

Abstract
The central event in protein misfolding disorders (PMDs) is the accumulation of a misfolded form of a naturally expressed protein. Despite the diversity of clinical symptoms associated with different PMDs, many similarities in their mechanism suggest that distinct pathologies may cross talk at the molecular level. The main goal of this study was to analyze the interaction of the protein misfolding processes implicated in Alzheimer's and prion diseases. For this purpose, we inoculated prions in an Alzheimer's transgenic mouse model that develop typical amyloid plaques and followed the progression of pathological changes over time. Our findings show a dramatic acceleration and exacerbation of both pathologies. The onset of prion disease symptoms in transgenic mice appeared significantly faster with a concomitant increase on the level of misfolded prion protein in the brain. A striking increase in amyloid plaque deposition was observed in prion-infected mice compared with their noninoculated counterparts. Histological and biochemical studies showed the association of the two misfolded proteins in the brain and in vitro experiments showed that protein misfolding can be enhanced by a cross-seeding mechanism. These results suggest a profound interaction between Alzheimer's and prion pathologies, indicating that one protein misfolding process may be an important risk factor for the development of a second one. Our findings may have important implications to understand the origin and progression of PMDs.
Introduction
Misfolding and aggregation of proteins is the central pathogenic event in protein misfolding disorders (PMDs), which include Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease, diabetes type 2, systemic amyloidosis, and transmissible spongiform encephalopathies (TSEs), among others (Soto, 2001; Chiti and Dobson, 2006). The main structural changes in the protein include an increase of the β-sheet structure, oligomerization, and formation of fibrillar amyloid-like polymers (Nelson and Eisenberg, 2006). As a result, protein aggregates become insoluble, resistant to proteolysis, and resilient to cellular clearance mechanisms.
PMDs share some common features such as their appearance late in life, the progressive and chronic nature of the disease, and the tissue deposition of misfolded protein aggregates. The misfolding and aggregation mechanisms and its structural intermediates are very similar in all PMDs (Glabe, 2006; Soto et al., 2006). The mechanism of aggregation follows a seeding–nucleation process, in which the critical step is the generation of a misfolded seed that acts as a nucleus to catalyze additional aggregation, leading to the formation of oligomeric and fibrillar species (Soto et al., 2006).
TSEs or prion disorders affect humans and several animal species and are the only PMDs described as transmissible so far. Despite their low incidence, TSEs have produced big health and economic problems because of the bovine spongiform encephalopathy epidemic and the emergence of the new variant form of Creutzfeldt–Jakob disease (CJD) in humans (Bradley and Liberski, 2004). Compelling evidence indicates that the infectious agent in TSEs is composed exclusively by the misfolded and aggregated prion protein (termed PrPSc), which transmits the disease by inducing the autocatalytic transformation of the normal host prion protein (termed PrPC) (Prusiner, 2004).
AD is characterized by the extracellular deposition of the amyloid-β (Aβ) peptide and intracellular accumulation of hyperphosphorylated tau protein, both leading to synaptic dysfunction and neuronal death (Cummings, 2004). Extensive biochemical, neuropathological, and genetic evidence suggests that the cerebral accumulation of misfolded Aβ is the central event in the pathogenesis (Selkoe, 2000).
The coexistence of various misfolded aggregates has been described in vivo for several proteins such as α-synuclein, tau, prion protein, and Aβ, among others (for references, see Giasson et al., 2003). Specifically, the coexistence of PrPSc and Aβ in patients with clinical manifestation of both AD and TSE has been extensively reported previously (Muramoto et al., 1992; Tsuchiya et al., 2004; Haraguchi et al., 2009; Yoshida et al., 2009). PrP has been identified in senile plaques of AD patients and Aβ aggregates have been found within PrP deposits in patients affected with CJD or Gertsmann–Straussler–Scheinker syndrome (Bugiani et al., 1993; Hainfellner et al., 1998). Furthermore, in a study of patients with early-onset familial AD, it was found that all patients exhibited neuropathological characteristics of TSEs, including spongiform degeneration and PrP accumulation in addition of the typical AD alterations (Leuba et al., 2000). Finally, it was reported that the incidence of CJD in inherited AD patients is way higher than the prevalence of the disease in the general population (Masters et al., 1981). These findings suggest a strong interaction between TSE and AD.
In spite of the reported co-occurrence of AD and TSEs in patients, it is unclear whether both pathologies interact or are simply coexisting. In the present study, we aimed to evaluate the interaction between AD and TSEs using mice models for both diseases.
Materials and Methods
Animal bioassays.
For these studies, we used the well characterized Tg2576 transgenic mice model of AD that overexpresses the human amyloid precursor protein bearing the Swedish mutation (Hsiao et al., 1996). Groups of 365- and 45-d-old Tg2576 mice were intraperitoneally inoculated with 50 μl of PBS or 1.5% brain homogenate prepared from scrapie sick mice. Prions were obtained from C57BL/6 mice affected by scrapie disease produced by inoculation with the RML prion strain. Brains containing prions were homogenized at 1.5% in PBS plus Complete mixture of protease inhibitors (Roche Diagnostics). As controls, age-matched wild-type (WT) mice (littermates) were intraperitoneally inoculated with the same stock of prions. For second infectivity passage, 11-week-old C57BL/6 mice were inoculated with brain homogenate preparations from Tg2576 mice of the 365-d-old inoculated group showing the clinical signs of scrapie. Animals were checked three times every week to evaluate health condition and appearance of scrapie clinical symptoms. The onset of clinical prion disease was measured by scoring the animals twice a week using the following scale: 1, normal animal; 2, rough coat on limbs; 3, extensive rough coat, hunchback, and visible motor abnormalities; 4, urogenital lesions; 5, terminal stage of the disease in which the animal presents cachexia and lies in the cage with little movement. Animals scoring level 5 were considered sick and were killed to avoid excessive pain using exposure to carbonic dioxide. Left brain hemispheres were extracted and analyzed histologically (see below). The right cerebral hemisphere was frozen and stored at −70°C for biochemical studies. For time course PrPSc accumulation studies, 365-d-old C57BL/6 mice were intraperitoneally inoculated with the same stock of prions mentioned above (RML from WT C57BL/6 mice). Mice were killed at 140, 153, 161, 169, 181, and 193 d postinoculation (dpi).
