinases are emerging as critical drug targets for a number of disease states. Although most work has focused on anti-cancer properties of PI3K inhibitors, here we address the two key challenges, one chemical and one biological, in the development of isoform selective inhibitors of PI3Kγ and PI3Kδ as a novel class of anti-inflammatory agents. The first challenge is how to selectively target PI3Kδ and PI3Kγ without inhibiting the ubiquitously expressed PI3Kα and PI3Kβ isoforms, which have nearly identical ATP binding pockets. We address this challenge by exploiting the first conformation specific binders to PI3Kδ and γ and develop the first panel of compounds with differing profiles against these critical inflammatory targets. The second challenge is determining which profile of PI3K inhibition is desirable: completely selective PI3-Kδ or PI3-Kγ inhibition, dual inhibition of PI3-Kδ and PI3-Kγ, or possibly pan-PI3K inhibition. Although full or conditional knockouts of each isoform have been generated in mice, such model systems only provide a steady state loss-of-function analysis. We therefore evaluated a panel of the newly discovered PI3Kδ/γ inhibitors, along with benchmark pan-selective inhibitors and a representative set of anti-inflammatory agents with broad mechanism of action, in primary human co-cultures stimulated with pro-inflammatory agents. The resulting map of anti-inflammatory drug action reveals an unanticipated similarity between dual-PI3Kδ/γ inhibitors, and anti-inflammatory glucocorticoids. Importantly, our studies also show that inhibition of PI3Ks beyond PI3Kδ/γ, result in a decrease of some anti-inflammatory effects, suggesting that pan-PI3K inhibitors will be less immunosuppressive. These studies contribute both chemical and biological insights into the emerging area of PI3K drug discovery for the treatment of inflammatory disease.
Methods
Chemical Synthesis
All compounds were synthesized from commercially available starting materials, and purified by RP-HPLC (MeCN:H2O:0.1% TFA). See SM for details.
Kinase Assays
IC50s were determined as previously described (Knight et al., 2007). See SM for details.
THP-1 Cell Culture
THP-1 monocytes (ATCC) were grown in RPMI-1640 media with 10% Fetal Bovine Serum (FBS), 0.004% beta mercaptoethanol (BME), and under 5% CO2 at 300,000-500,000 cells/mL.
THP-1 Signaling Assays
THP-1 monocytes were grown in serum-free RPMI-1640 with 0.004% BME under 5% CO2 at 1,000,000 cells/mL for 4 hours, incubated with inhibitors or DMSO for 10 minutes and stimulated with 100nM MCP-1 (R&D) for 5 minutes or 50ng/mL CSF-1 (Peprotec) for 7 minutes. Cells were fixed with paraformaldehyde; 1mL of cold methanol was added and cells were stored at -20°C overnight. 2mL of PBS was added, and cells were spun, resuspended in 1mL PBS with 5% FBS (FACS Buffer), and stored at 4°C. Buffer was aspirated, and 10μL of block solution was added. After 10 minutes, 30μL of Alexa conjugated antibody in FACS buffer (1/5 dilution) was added to cells. After 30 minutes, cells were resuspended in 2mL of PBS and spun. PBS was removed; cells were resuspended in 100μL of FACS buffer and read on the FACS.
Cell Culture
Human umbilical vein endothelial cells (HUVEC) were pooled from multiple donors, cultured according to standard methods, and plated into microtiter plates at passage 4. Peripheral blood mononuclear cells (PBMC) were prepared from buffy coats from normal human donors according to standard methods. Concentrations of agents added to confluent microtiter plates to build each system: cytokines (IL-1β, 1 ng/ml; TNFα, 5 ng/ml; IFN-γ, 20 ng/ml; IL-4, 5 ng/ml), activators (SAg, 20 ng/ml; histamine, 10 μM; or LPS, 2 ng/ml), and leukocytes (PBMC, 75,000 cells/well).
Compounds
Compounds were tested at the indicated concentrations. Compounds were added 1 hour before stimulation of the cells, and were present during the whole 24 hour stimulation period (or longer for proliferation assays).
