

Abstract
The recently developed murine model of smoke inhalation and burn (SB) injury was used to study the effect of the substance-P antagonist CP96345. C57BL/6 mice were pretreated with an i.v. dose of a specific NK-1 receptor antagonist, CP9635, or its inactive enantiomer, CP96344, (10 mg/Kg) 1 hr prior to SB injury per protocol (n = 5). Mice were anesthetized and exposed to cooled cotton smoke, 2X 30 sec, followed by a 40% total body surface area flame burn per protocol. At 48 hr after SB injury Evans Blue (EB) dye and myeloperoxidase (MPO) were measured in lung after vascular perfusion. Lungs were also analyzed for hemoglobin (Hb) and wet/dry weight ratio.
In the current study, CP96345 pretreatment caused a significant decrease in wet/dry weight ratio (23%, *p = 0.048), EB (31%, *p = 0.047), Hb (46%, *p = 0.002) and MPO (54%, *p = 0.037) levels following SB injury compared to animals with SB injury alone. CP-96344 pretreatment caused an insignificant decrease in wet/dry weight ratio (14%, p=0.18), EB (16%, p = 0.134), Hb (9%, p = 0.39) and an insignificant increase in MPO (4%, p =0.79) as compared to mice that received SB injury alone. As expected, levels of EB, Hb, MPO, and wet/dry weight ratios were all significantly (p < 0.05) increased 48 hr following SB injury alone compared to respective sham animals. In conclusion, the current study indicates that pretreatment with specific NK-1R antagonist CP-96345 attenuates the lung injury and inflammation induced by SB injury in mice.
Keywords: Smoke inhalation, Substance P, NK-1 receptor antagonist, CP-96345, CP-96344, neurogenic inflammation, plasma extravasation, vascular permeability, acute lung injury, myeloperoxidase, burn injury
INTRODUCTION
Severe injury due to burns is common worldwide. The morbidity and mortality increase when burn injury is associated with smoke inhalation (Shirani et al. 1987). Deposition of numerous toxic products of combustion often complicates burn injury leading to acute injury to the airways and lungs (Traber and Herndon 1990). It has been reported that the smoke produced by burning cotton, wood, and plastic materials contains multiple potentially injurious components including carbon monoxide, HCl, acrolein, and aliphatic aldehydes (Prien adn Traber 1988; Alarie et al. 1983; Alarie 1985; Barrow 1990, Dost 1991). Previous studies have morphologically characterized smoke inhalation injury in humans (Cox et al. 2008, LaLonde et al. 1996, Linares 1981).
Recent investigations in the ovine model have begun to clarify the separate roles of vascular leakage in the bronchial and pulmonary circulations, nitric oxide, and airway obstruction to excessive shunt flow and progressive pulmonary function (Cox et al. 2003, Enkhbaatar and Traber 2004, Enkhbaatar et al. 2006). Despite this productive research, the pathogenetic sequences involved in the toxic and inflammatory reactions in the airways and the factors that trigger the acute inflammatory response to toxic smoke are not well understood. In the sheep model of combined smoke and burn injury, a striking increase in tracheal and bronchial blood flow occurs almost immediately after smoke inhalation injury. Neutrophils generally are not seen in respiratory bronchioles and the lung parenchyma until approximately 48 hr after injury (Sakurai et al. 1998). Intense acute inflammation is seen in the proximal trachea 3–4 hr after SB injury, and then progressively in the smaller peripheral airways over the course of 48 hr (Cox et al. 2003). The ability to study mice with specific and/or conditional genetic deletions or additions would make possible substantial contributions to this ongoing research if a relevant murine model were available. Therefore, as a part of our overall goal of studying the mechanisms responsible for ALI, we compared quantitative measures of acute inflammation in the lungs of C57BL/6 mice. Previous studies have indicated the involvement of tachykinins in inflammatory conditions (Lau and Bhatia, 2006). Primary tachykinins such as substance P (SP), neurokinin A and neurokinin B show high binding affinity for neurokinin-1 receptor (NK1R) (Harrison and Geppetti, 2001). In the airways, these peptides induce a variety of biological effects including smooth muscle constriction (Shore and Drazen, 1991), increased vascular permeability (Lundberg et al. 1984), facilitation of cholinergic transmission (Tanaka and Grunstein 1984), and mucus secretion (Kuo et al. 1990). Release of neuropeptides from peripheral nerve endings of sensory neurons has been characterized as the major cause of airway neurogenic inflammation, and includes plasma protein extravasation, airway smooth muscle contraction, and elevated mucus secretion (Nadel 1991). In vivo the concentration of released neuropeptides depends on both release and degradation, and the principal degradative enzyme is neutral endopeptidase (NEP) which is abundantly expressed in epithelial cells of the lung (Johnson et al. 1985, Roques et al. 1993).