PrPSc detection.
Ten percent brain homogenates were prepared as previously described (Castilla et al., 2005). Brain homogenate aliquots were digested with 50 μg/ml proteinase K (PK) (Fisher BioReagents) at 37°C for 1 h, and the reaction was stopped by adding NuPAGE LDS Sample buffer (Invitrogen). Proteins were then fractionated by electrophoresis using 4–12% SDS-polyacrylamide gels (SDS-PAGE) (Invitrogen), electroblotted into Hybond-ECL nitrocellulose membrane (GE Healthcare), and probed with the 6H4 antibody (Prionics AG) (dilution, 1:5000). The immunoreactive bands were visualized by ECL Plus Western blotting detection system (GE Healthcare) using a UVP Bioimaging system EC3 apparatus (UVP).
Histopathological studies.
Brain samples were fixed in 10% formaldehyde solution, embedded in paraffin, and cut in sections. Eight-micrometer-thick serial sections from each block were stained with hematoxylin–eosin, thioflavin S, or incubated with the anti-Aβ monoclonal antibody 4G8 (Covance) or the glial fibrillary acidic protein (GFAP) antibody (Dako North America), using previously described protocols (Permanne et al., 2002; Castilla et al., 2005). Immunoreactions were developed using the peroxidase–antiperoxidase method, following the manufacturer's specifications. Antibody specificity was verified by absorption. Samples were visualized with a Nikon Eclipse 800 microscope. For coimmunolocalization, slides were incubated with fluorescently labeled 4G8 and 6H4 antibodies and visualized with a Leica DMI600 microscope. The vacuolation profile was estimated by considering both number and size of vacuoles. Each analyzed brain area was scored from 0 to 4 according to the extent of vacuolation in slides stained with hematoxylin–eosin and visualized at a 40× magnification, as previously described (Castilla et al., 2005). The area and number of Aβ plaques was assessed in cortex images of Tg2576 mice either inoculated or noninoculated with prions. Several images were taken from each animal using a Nikon Eclipse 800 microscope (40× magnification). Five zones of the cortex were submitted to image analysis using MetaMorph.
Coimmunoprecipitation studies.
To evaluate whether Aβ and PrP may interact in the brain, we performed coimmunoprecipitation studies from frozen brain samples. For these studies, we used magnetic beads (Dynabeads from Invitrogen) covalently coupled with affinity-purified sheep anti-mouse polyclonal antibodies raised against mouse IgG. The monoclonal 4G8 antibody, recognizing specifically Aβ, was bound to the beads using the manufacturer's specifications. Beads were incubated with 1% brain homogenates prepared in PBS for 60 min at 4°C with stirring. Thereafter, the liquid solution was removed, and beads were washed three times with PBS. Finally, beads were resuspended directly in electrophoresis sample buffer and boiled, and the solution was subjected to SDS-PAGE. The gel was transferred onto nitrocellulose, and the membrane was developed with the anti-PrP antibody 6D11 or the control anti-AKT antibody.
In vitro Aβ seeding assay.
PrPSc was highly purified from the brains of scrapie-affected animals produced by inoculation with RML prions, using previously described protocols (Hetz et al., 2003). Aβ1-42 peptide was synthesized using solid-phase chemistry by the Protein Core at Yale University. Synthetic Aβ1-42 was resuspended in acetonitrile (50% v/v), aliquoted, and lyophilized. Aliquots were resuspended in NaOH, pH 12, and filtrated using 30,000 kDa cutoff filters (Millipore). Filtrates containing soluble, seed-free Aβ were stored at −80°C until use. The formation of Aβ aggregates was quantified by a modified sedimentation assay previously described (Soto et al., 1995). Briefly, Aβ samples were incubated with different amounts of PrPSc. After incubation for different times at 25°C, samples were centrifuged at 14,000 rpm for 10 min, and the soluble peptide was quantified by ELISA, using 4G8 monoclonal antibody.
Full-length recombinant prion protein expression and purification.
Murine prnp gene (coding residues 23–230) was PCR-amplified from mouse blood using standard molecular biology techniques. The amplicon was inserted in a pET303/CT His vector (Invitrogen) and propagated in DH10B-T1R Escherichia coli cells (Invitrogen). The plasmid was purified and then transformed into BL21-Star E. coli cells (Invitrogen). For expression, freshly transformed cells were grown in 5 ml of TB medium supplemented with kanamycin (100 μg/ml) at 37°C for 6 h. The starter culture was then diluted into 50 ml of the same medium and grown for another 6 h. This culture was finally diluted into 750 ml of the same medium and grown until it reached 0.7 OD. One millimolar IPTG (isopropyl β-d-thiogalactopyranoside) was then added, and the cells were induced for 5 h. The culture was harvested by centrifugation and stored at −80°C. For purification, cell pellets were thawed and resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 1 mm EDTA, and 100 mm NaCl). Cells were lysed by adding 0.5 mg/ml lysozyme and subsequently sonicated. The released inclusion bodies were pelleted by centrifugation at 22,000 × g and then washed twice with buffer A supplemented with 0.05% (v/v) Triton X-100. The inclusion bodies containing recombinant prion protein (recPrP) were solubilized for 2 h with buffer B (10 mm Tris-HCl, 100 mm Na2HPO4, pH 8.0, 100 mm NaCl, 10 mm β-mercaptoethanol, 6 m GdnCl) and then purified by using standard immobilized metal affinity chromatography procedure. Briefly, the sample was bound to Ni Sepharose 6 Fast Flow resin (GE Healthcare) in batch mode for 1 h at room temperature and then washed with buffer B. Recombinant PrPC was on-column refolded for 6 h and eluted with buffer B supplemented with 500 mm imidazole. The main peak was collected and quickly filtered to remove aggregates. The sample was buffer exchanged using Zeba-desalting columns (Pierce), further concentrated to ∼0.5 mg/ml, and flash-frozen at −80°C. Protein inhibitors (Complete protease inhibitor mixture from Roche) were used throughout the purification to minimize degradation. The protein was confirmed to be monomeric and folded by SDS-PAGE, Western blotting, and circular dichroism.
