

Summary
Hereditary motor and sensory neuropathy Lom type (HMSNL), also called CMT 4D, a hereditary autosomal recessive neuropathy, caused by mutation in N-Myc downstream regulated gene 1 (NDRG1 gene), was first described in a Bulgarian Gypsy population near Lom and later has been found in Gypsy communities in Italy, Spain, Slovenia and Hungary. We present two siblings with HMSNL, female and male, aged 30 and 26, respectively in a Serbian non-consanguineous family of Gypsy ethnic origin. They had normal developmental milestones. Both had symptoms of lower limb muscle weakness and walking difficulties with frequent falls, which began at the age of seven. At the age of 12, they developed hearing problems and at the age of 15 hand muscle weakness. Neurological examination revealed sensorineural hearing loss, dysarthria, severe distal and mild proximal muscle wasting and weakness, areflexia and impairment of all sensory modalities of distal distribution. Electrophysiological study revealed denervation with severe and early axonal loss. Sensorineural hearing loss was confirmed on electrocochleography and brainstem evoked potentials. Molecular genetic testing confirmed homozygote C564t (R148X) mutation in NDRG1 gene.
Keywords: Hereditary motor and sensory neuropathy, Lom type, NDRG1,
Introduction
Autosomal recessive mutations in the N-Myc downstream regulated gene 1 (NDRG1) cause hereditary motor and sensory neuropathy Lom type (HMSNL). We have seen two members of a Serbian Gypsy family with neuropathy and have documented their cases with additional electrophysiological and genetic investigations.
Case report
Clinical findings
We present two siblings, 30 years old female and her 26 years old brother of a Serbian non-consanguineous family of Gypsy ethnic origin. They had normal developmental milestones. In both cases, the disease began at age of seven with lower limb muscles wasting and weakness, walking difficulties and frequent falls. The patients developed hearing problem at the age of 12. At the age of 14, male patient underwent orthopedic surgery - elongation of Achilles tendons. Both patients developed hand muscle weakness at the age of 15. The female patient had right knee pain since she was a teenager. Neurological examination revealed impaired pupillary response to light and accommodation, sensorineural hearing loss, dysarthria, weakness with severe distal and mild proximal wasting which had affected the lower limbs much more than the upper ones, areflexia and impairment of all sensory modalities of distal distribution (Fig. 1). Both patients had scoliosis and pes cavus.
Figure 1.

Severe distal muscle wasting.
Additional investigation
Routine hematological and blood chemistry findings were within normal range except high serum cholesterol concentration (female sibling: glycemia 5.2 mmol/L, urea 4.1 mmol/L, creatinine 61 μmol/L, uric acid 257 μmol/L, proteins 68 g/L, cholesterol 7.71 mmol/L, sodium 139 mmol/L, potassium 5.0 mmol/L, serum iron concentration 14.2 μmol/L, aspartate transaminase 15 U/L, alanine transaminase 13 U/L, alkaline phosphatase 60 U/L, creatine kinase 170 U/L; male sibling: glycemia 5.0 mmol/L, urea 2.6 mmol/L, creatinine 72 μmol/L , uric acid 302 μmol/L, proteins 72 g/L, cholesterol 8.87 mmol/L, sodium 139 mmol/L, potassium 4.6 mmol/L, serum iron concentration 16.4 μmol/L, aspartate transaminase 17 U/L, alanine transaminase 16 U/L, alkaline phosphatase 51 U/L, creatine kinase 113 U/L). Immunological analyses revealed normal serum concentration of circulating immune complexes (female sibling 0.199 nm, male sibling 0.302 nm). Antinuclear antibodies (ANA) and antineutrophil cytoplasmic antibodies (ANCA) were negative in both patients.