Previous studies from this laboratory have demonstrated increased vascular permeability in a mouse model of combined smoke inhalation and burn (SB) injury (Jacob et al. 2008). There was a significant (p < 0.05) increase in lung Evans Blue, MPO content, and wet/dry weight ratio at 24 h and 48 hr after SB injury in Balb/C and C57BL/6 mice, indicating increased plasma extravasation and edema.
More recently we have used a different approach, the inhibition of neutral endopeptidase (NEP) that is known to alter the physiological and pharmacological effects of tachykinins in vitro (Stimler-Gerard, 1987, Martins et al. 1990) and in vivo (Dusser et al. 1988, Borson et al. 1989). Our studies indicated that pretreatment with the specific NEP inhibitor CGS 24592 caused a substantial increase in plasma extravasation in mice with SB injury as compared to SB injury alone (Jacob et al. 2009). To further identify the specific neuropeptide involved in this phenomenon, in the current study we have used the specific nonpeptide antagonist for the NK-1 receptor CP-96345. Because neurogenic inflammation can result from the action of substance P on NK1 receptors, it is of interest to study the effects of selective blockade of NK1 receptors to further clarify the mechanism of SB induced ALI. The objective of the current report is to study the effects of the highly selective NK-1A receptor antagonist CP 96345 (Snider et al. 1990, Lowe et al. 1993, Sio et al. 2008) in neurogenic inflammation in a mouse model of SB injury.
MATERIALS AND METHODS
Animal Preparation and Treatment
Male C57BL/6 mice weighing 19–21 g (5–6 weeks old) were obtained from Charles River Laboratories (Wilmington, MA). Mice were acclimatized to the animal facility for at least one week before the experiments were started. All procedures have been reviewed and approved by the Animal Care and Use Committee of the University of Texas Medical Branch specifically for use in these research studies.
Tracheal intubation and cotton smoke exposure
Briefly, mice were anesthetized with i.p. injection of ketamine (50 mg/kg), and xylazine (10 mg/kg). Once adequately anesthetized, mice were suspended by their upper incisors on a custom made intubation stand that allows direct visualization of the mouse glottis. A 20 gauge plastic cannula with a Y-shaped adaptor was constructed from readily available parts and inserted into the trachea using a specially constructed laryngoscope made from an aluminum “mag light” (Mag Instrument Inc., Ontario, CA) flashlight fitted with a fiber optic illuminator, under direct vision using a head-mounted jeweler’s magnifier.
A custom-made miniature smoke generator was arranged so that smoldering cotton toweling could be kept burning by a regulated flow of air. Smoke emerging from a cooling coil was delivered through plastic tubing to a Y-connector attached to the endotracheal tube at near atmospheric pressure. A pressurized air source was attached to the smoke generation chamber to control the flow and density of smoke delivered to the Y-tube. Mice were allowed to breathe normally during smoke exposure. Mice were exposed to two 30-sec intervals of smoke inhalation, breathing room air between exposures, followed by 48 hours of recovery.