Seeding of recPrP with Aβ fibrils.
Pure recPrP was incubated with Aβ fibrils following a modified protocol from a previously reported procedure for in vitro PrP conversion (Atarashi et al., 2008). The 4.3 μm recPrP (10 μg of protein) was mixed with preformed Aβ fibrils (fibrillogenesis was confirmed by thioflavin T assay and electron microscopy) at different recPrP/Aβ fibrils ratios in conversion buffer (0.1% SDS, 0.1% Triton X-100, 1× PBS, pH 7.0). The mixture was incubated for 30 h at 37°C in an Eppendorf Thermomixer with cycles of 1 min agitation at 1500 rpm and 1 min incubation. For analysis, samples were treated with 7 μg/ml PK and incubated at 37°C for 1 h in a water bath. The reaction was stopped by quickly adding SDS sample buffer and heating at 95°C for 10 min. Samples were analyzed by Western blotting using 6D11 anti-PrP antibody.
Statistical analysis.
Survival curves, in vitro Aβ seeding assay, profile of spongiform degeneration, and Aβ plaque quantifications were analyzed by GraphPad Prism, version 5.0, software (GraphPad Software).
Results
To assess the in vivo interaction between the pathological processes implicated in AD and TSEs, we inoculated prions intraperitoneally into Tg2576 mice at different stages of AD progression. One group of animals was inoculated at 45 d of age when Aβ accumulation is not yet detectable, and a second group was inoculated at the age at which amyloid deposition is extensive in these animals (365 d of age). Age-matched wild-type mice (nontransgenic littermates) were treated in the same way. Evaluation of the onset of prion disease showed that Tg2576 mice develop clinical symptoms significantly faster than WT littermates (Fig. 1). Interestingly, the acceleration of the disease depended on the stage of AD-like pathology, since transgenic mice inoculated at 365 d of age showed a substantially shorter incubation period than animals inoculated at 45 d of age (Fig. 1). These differences are not an effect of the animal age, because WT mice inoculated at 45 or 365 d of age did not show any statistically significant difference in the onset of prion symptoms. To assess whether the accelerated disease produced in Tg2576 animals kept the infectious characteristics, we inoculated WT mice with brain homogenate of sick animals from the group of Tg2576 injected at 365 d of age. The results of this second passage showed an average incubation period of 202.4 d, characteristic of RML prions inoculated intraperitoneally and similar to the incubation time observed in our first passage in WT animals (Fig. 1). Additional biochemical (glycosylation profile and electrophoretic mobility of PrPSc) and pathological (spongiform degeneration and astrogliosis) studies further showed that the RML-Tg2576 properties were the same as expected for RML prions (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results indicate that the strain characteristics of RML prions are likely maintained after passage in AD transgenic mice.
Figure 1.

Alzheimer's pathology accelerates prion disease in mice models. A, To assess the effect of AD neuropathology in the onset of prion disease, we inoculated intraperitoneally Tg2576 mice with RML prions at 365 (orange line) and 45 (green line) days of age. As control, age-matched WT mice (nontransgenic littermates) were inoculated with the same stock and quantity of the infectious agent (black line). WT animals infected at 45 or 365 d of age developed prion disease at very similar times, and thus the data for these two groups are presented together in the graph. Clinical signs were assessed as described in Materials and Methods. When animals were definitively diagnosed with prion disease, they were killed to avoid additional pain. The data show that both groups of Tg2576-inoculated mice develop prion disease in a shorter time compared with age-matched WT controls. In addition, we performed a second infectivity passage in WT mice by inoculating infectious material from the brain of a sick Tg2576 animal injected at 365 d of age (blue line). B, Average of incubation periods of the different groups shown in A, including the statistical comparison between each experimental group with the WT control mice. The statistical analysis was done using the Student t test.
Histopathological analyses of brains from Tg2576 prion-infected mice showed the coexistence of both TSE and AD pathologies. The brain exhibited extensive spongiform degeneration (Fig. 2 A), reactive astrogliosis (Fig. 2 B), Aβ deposition (Fig. 2 C,D), and PrPSc accumulation (see Fig. 4). Conversely, Aβ deposits were not detected in the brains of WT mice inoculated with prions and no vacuolation or PrPSc accumulation was seen in old noninfected Tg2576 mice (Fig. 2).
Figure 2.

A–D, Brain histopathological studies. Representative animals from different groups were studied histopathologically for spongiform brain degeneration after hematoxylin–eosin staining (A), reactive astrogliosis by GFAP staining (B), Aβ deposition by immunohistochemistry using the 4G8 anti-Aβ antibody (C), and staining with the amyloid-specific dye thioflavin S (D). It is important to emphasize that the prion deposits in mice affected by RML prions are not thioflavin S positive but are rather diffuse prefibrillar aggregates. The images in A and B correspond to the medulla, C to the hippocampus or cortex as indicated, and D to the cortex. (See also supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material.)