EMNG examination showed denervation with severe axonal loss. Motor and sensory evoked potentials were not detectable. Motor nerve conduction velocity could not be measured due to a total denervation of almost all muscles innervated by ulnar, median and peroneal nerves. The extensive investigations of auditory system were performed by using pure tone audiometry, transient evoked otoacoustic emissions (TEOAE), brainstem auditory evoked potentials (BAEP). Vestibular testing was performed by caloric stimulation with warm and cold water. Audiogram in female patient showed a profound pure tone loss. Transient evoked otoacoustic emissions (TEOAE) were present in the right ear only (average correlation was 72% on the right ear, and 9% on the left ear). She had no BAEP waves, but cochlear microphonics was present in the right ear. Vestibular testing showed absence of caloric responsiveness, although she has never had dizziness. Her 26 years old brother had a mild sensorineural hearing loss, with the pure tone level at 20-25 dBHL bilaterally. Transient evoked otoacoustic emissions were present bilaterally (average correlation on the right ear was 83%, and on the left ear was 84%). He had no BAEP waves, and had cochlear microphonics bilaterally. Vestibular testing showed symmetric caloric response.
DNA isolated from peripheral blood was used PCR to amplify exon 7 of NDRG1. 176 bp long PCR products were digested using TaqI restriction endonuclease, and restriction products were separated in 3% agarose gels stained with ethidium bromide. C564T transition abolishes TaqI restriction site in exon 7 of NDRG1, so analysis of HMSNL patients on agarose gel showed one band of 176 bp instead of two bands of 104 and 72 bp (Fig. 2). A sequencing reaction was used to confirm the identity of mutation.
Figure 2.
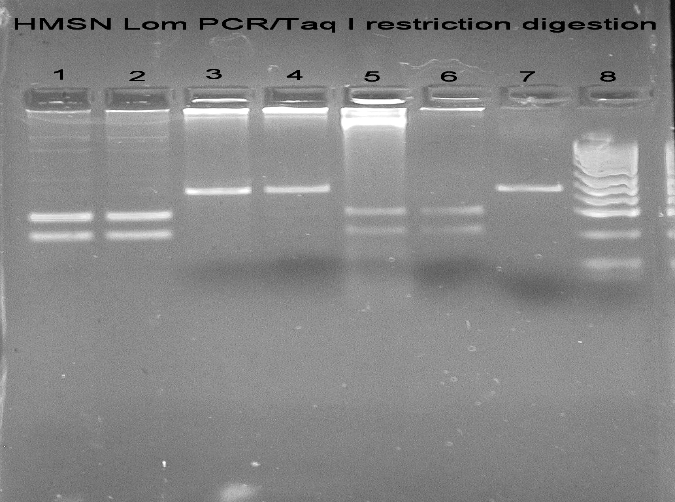
HMSN Lom PCR/Taq I restriction digestion: (1) Lanes 1, 2, 5, 6: patients negative for C564T (R148X) mutation; (2) Lanes 3, 4: patients positive for C564T (R148X) mutation; (3) Lane 7: HMSN Lom PCR; (4) Lane 8: pUC19/MSPI ladder.