Burn Injury
After smoke exposure, the back and bilateral flanks of each mouse were shaved, and 0.9 ml of normal saline was injected subcutaneously along the vertebral column. This subcutaneous liquid prevents thermal injury to internal organs such as the spinal cord and liver (Stephenson 1975). A Zetex cloth with a square window was placed over each mouse during the burning procedure to confine the burn area. Each mouse received a brief (less than10 second) exposure to a Bunsen burner flame, applied just long enough to induce retraction of the skin. This method allows for administration of a 40% total body surface area full thickness burn.
Mice were resuscitated with normal saline (4 ml/kg/% TBSA burn, i.p.). The endotracheal tube was removed as the mice recovered. Animals were given a daily dose of buprenorphene (0.1 mg/kg, i.p.) starting immediately after the injury for analgesia. At the end of the experimental time period, mice were killed by isoflurane overdose followed by cervical dislocation. Histologic study demonstrated full thickness dermal injury and showed no signs of necrosis or other recognizable changes in the portion of the liver under the site of injury. Sham control animals were anesthetized and intubated in the same manner as experimental animals but were allowed to breathe room air rather than given smoke exposure. They were also shaved and injected along their spines with saline but not burned. The sham control mice received the same volume of saline resuscitation and the same routine analgesia as the animals injured by burn and smoke inhalation.
Preparation and Administration of Drug
The animals were injected intravenously 1 hr prior to SB injury and then every 12 hours over the 48 hr period with the nonpeptide selective NK-1 receptor antagonist, CP-96345 (10 mg/kg), (2S,3S ) - cis-2- (diphenylmethyl) -N- [ (2-methoxyphenyl ) -methyl ] -1-azabicyclo-[2.2.2] octan-3-amine (Pfizer, Inc., New York, NY). Control animals received the inactive stereoisomer CP-96344 using the same dosing regimen. The drug was dissolved in lactated Ringer solution filtered through a 0.22-μm filter (Millipore), and stored at −70°C. Sham animals received an equal volume of lactated Ringer solution used for preparation of antagonist.
Tissue sampling
Mice were deeply anesthetized at 48 hr after SB injury, the chest of each mouse was opened, and a median sternotomy was performed. After excising the lung in one block the left and right lungs were separated. Both lungs were immediately flash-frozen in liquid nitrogen for determination of wet/dry weight ratio.
Evans Blue Extravasation
Thirty min. prior to the termination of the experiment mice were injected i.v. with 20 mg/kg Evans Blue (EB) via the tail vein and vascular leakage was assessed (Wang et al. 2002). The lungs were excised en bloc and dried on filter paper immediately after perfusion of the lungs with PBS as described previously (Jacob et al. 2008). After weighing, the lung tissue was snap frozen in liquid nitrogen and stored at −80°C. Lung tissue was manually homogenized using a bead beater (Biospec Products, Bartlesville, OK) and incubated with double the volume of formamide (Fisher Scientific, Fair Lawn, NJ) for 18 hr at 60°C. After centrifugation at 5,000 g for 30 min, the supernatant was collected. The optical density of the supernatant was determined spectrophotometrically at a wavelength of 620 nm. Levels of EB in samples were quantitated using a standard curve prepared with known amounts of EB, and the data are expressed as nanograms EB/mg lung.
Wet-to-Dry-Weight Ratio
After removing the lung tissue, samples were placed in pre-weighed glass vials, then dried for 48 hr in an oven at 65°C and reweighed, using a balance capable of indicating the weight to the nearest 0.1 mg, to determine the ratio of wet to dry weight, a measure of total tissue water content (Zhao et al. 2006).
Determination of Hemoglobin
Lung tissues were homogenized in TBS-T buffer and supernatant was collected as described above. To each 100 μl supernatant, 900 μl Drabkin’s reagent (Sigma, St. Louis, MO) was added, and the samples were incubated at room temperature for 15 min. Cyanmethemoglobin, the product of the reaction, was measured by its absorption at 550 nm using a Beckman DU-650 spectrophotometer. Hemoglobin levels were quantitated using a standard curve with known quantities of bovine erythrocyte hemoglobin (Sigma, St. Louis, MO).