Figure 4.
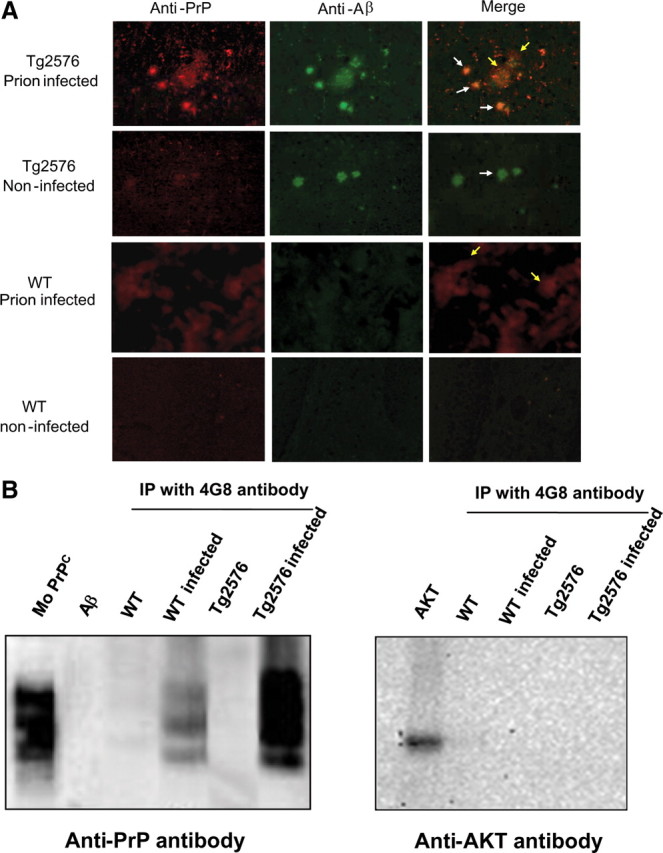
Interaction between Aβ and PrP in the brain. A, Sections from the cortex of animals in different groups were stained with fluorescent antibodies against Aβ (4G8; green) and PrP (6H4; red), and colocalization was evaluated by confocal microscopy. The white arrows point to the typical Aβ amyloid plaques seen in transgenic mice and AD patients; the yellow arrows point to the typical diffuse accumulations of PrPSc aggregates in the brain of TSE-affected individuals. B, Coimmunoprecipitation experiments. Aliquots from the brain of animals from different groups were immunoprecipitated by 4G8 antibody, which specifically recognize Aβ. The immunoprecipitated material was separated by electrophoresis and analyzed by Western blot using the 6H4 anti-prion antibody and, as control, the AKT antibody.
The degree of spongiform degeneration in animals with the double pathology did not differ from those affected only by TSE (Fig. 2 A). The extent of vacuolation in diverse areas of the brain is widely used to characterize TSEs. Indeed, different prion strains often produce a distinct pattern of spongiform degeneration (Morales et al., 2007). Evaluation of the lesion profile in different brain areas of Tg2576 mice infected with prions showed a similar pattern in all inoculated groups (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Vacuolation profiles were not statistically significantly different between Tg2576 and WT prion-infected mice, indicating that spongiform degeneration did not change because of the presence of Aβ deposits.
Conversely, it is clear that both brain inflammation (Fig. 2 B) and Aβ deposition (Fig. 2 C,D) were substantially higher on animals bearing the double pathology. The increase on brain inflammation may be an additive result, since both pathologies are associated with astrogliosis (Diedrich et al., 1987) (Fig. 2 B). More remarkably is the dramatic increase on Aβ deposition observed in the Tg2576-inoculated mice compared with noninfected animals. Indeed, some of the Tg2576 mice (2 of 8) inoculated at 45 d of age and killed when prion disease was evident (∼185 d later), showed Aβ diffuse amyloid deposits at an age (∼230 d) when these animals never show amyloid lesions (Fig. 2 C). These deposits were recognized with anti-Aβ specific antibodies in both hippocampus and cortex (Fig. 2 C), but were negative for thioflavin S (Fig. 2 D). The latter is not surprising since early, diffuse, preamyloid deposits in Alzheimer's brain are usually not detectable by this amyloid-binding dye (Tagliavini et al., 1988). However, a more detailed study of thioflavin S staining at higher magnification showed that these lesions were indeed slightly stained by thioflavin S (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Moreover, the size, number, and maturity of Aβ plaques in the Tg2576 group inoculated at 365 d of age was dramatically higher than in the age-matched control inoculated with PBS (Fig. 2 C). To quantitatively analyze the extent of Aβ aggregation in prion-infected Tg2576 mice, we determined both the percentage of brain area covered by thioflavin S-positive Aβ aggregates and the number of plaques between 365-d-old prion-infected and age-matched noninfected animals. The results showed that Aβ plaque area as well as the number of plaques was significantly higher in Tg2576 mice infected with prions (p < 0.01). Indeed, infected animals have >2-fold higher number of plaques and strikingly >10-fold greater plaque area than noninfected Tg2576 (data not shown). These data strongly support an interaction between the prion and AD pathologies, leading to a dramatic increase on the misfolding, aggregation, and cerebral accumulation of Aβ in the presence of PrPSc.