Readout Measurements
The expression of many readouts was measured by cell-based ELISAs. For the ELISAs, microtiter plates are treated, blocked, and then incubated with primary antibodies or isotype control antibodies (0.01-0.5 μg/ml) for 1 hr. After washing, plates were incubated with a peroxidase-conjugated anti-mouse IgG secondary antibody or a biotin-conjugated anti-mouse IgG antibody for 1 hr followed by streptavidin-HRP for 30 min. Plates were washed and developed with TMB substrate and the absorbance (OD) was read at 450 nm (subtracting the background absorbance at 650 nm). Quantitation of TNF-α and PGE2 was done using commercially available kits according to the manufacturer's directions. Proliferation of PBMCs (T cells) was quantified by Alamar blue reduction. Proliferation of adherent cell types was quantified by SRB staining.
Supplementary Material
Acknowledgments
This material is based on work supported under an NSFGRF to O.W. and was supported by the Graduate Research and Education in Adaptive bi-Technology Training Program of the UC Systemwide Biotechnology Research and Education Program, grant # 2008-005 to O.W. We would like to thank C Kasap and N Shah for help with FACS and S. Tong, M. Rha, and D. Nguyen for technical assistance, and C Rommel for helpful advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali K, Bilancio A, Thomas M, Pearce W, Gilfillan A, Tkaczyk C, Kuehn N, Gray A, Giddings J, Peskett E, et al. Essential role for the p110 delta phosphoinositide 3-kinase in the allergic response. Nature. 2004;431:1007–1011. doi: 10.1038/nature02991. [DOI] [PubMed] [Google Scholar]
- Ali K, Camps M, Pearce W, Ji H, Ruckle T, Kuehn N, Pasquali C, Chabert C, Rommel C, Vanhaesebroeck B. Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J Immunol. 2008;180:2538–2544. doi: 10.4049/jimmunol.180.4.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhouse C, Engler C, English J. Naproxen Sodium and Indomethacin in Acute Musculoskeletal Disorders. Rheumatol Rehabil. 1980;19:113–119. doi: 10.1093/rheumatology/19.2.113. [DOI] [PubMed] [Google Scholar]
- Barbi J, Cummings H, Lu B, Oghumu S, Ruckle T, Rommel C, Lafuse W, Whitacre C, Satoskar A. PI3Kgamma (PI3K gamma) is essential for efficient induction of CXCR3 on activated T cells. Blood. 2008;112:3048–3051. doi: 10.1182/blood-2008-02-135715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg E, Kunkel E, Hytopoulos E, Plavec I. Characterization of compound mechanisms and secondary activities by BioMAP analysis. Journal of Pharmacological and Toxicological Methods. 2006;53:67–74. doi: 10.1016/j.vascn.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Berndt A, Miller S, Williams O, Le D, Houseman B, Pacold J, Gorrec F, Hon W, Liu Y, Rommel C, et al. The crystal structures of the class IA PI3K p110delta uncover mechanisms for specificity and potency of novel PI3Kdelta inhibitors. Nature Chemical Biology. 2010 doi: 10.1038/nchembio.293. advance online publication, 10 January 2010. [DOI] [PubMed] [Google Scholar]
- Calippe B, Douin-Echinard V, Laffargue M, Laurell H, Rana-Poussine V, Pipy B, Guery J, Bayard F, Arnal J, Gourdy P. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: Involvement of the phosphatidylinositol 3-kinase pathway. J Immunol. 2008;180:7980–7988. doi: 10.4049/jimmunol.180.12.7980. [DOI] [PubMed] [Google Scholar]
- Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, et al. Blockade of PI3K gamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Cantley L. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Clayton E, Bardi G, Bell S, Chantry D, Downes C, Gray A, Humphries L, Rawlings D, Reynolds H, Vigorito E, Turner M. A crucial role for the p110 delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196:753–763. doi: 10.1084/jem.20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condliffe A, Davidson K, Anderson K, Ellson C, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann M, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- Crackower M, Oudit G, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Daly C, Rollins B. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: Therapeutic opportunities and controversies. Microcirculation. 2003;10:247–257. doi: 10.1038/sj.mn.7800190. [DOI] [PubMed] [Google Scholar]