Determination of Myeloperoxidase
Polymorphonuclear neutrophil (PMN) sequestration as indicated by myeloperoxidase (MPO) activity was measured in lung tissue homogenates using an MPO-EIA kit (Oxis Inc, Portland, OR), following the manufacturers protocol. Briefly, lungs were perfused with PBS as described earlier and homogenized in TBS-T buffer using a bead beater and centrifuged at 13,000g for 20 min. The protein content of the supernatant was assessed using the Bradford assay (Bradford 1976). To assess MPO, 100 μl of the supernatant was added to the micro wells in triplicate and incubated for 1 hr at room temperature. Plates were washed with buffer and diluted anti-MPO was added to each well. After 30 min incubation, plates were washed and avidin alkaline phosphatase was added followed by p-nitrophenol phosphate (pNPP) solution. The reaction was allowed to develop a pale yellow color for 30 min and was then terminated by adding 50 μl stop solution, and absorbance was immediately read at 405 nm using a TECAN-GENios microplate reader (Durham, NC). Myeloperoxidase activity was quantitated using a standard curve with known amounts of MPO, taking care that samples were compared to the linear portion of the standard curve.
Statistical Analysis
Values are shown as the mean ± SE of at least 5 determinations (n = 5). Statistical significance of the results was conducted by one-way analysis of variance followed by Dunnette’s post hoc test. The level of significance in all experiments was defined as p < 0.05.
RESULTS
Plasma Leakage
Evans Blue (EB) levels were quantitated in SB injured mice pretreated with specific NK-1R antagonist CP-96345 and compared to those with SB injury alone. The data indicate a statistically significant decrease (31%, *p = 0.047) in levels of EB in CP-96345 pretreated and SB injured mice as compared to untreated animals with SB injury at 48 hr after injury (Fig. 1). Pretreatment with the inactive stereoisomer CP-96344 showed only a minimal decrease in EB dye content (16%, *p=0.1344). When compared to sham animals, the groups injured and pretreated with both the active antagonist and its inactive isomer showed significantly higher levels of EB (110%, *p= 0.011, and 78%, *p=0.049) respectively.
Fig. 1. Effect of NK-1 antagonist pretreatment on plasma extravasation.

A significant decrease (31%, *p = 0.047) in EB levels was observed in NK-1 antagonist pretreated mice (checked Bar) 48 hr following SB injury compared to animals with SB injury alone (black filled bar). EB levels were significantly (*p =0.0356) increased 48 hr following SB injury alone (black filled bar) compared to respective sham animals (open bar). The group treated with CP-96345 (speckled bar) had a statistically insignificant increase (16%, *p = 0.134) in EB level as compared to respective sham animals. EB levels were quantitated from an authentic standard curve with known concentrations of EB. Values are means ±SE of at least 5 mice/group.
Edema
Wet/dry weight ratios were calculated as described earlier. In groups of mice with SB injury wet/dry weight ratios were significantly elevated (47%, *p=0.018) as compared to sham animals. However, there was a statistically significant decrease (23%, *p = 0.048) in mice pretreated with CP-96345 followed by SB injury as compared to corresponding SB control animals (Fig. 2). Even though there was a slight (14%) decrease in wet/dry weight ratios between mice with SB injury alone vs. those that received CP-96344 prior to SB injury, these changes were found to be insignificant (p=0.354). There was a significant increase (19.4% *p=0.043) in the wet-dry weight ratios of those treated with the inactive isomer CP-96344 compared to the sham group and an insignificant increase (13%) compared to sham animals.
Fig. 2. Effect of NK-1 antagonist pretreatment on edema.