To evaluate whether PrPSc accumulation was also increased in the presence of AD pathology, we measured the quantity of PrPSc in the brain by Western blot analysis (Fig. 3). Infected Tg2576 mice were killed when clinical signs of scrapie were confirmed, on average 186 and 165 dpi in animals injected at 45 and 365 d of age, respectively. As shown in Figure 3 A, the quantity of PrPSc in the brain was high and similar in these two groups (one representative animal is shown in each group). No PrPSc was detected in AD transgenic mice noninfected with prions. As comparison, we analyzed by Western blot the levels of PrPSc in 365-d-old prion inoculated WT mice at different time points after intraperitoneal inoculation (Fig. 3 B; supplemental Table 1, available at www.jneurosci.org as supplemental material). In animals killed at 140, 153, and 161 dpi, no PrPSc was detected even in concentrated samples (Fig. 3 C). In contrast, at these times, most of the Tg2576 mice inoculated at 365 d of age showed clear clinical signs and strong deposition of PrPSc in their brains (Figs. 1, 3 A,C). At 169 dpi, only one of three WT mice showed a very faint PrPSc signal (in absence of clinical signs) (Fig. 3 B). The quantity of PrPSc in the brain of infected WT mice became high and similar to the one in Tg2576 groups after ∼225 dpi (Fig. 3 B,C). These results indicate that PrPSc formation and accumulation in the brain is accelerated in mice simultaneously affected by AD brain pathology.
Figure 3.

PrPSc levels in Tg2576 or WT mice inoculated with prions. A, Brain homogenates from clinically sick Tg2576 mice inoculated with prions at 45 or 365 d of age or, as a control, Tg2576 mice noninoculated with prions (right panel) were PK digested and Western blotted to analyze PrPSc burden. The result shown corresponds to one animal representative of all mice in the group. The animal of the 45 d group shown in the figure was killed at 190 dpi, and the animal of the 365 d at 159 dpi. B, For comparison, we measured the PrPSc levels in the brain of WT mice challenged with RML prions and killed at 140, 169, 193, and 225 dpi. Only the animal at 225 dpi was exhibiting signs of prion disease. Numbers at the top of each gel represent brain dilution. Brain dilutions were performed from a 10% brain homogenates, and various dilutions are shown to facilitate the comparisons. C, Densitometric analysis of PrPSc quantity in different animals. Western blot data as those shown in A and B were evaluated densitometrically to estimate the extent of the PrPSc signal. The data represent the average and SE of five different animals in each group. As seen, Tg2576 mice inoculated with prions at 45 and 365 d of age accumulate a similar quantity of PrPSc as WT mice but in a much shorter time. **p < 0.01. (See also supplemental Table 1, available at www.jneurosci.org as supplemental material.)
One putative explanation for the acceleration of AD and TSE pathologies in animals affected by both diseases could be a direct interaction between Aβ and PrP misfolded proteins leading to speeding up the process of misfolding and aggregation. To assess this possibility, we studied the colocalization of both proteins in pathological aggregates. In Tg2576 mice injected with prions at 365 d of age, we detected signals reactive against Aβ and PrP antibodies in both compacted amyloid plaques, typical of AD, and large diffuse deposits, characteristics of prion-affected animals (Fig. 4 A). Conversely, no colocalization was observed in noninfected Tg2576 or WT infected with RML, in which only the characteristics aggregates associated with AD or prion disease were observed, respectively (Fig. 4 A). These results suggest that misfolded proteins interact in the brain when both pathological processes are occurring simultaneously. To further assess the interaction between Aβ and PrP in the brain, we performed coimmunoprecipitation studies. As shown in Figure 4 B, immunoprecipitation using the 4G8 antibody that specifically recognize Aβ, coimmunoprecipitated a large amount of PrP in T2576 animals inoculated with prions. A fainter band was also observed in prion-infected WT animals, but no signal was observed in transgenic or WT mice noninfected with prions. A negative control developed with the antibody against the AKT protein showed no coimmunoprecipitation (Fig. 4 B).
To evaluate whether the interaction between Aβ and PrP may have functional consequences for the processes of protein misfolding and aggregation, we performed in vitro aggregation experiments in mixtures of both proteins. It is widely accepted that proteins misfold and aggregate by a seeding–nucleation mechanism, in which the formation of an oligomeric nuclei is the key step that control the kinetics of aggregation (Jarrett and Lansbury, 1993; Soto et al., 2006). It has been described that seeds composed of one protein can accelerate a second misfolding process through a process known as cross-seeding (Harper and Lansbury, 1997; O'Nuallain et al., 2004). To test the possibility that Aβ and PrP may cross-seed each other to accelerate protein misfolding and aggregation, we evaluated the seeding capability of purified PrPSc in the aggregation of synthetic Aβ (Fig. 5 A). Addition of various small quantities of prion seeds produced a clear and dramatic acceleration of Aβ aggregation, measured as a shortening of the lag phase for polymerization. The acceleration of Aβ aggregation was directly proportional to the quantity of PrPSc seeds added to the sample (Fig. 5 A). These results may explain in part the large increase of Aβ deposition observed in animals inoculated with murine prions. To evaluate whether Aβ aggregates alter PrP misfolding, we studied the aggregation of recPrP in the presence of different quantities of Aβ fibrils. The formation of misfolded recPrP was studied by the standard PK degradation assay followed by Western blot. In the absence of Aβ fibrils, only faints bands of PK resistance recPrP were observed with molecular weights of 17 and 12 kDa, similar to previously reported results (Atarashi et al., 2008) (Fig. 5 B). However, in the presence of various quantities of Aβ aggregates, a prominent PK-resistant band was observed with an apparent molecular weight of ∼17 kDa. The switch on the molecular weight is indicative of bona fide PrP conversion and is similar to the size expected for the unglycosylated PK-resistant PrPSc core. We are currently testing whether misfolded recPrP produced on incubation with Aβ fibrils is infectious to animals. Interestingly, the extent of PK-resistant recPrP was directly proportional to the quantity of Aβ fibrils added to the reaction (Fig. 5 B). These data provide evidence for a functional interaction between Aβ and PrP resulting in mutual acceleration of protein misfolding and aggregation by cross-seeding.