Discussion
We presented clinical and electrophysiological findings in the first Serbian non-consanguineous family of Gypsy ethnic origin with hereditary motor and sensory neuropathy Lom type confirmed by genetic finding of homozygote C564t (R148X) mutation in NDRG1 gene. The clinical phenotype of our patients corresponded to that of HMSNL. The onset was in the first decade of life, with weakness in their lower limb muscles and gait difficulties. The weakness of the upper limbs and hearing problems occurred in the second decade. Nerve conduction velocities were not measurable due to a total denervation of limb muscles and severe axonal loss. Hearing loss of our patients was caused by an auditory nerve dysfunction in the presence of preserved cochlear outer hear cell function. The patients had an absence of all neural components of the auditory brainstem potentials beginning with wave I. In the three of the four ears, a positive correlation of otoacoustic emission was found as well as evidence of cochlear microphonic potentials, which are receptor potentials generated by both inner and outer hear cells (1). All of these findings suggested that the hearing loss in these patients was of neural origin, as cochlear hear cell function was preserved, but auditory nerve response was abnormal. The absence of caloric responsiveness in one patient was most likely the result of a neuropathy of the vestibular portion of the cochleovestibular nerve rather than a receptor disorder. The absence of vestibular disorder symptoms may have been the result of the gradual occurrence of a bilateral vestibular disorder, allowing the development of mechanisms that would compensate for altered vestibular inputs (2). The clinical and electrophysiological findings in our cases correlated with those already published (3, 4). HMSNL was first described in a Bulgarian Gypsy population near Lom (5), and later has been found in Gypsy communities in Italy, Spain, Slovenia, and Hungary (6, 7, 2, 8). Lower limb muscle wasting and weakness characterized the phenotype in the first decade of life followed by upper limb weakness and wasting in the second decade and hearing problems in the third decade (3). Electrophysiological studies revealed severely reduced motor nerve conduction velocity in younger patients and unobtainable in older patients. Biopsy findings showed demyelinating disorder and significant loss of large axons. Brainstem auditory evoked potentials did not contain neural component (2).
Eight to ten million Gypsies who live in Europe today are described as a conglomerate of genetically isolated founder populations. To date, a number of rare autosomal recessive disorders caused by “private Gypsy" mutations have been described (9). Autosomal recessive forms of demyelinating Charcot-Marie-Tooth (CMT4) disease among European Gypsies are caused by private founder mutations. So far, demyelinating peripheral neuropathy in the European Gypsy population has been reported to be associated with four founder mutations: hereditary motor and sensory neuropathy Lom type (HMSNL), hereditary motor and sensory neuropathy Russe (HMSNR), congenital cataracts facial dysmorphism neuropathy (CCFDN) syndrome and neuropathy with the p.R1109X mutation in SH3TC2 gene (CMT4C). HMSNL is the most common and widespread neuropathy among European Gypsies (10). Autosomal recessive nonsense mutation in the NDRG1 gene on chromosome 8q24 has been reported to be causative for HMSNL. Founder mutation is C to T transition in exon 7 at mRNA nucleotide position 564 that results in replacement of arginine by translation termination signal at codon position 148 (R148) (11). NDRG1 expression is induced by differentiation or stress stimuli. NDRG1 encodes protein with molecular mass 43 kDa, which is broadly expressed and implicated in cell growth and differentiation during development and maintenance of the differentiated state of the adult (12, 13). It is also implicated in tumor suppression, stress and hormonal response (14, 15). NDRG1 protein expressed in peripheral nerve is localized in the cytoplasm of myelinated Schwann cells, including the paranodes and Schmidt–Lanterman incisures. NDRG1 is not found in sensory or motor neurons. Oligodendrocytes also express NDRG1 (16). In Schwann cells this protein is localized in cytoplasm and interacting with apoA1, apoA2, reticulin 1c and several other proteins may also be involved in the regulation of lipid trafficking and Schwann cell-axon communication (17, 18) Cytoplasmic expression and phosphorylation of NDRG1 implies its association with intracellular signal transduction in Schwann cells. The NDRG1-deficient mice exhibited a progressive demyelinating disorder of the peripheral nerves leading to muscle weakness, indicating that NDRG1 function is important for the maintenance of myelin sheaths and axonal survival (19).
The patients in Serbian family are presented with the typical phenotype, severe denervation and severe affection of cochlear nerve.