A significant decrease (23%, *p = 0.048) in wet/dry weight ratios was indicated in NK-1 antagonist CP-96345 pretreated mice (checked Bar) 48 hr following SB injury compared to animals with SB injury alone. (black filled bar). Wet/dry weight ratios were significantly increased (47%, *p = 0.018) 48 hr following SB injury alone (black filled bar) compared to respective sham animals (open bar). Pretreatment with NK-1 inactive enantiomer CP-96344 followed by SB injury (speckled bar) caused a statistically insignificant decrease (14%, *p=0.354) in wet/dry weight ratios compared to mice that received SB injury alone (black filled bar). Values are means ±SE of at least 6 mice/group.
Levels of Hemoglobin in mouse lung
Hemoglobin content in the lung was measured using Drabkin’s reagent as described before. There was a significant increase (68%, *p = 0.037) in hemoglobin levels 48 hr following SB injury as compared to sham (Fig. 3). However, the hemoglobin levels in mice pretreated with specific NK-1 antagonist CP-96345 were significantly lower (46%, *p=0.0019) as compared to those who received SB injury alone (Fig. 3). The slight decrease in lung levels of hemoglobin observed in mice pretreated with NK-1 stereo isomer CP-96344 following SB injury could not be shown to be of statistical significance (9%, p = 0.39). There were significantly higher levels of hemoglobin (34%, *p = 0.005) in mice treated with CP-96344 as compared to those that received specific NK-1 antagonist CP-96345. When compared to sham animals, the groups pretreated with both the active antagonist and its inactive isomer showed higher levels (14%, p = 0.35 and 54%, *p = 0.0031) of Hb respectively.
Fig. 3. Effect of NK-1 antagonist pretreatment on Hemoglobin (Hb).

Significantly decreased Hb levels (46%, *p = 0.002) were seen in NK-1R antagonist pretreated mice after SB injury (checked bar). Increased Hb levels (68%, *p=0.037) were observed in SB injury alone (black filled bar) compared to sham (open bar). There was a low level insignificant decrease (9%, *p=0.39) in CP-96344 pretreated mice (specked bar) compared to the SB injury alone group (black filled bar). Values are means ±SE of at least 6 mice/group.
Myeloperoxidase Activity
Levels of myeloperoxidase (MPO) were determined using a sandwich ELISA kit as described, following thorough perfusion of the pulmonary arterial system with normal saline. This assay includes neutrophils that are firmly adherent to pulmonary endothelium as well as those that have extravasated from vessels. As expected there was a significant increase (227%, *p = 0.005) in myeloperoxidase content 48 hr following SB injury as compared to sham (Fig. 3). However, in mice pretreated with specific NK-1 antagonist CP-96345 followed by SB injury there was a significant decrease (54%, *p = 0.037) in MPO levels as compared to untreated mice with SB injury alone (Fig. 4). Pretreatment with inactive isomer CP-96344 before SB injury caused a slight insignificant increase (4%, p= 0.79) in MPO activity as compared to injured but untreated mice. There were significantly higher levels of myeloperoxidase activity (61%, p = 0.05) in mice treated with CP-96344 as compared to those that received specific NK-1 antagonist CP-96345.
Fig. 4. Effect of NK-1 antagonist pretreatment on neutrophil infiltration and inflammation.

Significantly decreased MPO levels (54%, * p = 0.037) were found in NK-1 antagonist pretreated mice (checked bar) following SB injury compared to animals with SB injury alone (black filled). Significantly increased MPO levels were indicated (227%, *p =0.005) 48 hr following SB injury alone (black filled bar) compared to respective sham animals (open bars). Pretreatment with inactive isomer CP-96344 followed by SB injury (speckled bar) led to a slight insignificant increase (4%, p=0.79) compared to animals pretreated with CP-96345 followed by SB injury (black filled bar). MPO levels were quantitated from an authentic standard curve with known amounts of MPO. Values are means ±SE of at least 5 mice/group.