Figure 5.

Cross-seeding of PrP and Aβ misfolding and aggregation in vitro. A, The effect of purified PrPSc on Aβ aggregation was measured over time by sedimentation followed by sensitive ELISA. Seed-free soluble Aβ1-42 (0.01 mg/ml) was incubated with different concentrations of purified PrPSc seeds or PBS (control). The concentration of PrPSc is expressed as a percentage of oligomers per Aβ monomer and was calculated assuming that a PrPSc oligomer has an average molecular weight of 7700 kDa. The latter was based on data coming from flow field fractionation of PrPSc and corresponds to the fraction with the highest concentration of PrPSc (Silveira et al., 2005). Samples were incubated at 25°C with shaking for the indicated times. Thereafter, soluble and aggregated Aβ were separated by centrifugation at 14,000 rpm for 10 min, and the quantity of peptide in the supernatant was measured by ELISA. The experiment was done by triplicate, and results represent the average ± SE. Analysis by two-way ANOVA (using condition and time as the variables) show that the kinetics of Aβ aggregation in the presence of PrPSc is highly significantly different from the control (p < 0.0001). B, The effect of Aβ aggregates on PrP misfolding was studied by incubating 10 μg of recombinant mouse PrP in the presence of increasing concentrations of preformed Aβ fibrils. Fibrils were prepared as indicated in Materials and Methods, and aliquots corresponding to 0.14% (1.2 μg of total Aβ), 0.28% (2.4 μg of total Aβ), 0.56% (4.8 μg of total Aβ), and 1.1% (9.6 μg of total Aβ) were added to monomeric recPrP (lines 1, 2, 3, and 4, respectively). The concentration of Aβ fibrils is expressed as a molar percentage per recPrP monomer and was calculated assuming that the average molecular weight of Aβ fibrils is 2000 kDa, as estimated by a combination of size-exclusion chromatography, atomic force microscopy, and electron microscopy (Goldsbury et al., 2000). The mixture was incubated for 30 h at 37°C in an Eppendorf Thermomixer with cycles of 1 min agitation at 1500 rpm and 1 min incubation. To assess PrP misfolding, samples were incubated at 37°C with 7 μg/ml PK and PrPres signal analyzed by Western blot. Line 5 is the control with the same quantity of recPrP incubated in the absence of Aβ fibrils. Line 6 corresponds to recPrP nontreated with PK to display the migration of the full-length protein. The numbers at the right side correspond to the molecular weight standards.
Discussion
PMDs include >25 clinically diverse human disorders (Soto, 2001; Chiti and Dobson, 2006). Despite the obvious differences among these diseases, the molecular mechanism triggering the pathology appears to be the same: the misfolding, aggregation, and tissue accumulation of a protein (Soto, 2001; Chiti and Dobson, 2006). Although the protein involved in each disease is different, the structure of the misfolded aggregates as well as the mechanism and intermediates in the process are similar (Glabe, 2006; Soto et al., 2006). Indeed, a typical signature of most PMDs is the accumulation of amyloid-like fibrils, folded in a cross-β conformation (Sunde et al., 1997). To reach this stage, a series of events of misfolding and protein–protein interaction occur to form oligomers and protofibrils in a process of seeded/nucleated polymerization (Soto et al., 2006). The cellular consequences of the accumulation of misfolded aggregates in distinct diseases are also similar and include extensive tissue inflammation, cellular stress, and activation of the unfolded-protein response (Rutkowski and Kaufman, 2004). The permanent accumulation of aggregates, as well as the failure of the clearance system to eliminate them, leads to chronic endoplasmic reticulum stress, saturation of the proteasome system, and activation of apoptotic pathways, resulting in cellular dysfunction and death (Kopito, 2000; Morimoto, 2008).
The mechanistic and pathological similarities among these diseases suggest that protein-misfolding processes occurring simultaneously may synergistically interact among each other leading to an acceleration of the disease. Indeed, it has been reported extensively the coexistence of two PMDs in a single patient, including cases of AD, PD, TSEs, ALS, diabetes type 2, systemic amyloidosis, etc. (Moss et al., 1988; Brown et al., 1990, 1998; Rajput et al., 1993; Fernandez-Alonso et al., 1994; Giasson et al., 2003; Popescu et al., 2004). Moreover, some PMDs involve the presence of more than one type of misfolded aggregated protein, the archetype case being AD, in which intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau are present simultaneously with extracellular Aβ amyloid plaques (Dickson, 2001). Although it is possible that NFTs and senile plaques are formed independently, a recent study showed that Aβ and tau form soluble complexes that may promote their self-aggregation into the insoluble forms observed in AD (Guo et al., 2006).
Our findings suggest that AD and TSE pathologies synergistically interact to accelerate the onset of both diseases. Indeed, scrapie clinical signs appear much faster after infection of animals that are simultaneously accumulating Aβ aggregates. The rate of acceleration depended on the stage of AD and correlated with a rapid accumulation of PrPSc. However, AD transgenic mice infected with prions developed a strikingly higher load of cerebral amyloid plaques that appeared much faster than in noninfected mice. We cannot completely rule out that the increased Aβ amyloid accumulation in prion-infected animals might be attributable in part to an inflammatory response produced by other factors present in the injected brain homogenate. However, we believe this is unlikely considering that a similar result in terms of exacerbation of protein misfolding and aggregation of PrPSc was induced by the presence of the amyloid-β aggregates, which were produced by transgenesis and not by injection of an exogenous substance. Furthermore, recent studies, aimed to assess the possibility that Alzheimer's pathology could be transmissible, showed that inoculation of brain homogenate from humans (not affected by AD) or wild-type animals into AD transgenic mice did not produce any increase or acceleration of amyloid deposition (Meyer-Luehmann et al., 2006).