- Deane J, Fruman D. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- Del Prete A, Vermi W, Dander E, Otero K, Barberis L, Luini W, Bernasconi S, Sironi M, Santoro A, Garlanda C, et al. Defective dendritic cell migration and activation of adaptive immunity in PI3K gamma-deficient mice. EMBO J. 2004;23:3505–3515. doi: 10.1038/sj.emboj.7600361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Maini R. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- Frey R, Gao X, Javaid K, Siddiqui S, Rahman A, Malik A. Phosphatidylinositol 3-kinase gamma signaling through protein kinase C zeta induces NADPH oxidase-mediated oxidant generation and NF-kappa B activation in endothelial cells. J Biol Chem. 2006;281:16128–16138. doi: 10.1074/jbc.M508810200. [DOI] [PubMed] [Google Scholar]
- Gray R, Doherty S, Galloway J, Coulton L, Debroe M, Kanis J. A Double-Blind Study of Deflazacort and Prednisone in Patients with Chronic Inflammatory Disorders. Arthritis Rheum. 1991;34:287–295. doi: 10.1002/art.1780340306. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Ciraolo E, Ghigo A, Costa C. Taming the PI3K team to hold inflammation and cancer at bay. Pharmacol Therapeut. 2008;118:192–205. doi: 10.1016/j.pharmthera.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev V, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann M. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Imperato A, Bingham C, Abramson S. Overview of benefit/risk of biological agents. Clin Exp Rheumatol. 2004;22:S108–S114. [PubMed] [Google Scholar]
- Ji H, Rintelen F, Waltzinger C, Meier D, Bilancio A, Pearce W, Hirsch E, Wymann M, Ruckle T, Camps M, et al. Inactivation of PI3K gamma and PI3K delta distorts T-cell development and causes multiple organ inflammation. Blood. 2007;110:2940–2947. doi: 10.1182/blood-2007-04-086751. [DOI] [PubMed] [Google Scholar]
- Jones G, Prigmore E, Calvez R, Hogan C, Dunn G, Hirsch E, Wymann M, Ridley A. Requirement for PI 3-kinase gamma in macrophage migration to MCP-1 and CSF-1. Exp Cell Res. 2003;290:120–131. doi: 10.1016/s0014-4827(03)00318-5. [DOI] [PubMed] [Google Scholar]
- Jou S, Carpino N, Takahashi Y, Piekorz R, Chao J, Carpino N, Wang D, Ihle J. Essential, nonredundant role for the phosphoinositide 3-kinase p110 delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22:8580–8591. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield M. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- Kelley T, Graham M, Doseff A, Pomerantz R, Lau S, Ostrowski M, Franke T, Marsh C. Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J Biol Chem. 1999;274:26393–26398. doi: 10.1074/jbc.274.37.26393. [DOI] [PubMed] [Google Scholar]
- Kerfant B, Zhao D, Lorenzen-Schmidt I, Wilson L, Cai S, Chen S, Maurice D, Backx P. PI3K gamma is required for PDE4, not PDE3, activity in subcellular Microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res. 2007;101:400–408. doi: 10.1161/CIRCRESAHA.107.156422. [DOI] [PubMed] [Google Scholar]
- Kjoller L, Hall A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J Cell Biol. 2001;152:1145–1157. doi: 10.1083/jcb.152.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z, Chiang G, Alaimo P, Kenski D, Ho C, Coan K, Abraham R, Shokat K. Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg Med Chem. 2004;12:4749–4759. doi: 10.1016/j.bmc.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Knight Z, Feldman M, Balla A, Balla T, Shokat K. A membrane capture assay for lipid kinase activity. Nat Protoc. 2007;2:2459–2466. doi: 10.1038/nprot.2007.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z, Gonzalez B, Feldman M, Zunder E, Goldenberg D, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110 alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z, Shokat K. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Knight Z, Shokat K. Chemical genetics: Where genetics and pharmacology meet. Cell. 2007;128:425–430. doi: 10.1016/j.cell.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Konrad S, Ali S, Wiege K, Syed S, Engling L, Piekorz R, Hirsch E, Nurnberg B, Schmidt R, Gessner J. Phosphoinositide 3-Kinases gamma and delta, Linkers of Coordinate C5a Receptor-Fc gamma Receptor Activation and Immune Complex-induced Inflammation. J Biol Chem. 2008;283:33296–33303. doi: 10.1074/jbc.M804617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel E, Dea M, Ebens A, Hytopoulos E, Melrose J, Nguyen D, Ota K, Plavec I, Wang Y, Watson S, et al. An integrative biology approach for analysis of drug action in models of human vascular inflammation. Faseb J. 2004;18:1279–1281. doi: 10.1096/fj.04-1538fje. [DOI] [PubMed] [Google Scholar]