References
- 1. Butinar D, Starr A, Vatovec J. Brainstem auditory evoked potentials and cochlear microphonics in the HMSN family with auditory neuropathy. Pflugers Arch 2000;439:204-5. [PubMed] [Google Scholar]
- 2. Butinar D, Zidar J, Leonardis L, et al. Hereditary auditory, vestibular, motor and sensory neuropathy in a Slovenian Roma (Gypsy) kindred. Ann Neurol 1999;46:36-44. [DOI] [PubMed] [Google Scholar]
- 3. Kalaydjieva L, Nikolova A, Tournev I, et al. Hereditary motor and sensory neuropathy - Lom, a novel demyelinating neuropathy associated with deafness in Gypsies: clinical, electrophysiological and nerve biopsy findings. Brain 1998;121:399-408. [DOI] [PubMed] [Google Scholar]
- 4. Ishpekova BA, Christova LG, Alexandrov AS et al. The electrophysiological profile of hereditary motor and sensory neuropathy-Lom. J Neurol Neurosurg Psychiatry 2005;76:875-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kalaydjieva L, Hallmayer J, Chandler D, et al. Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nat Genet 1996;14:214-7. [DOI] [PubMed] [Google Scholar]
- 6. Merlini L, Villanova M, Sabatelli P, et al. Hereditary motor and sensory neuropathy Lom type in an Italian Gypsy family. Neuromuscul Disord 1998;8:182-5. [DOI] [PubMed] [Google Scholar]
- 7. Colomer J, Iturriaga J, Kalaydjieva L, et al. Hereditary motor and sensory neuropathy-Lom (HMSNL) in a Spanish family: clinical, electrophysiological, pathological and genetic studies. Neuromuscul Disord 2000;10:578-83. [DOI] [PubMed] [Google Scholar]
- 8. Szabó A, Siska E, Molnár MJ. Hereditary motor and sensory Lom-neuropathy-first Hungarian case report. Ideggyogy Sz 2007;60:51-5. [PubMed] [Google Scholar]
- 9. Kalaydjieva L, Gresham D, Calafell F. Genetic studies of the Roma (Gypsies): a review. BMC Med Genet 2001;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Claramunt R, Sevilla T, Lupo V, et al. The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin Genet 2007;71:343-9. [DOI] [PubMed] [Google Scholar]
- 11. Kalaydjieva L, Gresham D, Gooding R, et al. N-myc downstream regulated gene 1 is mutated in hereditary motor and sensory neuropathy - Lom. Am J Hum Genet 2000;67:47-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piquemal D, Joulia D, Balaguer P, et al. Differential expression of the RTP/Drg1/Ndr1 gene product in proliferating and growth arrested cells. Biochim Biophys Acta 1999;1450:364-73. [DOI] [PubMed] [Google Scholar]
- 13. Lachat P, Shaw P, Gebhard S, et al. Expression of NDGR1, a differentiation-related gene, in human tissues. Histochem Cell Biol 2002;118:399-408. [DOI] [PubMed] [Google Scholar]
- 14. Chen B, Nelson DM, Sadovsky Y. N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J Biol Chem 2006;281:2764-72. [DOI] [PubMed] [Google Scholar]
- 15. Kurdistani SK, Arizti P, Reimer CL, et al. Inhibition of tumor cell growth by RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res 1998;58:4439-44. [PubMed] [Google Scholar]
- 16. Berger P, Sirkowski EE, Scherer SS, et al. Expression analysis of the N-Myc downstream-regulated gene 1 indicates that myelinating Schwann cells are the primary disease target in hereditary motor and sensory neuropathy-Lom. Neurobiol Dis 2004;17:290-9. [DOI] [PubMed] [Google Scholar]
- 17. Berger P, Niemann A, Suter U. Schwann cells and the pathogenesis of inherited motor and sensory neuropathies (Charcot-Marie-Tooth disease). Glia 2006;54:243-57. [DOI] [PubMed] [Google Scholar]
- 18. Hunter M, Angelicheva D, Tournev I, et al. NDRG1 interacts with APO A-I and A-II and is a functional candidate for the HDL-C QTL on 8q24. Biochem Biophys Res Commun 2005;332:982-92. [DOI] [PubMed] [Google Scholar]
- 19. Okuda T, Higashi Y, Kokame K, et al. NDRG1-deficient mice exhibit a progressive demyelinating disorder of peripheral nerves. Mol Cell Biol 2004;24:3949-56. [DOI] [PMC free article] [PubMed] [Google Scholar]