There are at least three possible explanations for the synergistic effects observed in our study: (1) The clearance mechanisms already impaired by one misfolded protein may be further diminished by a second misfolding event, leading to the faster and higher accumulation of both type of aggregates and subsequent brain damage. (2) Nerve cells stressed and injured by exposure to one toxic misfolded aggregated protein might be more easily damaged by an additional toxic aggregate. This would lead to accelerated and more severe cellular dysfunction and death. (3) A direct interaction between misfolded proteins may result in acceleration of protein misfolding and aggregation through a cross-seeding mechanism, leading to higher and faster accumulation of toxic aggregates. The latter possibility is supported by our coimmunolocalization and coimmunoprecipitation studies, which provide evidence for a direct interaction between Aβ and PrP in the brain. This idea is further supported by our in vitro experiments in which purified PrPSc can accelerate aggregation of synthetic Aβ and preformed Aβ aggregates can induce the formation of misfolded protease-resistance PrPSc-like. Therefore, our findings lend support for the possibility of a direct interaction between the proteins leading to cross-seeding and increased pathogenesis. Several studies have demonstrated the cross-seeding of misfolded aggregates both in vitro and in vivo (Johan et al., 1998; O'Nuallain et al., 2004). These data, added to the now well accepted idea that seeding is the general mechanism by which these proteins aggregate and the basis for disease propagation in TSEs (Soto et al., 2006), determine that cross-seeding between diverse misfolded proteins is a feasible mechanism. However, given the complexity of the signaling events and changes produced in the disease brain, it cannot be ruled out that other pathways also contribute to explain the effects observed in our experiments. Furthermore, two recent and interesting reports indicate that PrPC may be involved in regulating the β-secretase cleavage of APP to produce Aβ (Parkin et al., 2007) or acts as a receptor for Aβ oligomers in cells (Laurén et al., 2009). These studies provide additional pathways by which these diseases may interact at the molecular level.
Regardless of which is the molecular mechanism explaining the acceleration and exacerbation of AD and TSE pathologies in animals affected by both diseases, our results suggest that one PMD is a significant risk factor for the emergence of a second disease. Whether this conclusion can be extrapolated to the diseases in humans will require additional epidemiological studies. In this sense, it is important to highlight that it has been shown previously that the two most prevalent PMDs (AD and diabetes type 2) are a risk factor for each other (Arvanitakis et al., 2004; Janson et al., 2004). In the case of AD and TSEs, it would be important to study whether the rare, but infectious prion diseases may contribute to increase the risk for AD in patients infected with prions. Since human TSEs have a very long incubation period, which can span several decades (Collinge et al., 2006), it can be speculated that people infected with prions may develop AD or other PMDs before showing the symptoms of prion disease, contributing to explain the low incidence of TSEs in humans. Therefore, our findings may have profound implications to understand the origin and progression of human disorders associated with the misfolding and aggregation of proteins.
Footnotes
This work was supported in part by National Institutes of Health Grants AG028821 and NS050349 and an award from the Mitchell Foundation (C.S.). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of this manuscript.
References
- Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5:211–212. doi: 10.1038/nmeth0308-211. [DOI] [PubMed] [Google Scholar]
- Bradley R, Liberski PP. Bovine spongiform encephalopathy (BSE): the end of the beginning or the beginning of the end? Folia Neuropathol. 2004;42(Suppl A):55–68. [PubMed] [Google Scholar]
- Brown DF, Dababo MA, Bigio EH, Risser RC, Eagan KP, Hladik CL, White CL., 3rd Neuropathologic evidence that the Lewy body variant of Alzheimer disease represents coexistence of Alzheimer disease and idiopathic Parkinson disease. J Neuropathol Exp Neurol. 1998;57:39–46. doi: 10.1097/00005072-199801000-00006. [DOI] [PubMed] [Google Scholar]
- Brown P, Jannotta F, Gibbs CJ, Jr, Baron H, Guiroy DC, Gajdusek DC. Coexistence of Creutzfeldt-Jakob disease and Alzheimer's disease in the same patient. Neurology. 1990;40:226–228. doi: 10.1212/wnl.40.2.226. [DOI] [PubMed] [Google Scholar]
- Bugiani O, Giaccone G, Verga L, Pollo B, Frangione B, Farlow MR, Tagliavini F, Ghetti B. Beta PP participates in PrP-amyloid plaques of Gerstmann-Straussler-Scheinker disease, Indiana kindred. J Neuropathol Exp Neurol. 1993;52:64–70. doi: 10.1097/00005072-199301000-00008. [DOI] [PubMed] [Google Scholar]
- Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–2074. doi: 10.1016/S0140-6736(06)68930-7. [DOI] [PubMed] [Google Scholar]
- Cummings JL. Alzheimer's disease. N Engl J Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Neuropathology of Alzheimer's disease and other dementias. Clin Geriatr Med. 2001;17:209–228. doi: 10.1016/s0749-0690(05)70066-5. [DOI] [PubMed] [Google Scholar]