- Laffargue M, Calvez R, Finan P, Trifilieff A, Barbier M, Altruda F, Hirsch E, Wymann M. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity. 2002;16:441–451. doi: 10.1016/s1074-7613(02)00282-0. [DOI] [PubMed] [Google Scholar]
- Lee J, Nauseef W, Moeenrezakhanlou A, Sly L, Noubir S, Leidal K, Schlomann J, Krystal G, Reiner N. Monocyte p110 alpha phosphatidylinositol 3-kinase regulates phagocytosis, the phagocyte oxidase, and cytokine production. J Leukoc Biol. 2007;81:1548–1561. doi: 10.1189/jlb.0906564. [DOI] [PubMed] [Google Scholar]
- Li X, Cao W, Li T, Zeng A, Hao L, Zhang X, Mei Q. Amlodipine inhibits TNF-alpha production and attenuates cardiac dysfunction induced by lipopolysaccharide involving PI3K/Akt pathway. Int Immunopharm. 2009;9:1032–1041. doi: 10.1016/j.intimp.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Li X, Stark G. NF kappa B-dependent signaling pathways. Exp Hematol. 2002;30:285–296. doi: 10.1016/s0301-472x(02)00777-4. [DOI] [PubMed] [Google Scholar]
- Lisby S, Faurschou A, Gniadecki R. The autocrine TNF alpha signalling loop in keratinocytes requires atypical PKC species and NF-kappa B activation but is independent of cholesterol-enriched membrane microdomains. Biochem Pharmacol. 2007;73:526–533. doi: 10.1016/j.bcp.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Marone R, Cmijanovic V, Giese B, Wymann M. Targeting phosphoinositide 3-kinase - Moving towards therapy. Biochim Biophys Acta Protein Proteomics. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- McEver R. Selectins - Novel Receptors That Mediate Leukocyte Adhesion During Inflammation. Thromb Haemostasis. 1991;65:223–228. [PubMed] [Google Scholar]
- Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek S, Salpekar A, Waterfield M, et al. Impaired B and T cell antigen receptor signaling in p110 delta PI 3-kinase mutant mice. Science. 2002;297:1031–1034. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, Vanhaesebroeck B. PI3K-signalling in B- and T-cells: insights from gene-targeted mice. Biochem Soc Trans. 2003;31:270–274. doi: 10.1042/bst0310270. [DOI] [PubMed] [Google Scholar]
- Onishi K, Higuchi M, Asakura T, Masuyama N, Gotoh Y. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Gene Cell. 2007;12:535–546. doi: 10.1111/j.1365-2443.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- Palanki M, Dneprovskaia E, Doukas J, Fine R, Hood J, Kang X, Lohse D, Martin M, Noronha G, Soll R, et al. Discovery of 3,3-(2,4-Diaminopteridine-6,7-diyl)diphenol as an isozyme-selective inhibitor of PI3K for the treatment of ischemia reperfusion injury associated with myocardial infarction. J Med Chem. 2007;50:4279–4294. doi: 10.1021/jm051056c. [DOI] [PubMed] [Google Scholar]
- Patrucco E, Notte A, Barberis L, Selvetella G, Maffei A, Brancaccio M, Marengo S, Russo G, Azzolino O, Rybalkin S, et al. PI3K gamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Plavec I, Sirenko O, Privat S, Wang Y, Dajee M, Melrose J, Nakao B, Hytopoulos E, Berg E, Butcher E. Method for analyzing signaling networks in complex cellular systems. Proc Natl Acad Sci USA. 2004;101:1223–1228. doi: 10.1073/pnas.0308221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomel V, Klicic J, Covini D, Church D, Shaw J, Roulin K, Burgat-Charvillon F, Valognes D, Camps M, Chabert C, et al. Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase gamma. J Med Chem. 2006;49:3857–3871. doi: 10.1021/jm0601598. [DOI] [PubMed] [Google Scholar]
- Puri K, Doggett T, Douangpanya J, Hou Y, Tino W, Wilson T, Graf T, Clayton E, Turner M, Hayflick J, Diacovo T. Mechanisms and implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue. Blood. 2004;103:3448–3456. doi: 10.1182/blood-2003-05-1667. [DOI] [PubMed] [Google Scholar]