- Diedrich J, Wietgrefe S, Zupancic M, Staskus K, Retzel E, Haase AT, Race R. The molecular pathogenesis of astrogliosis in scrapie and Alzheimer's disease. Microb Pathog. 1987;2:435–442. doi: 10.1016/0882-4010(87)90050-7. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alonso J, Rios-Camacho C, Valenzuela-Castaño A, Hernanz-Mediano W. Mixed systemic amyloidosis in a patient receiving long term haemodialysis. J Clin Pathol. 1994;47:560–561. doi: 10.1136/jcp.47.6.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Lee VM, Trojanowski JQ. Interactions of amyloidogenic proteins. Neuromolecular Med. 2003;4:49–58. doi: 10.1385/NMM:4:1-2:49. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Goldsbury CS, Wirtz S, Müller SA, Sunderji S, Wicki P, Aebi U, Frey P. Studies on the in vitro assembly of a beta 1–40: implications for the search for a beta fibril formation inhibitors. J Struct Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- Guo JP, Arai T, Miklossy J, McGeer PL. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:1953–1958. doi: 10.1073/pnas.0509386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998;96:116–122. doi: 10.1007/s004010050870. [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Terada S, Ishizu H, Sakai K, Tanabe Y, Nagai T, Takata H, Nobukuni K, Ihara Y, Kitamoto T, Kuroda S. Coexistence of Creutzfeldt-Jakob disease, Lewy body disease, and Alzheimer's disease pathology: an autopsy case showing typical clinical features of Creutzfeldt-Jakob disease. Neuropathology. 2009;29:454–459. doi: 10.1111/j.1440-1789.2008.00964.x. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Johan K, Westermark G, Engström U, Gustavsson A, Hultman P, Westermark P. Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci U S A. 1998;95:2558–2563. doi: 10.1073/pnas.95.5.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba G, Saini K, Savioz A, Charnay Y. Early-onset familial Alzheimer disease with coexisting beta-amyloid and prion pathology. JAMA. 2000;283:1689–1691. doi: 10.1001/jama.283.13.1689-a. [DOI] [PubMed] [Google Scholar]
- Masters CL, Gajdusek DC, Gibbs CJ., Jr The familial occurrence of Creutzfeldt-Jakob disease and Alzheimer's disease. Brain. 1981;104:535–558. doi: 10.1093/brain/104.3.535. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Morales R, Abid K, Soto C. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta. 2007;1772:681–691. doi: 10.1016/j.bbadis.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RJ, Mastri AR, Schut LJ. The coexistence and differentiation of late onset Huntington's disease and Alzheimer's disease. A case report and review of the literature. J Am Geriatr Soc. 1988;36:237–241. doi: 10.1111/j.1532-5415.1988.tb01807.x. [DOI] [PubMed] [Google Scholar]
- Muramoto T, Kitamoto T, Koga H, Tateishi J. The coexistence of Alzheimer's disease and Creutzfeldt-Jakob disease in a patient with dementia of long duration. Acta Neuropathol. 1992;84:686–689. doi: 10.1007/BF00227747. [DOI] [PubMed] [Google Scholar]
- Nelson R, Eisenberg D. Structural models of amyloid-like fibrils. Adv Protein Chem. 2006;73:235–282. doi: 10.1016/S0065-3233(06)73008-X. [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci U S A. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, Dewachter I, Banks WA, Van Leuven F, Soto C. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer's disease by treatment with a beta-sheet breaker peptide. FASEB J. 2002;16:860–862. doi: 10.1096/fj.01-0841fje. [DOI] [PubMed] [Google Scholar]
- Popescu A, Lippa CF, Lee VM, Trojanowski JQ. Lewy bodies in the amygdala: increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol. 2004;61:1915–1919. doi: 10.1001/archneur.61.12.1915. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Early evidence that a protease-resistant protein is an active component of the infectious prion. Cell. 2004;116(2) Suppl doi: 10.1016/s0092-8674(03)01032-8. [DOI] [PubMed] [Google Scholar]
- Rajput AH, Rozdilsky B, Rajput A. Alzheimer's disease and idiopathic Parkinson's disease coexistence. J Geriatr Psychiatry Neurol. 1993;6:170–176. doi: 10.1177/089198879300600306. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C. Protein misfolding and disease; protein refolding and therapy. FEBS Lett. 2001;498:204–207. doi: 10.1016/s0014-5793(01)02486-3. [DOI] [PubMed] [Google Scholar]
- Soto C, Castaño EM, Prelli F, Kumar RA, Baumann M. Apolipoprotein E increases the fibrillogenic potential of synthetic peptides derived from Alzheimer's, gelsolin and AA amyloids. FEBS Lett. 1995;371:110–114. doi: 10.1016/0014-5793(95)00863-5. [DOI] [PubMed] [Google Scholar]
- Soto C, Estrada L, Castilla J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci. 2006;31:150–155. doi: 10.1016/j.tibs.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Tagliavini F, Giaccone G, Frangione B, Bugiani O. Preamyloid deposits in the cerebral cortex of patients with Alzheimer's disease and nondemented individuals. Neurosci Lett. 1988;93:191–196. doi: 10.1016/0304-3940(88)90080-8. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, Yagishita S, Ikeda K, Sano M, Taki K, Hashimoto K, Watabiki S, Mizusawa H. Coexistence of CJD and Alzheimer's disease: an autopsy case showing typical clinical features of CJD. Neuropathology. 2004;24:46–55. doi: 10.1111/j.1440-1789.2003.00513.x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Terada S, Ishizu H, Ikeda K, Hayabara T, Ikeda K, Deguchi K, Touge T, Kitamoto T, Kuroda S Advance online publication. An autopsy case of Creutzfeldt-Jakob disease with a V180I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology. 2009 doi: 10.1111/j.1440-1789.2009.01048.x. Retrieved February 21, 2010. [DOI] [PubMed] [Google Scholar]