- Puri K, Doggett T, Huang C, Douangpanya J, Hayflick J, Turner M, Penninger J, Diacovo T. The role of endothelial PI3K-gamma activity in neutrophil trafficking. Blood. 2005;106:150–157. doi: 10.1182/blood-2005-01-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainsford K. Leukotrienes in the Pathogenesis of NSAID-Induced Gastric and Intestinal Mucosal Damage. Agents Actions. 1993;39:C24–C26. doi: 10.1007/BF01972709. [DOI] [PubMed] [Google Scholar]
- Reif K, Okkenhaug K, Sasaki T, Penninger J, Vanhaesebroeck B, Cyster J. Cutting edge: Differential roles for phosphoinositide 3-kinases, p110 gamma and p110 delta, in lymphocyte chemotaxis and homing. J Immunol. 2004;173:2236–2240. doi: 10.4049/jimmunol.173.4.2236. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Borlado L, Barber D, Hernandez C, Rodriguez-Marcos M, Sanchez A, Hirsch E, Wymann M, Martinez C, Carrera A. Phosphatidylinositol 3-kinase regulates the CD4/CD8 T cell differentiation ratio. J Immunol. 2003;170:4475–4482. doi: 10.4049/jimmunol.170.9.4475. [DOI] [PubMed] [Google Scholar]
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- Rothbard J, Lechler R, Howland K, Bal V, Eckels D, Sekaly R, Long E, Taylor W, Lamb J. Structural Model of HLA-DR1 Restricted T-Cell Antigen Recognition. Cell. 1988;52:515–523. doi: 10.1016/0092-8674(88)90464-3. [DOI] [PubMed] [Google Scholar]
- Ruckle T, Schwarz M, Rommel C. PI3K gamma inhibition: towards an ‘aspirin of the 21st century’? Nat Rev Drug Discov. 2006;5:903–918. doi: 10.1038/nrd2145. [DOI] [PubMed] [Google Scholar]
- Sadhu C, Dick K, Tino W, Staunton D. Selective role of PI3K delta in neutrophil inflammatory responses. Biochem Biophys Res Commun. 2003;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones R, Oliveira-dos-Santos A, Stanford W, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, et al. Function of PI3K gamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- Schaecke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): Novel ligands with an improved therapeutic index. Mol Cell Endorcinol. 2007;275:109–117. doi: 10.1016/j.mce.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Schindler U, Baichwal V. 3 NF-kappa-B Binding Sites in the Human E-Selectin Gene Required for Maximal Tumor Necrosis Factor Alpha-Induced Expression. Mol Cell Biol. 1994;14:5820–5831. doi: 10.1128/mcb.14.9.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoncini T, Hafezl-Moghadam A, Brazil D, Ley K, Chin W, Liao J. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg H, Ploug M, Hoyer-Hansen G, Nielsen B, Lund L. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J Histochem Cytochem. 2001;49:237–246. doi: 10.1177/002215540104900211. [DOI] [PubMed] [Google Scholar]
- Souness J, Griffin M, Maslen C, Ebsworth K, Scott L, Pollock K, Palfreyman M, Karlsson J. Evidence that cyclic AMP phosphodiesterase inhibitors suppress TNF alpha generation from human monocytes by interacting with a ‘low-affinity’ phosphodiesterase 4 conformer. Br J Pharmacol. 1996;118:649–658. doi: 10.1111/j.1476-5381.1996.tb15450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swat W, Montgrain V, Doggett T, Douangpanya J, Puri K, Vermi W, Diacovo T. Essential role of PI3K delta and PI3K gamma in thymocyte survival. Blood. 2006;107:2415–2422. doi: 10.1182/blood-2005-08-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira M, Gristwood R, Cooper N, Hellewell P. Phosphodiesterase (PDE)4 inhibitors: Anti-inflammatory drugs of the future? Trends Pharmacol Sci. 1997;18:164–170. doi: 10.1016/s0165-6147(97)01049-3. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas L. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Walker E, Pacold M, Perisic O, Stephens L, Hawkins P, Wymann M, Williams R. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- Webb L, Vigorito E, Wymann M, Hirsch E, Turner M. Cutting edge: T cell development requires the combined activities of the p110 gamma and p110 delta catalytic isoforms of phosphatidylinositol 3-kinase. J Immunol. 2005;175:2783–2787. doi: 10.4049/jimmunol.175.5.2783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.