DISCUSSION
Following burn trauma, toxic smoke inhalation has been recognized as a major cause of mortality (Crapo 1981). Previous reports from this and other laboratories have indicated that toxic smoke causes progressive injury to the airways, followed by injury to lung parenchyma (Barrow et al. 1990; Traber and Herndon 1991; Bidani et al. 1999; Dost 1991). Even though rapid release of chemical mediators in the airways has been suggested as a mechanism for injury, there is limited understanding of the underlying processes
Chemical irritants that activate unmyelinated sensory nerves cause plasma leakage in the skin, respiratory tract, and other organs by triggering the release of substances from sensory nerve fibers (Lundberg 1984, De Swert and Joos, 2006). Substance P, which is synthesized and released by some sensory neurons, appears to be a major mediator, although other tachykinins, calcitonin gene-related peptide, and perhaps other peptides may also participate.
Neurogenic inflammation is regarded as a first line of defenseand protects tissues when noxious conditions threaten normal body functions. However, severe or prolongednoxious stimulation may result in the inflammatory responsemediating injury rather than facilitating repair (Day et al. 2005). Proinflammatory effects of NEP antagonists including the specific NEP inhibitor CGS 24592 have been studied (Pham et al. 2000, Nicolau et al. 2003, Damas et al. 1996). These agents caused increased plasma extravasation and vascular permeability in various animal models. Recent studies from this laboratory have indicated that the inhibition of neutral endopeptidase (NEP) using specific NEP inhibitor, CGS 24592, exacerbated smoke and burn (SB) injury leading to more plasma extravasation and more severe airway inflammation (Jacob et al. 2009).
Because neurogenic inflammation can result from the action of substance P on NK1 receptors, it is of interest to study the effect of blockage of selective NK1 receptor using specific NK-1 antagonist CP-96345 would decrease the injury. The NK1 receptors involved in plasma leakage are located on the endothelial cells of post capillary venules and collecting venules. Within seconds of the activation of NK1 receptors by substance P, gaps form in the endothelium of target vessels (Wong et al. 2003 and 2004). The endothelial gaps are transient, and the leak normally ends in a few minutes (McDonald et al. 1996).
The current study indicates that pretreatment with CP-96345 caused a significant decrease (p < 0.05) in plasma extravasation and wet/dry weight ratios 48 h after SB injury as compared to animals with SB injury alone(Figs. 1 &4). Sham animals showed an insignificant increase (32%, p = 0.137) in wet/dry weight ratio as compared to untouched control animals (data not shown). It should be emphasized that the method used in these studies to measure lung MPO content and extravasation of EB dye involves extensive vascular perfusion of the lungs with an excess of buffered saline, removing essentially all unattached intravascular red blood cells. Thus the neutrophils detected by the myeloperoxidase assay must be those that were adherent to vascular endothelium at the time of perfusion, as well as those that have emigrated from blood vessels into the interstitium, the airways, or the alveoli. Similar findings regarding the role of substance P and other tachykinins have been reported previously by Wong et al in rat models of smoke inhalation injury, and by Sio et al in a murine model of burn injury alone (Wong et al. 2003, 2004, Sio 2008).
The results of the study can be explained based on the proposed pathway summarized in Fig. 5. There are numerous toxic compounds in the smoke produced by burning wood, textiles and upholstery.(Prien and Traber 1988; Alarie et al 1983; Alarie 1985). Several of these are potent respiratory irritants, and include numerous free radicals.(Vinggaard et al. 1989; Pryor 1992) The pathogenesis of the response to deposition of these toxins in the airways and lungs remains under investigation and is complex and incompletely understood.(Demling, 2008) Irritation of sensory nerves in the airways leads to release of substance P and other neurokinins and the calcitonin gene-related peptide CGRP, which is a potent vasodilator, likely both due to direct stimulation of nerve endings and also as a consequence of depolarization of the cell bodies giving rise to the sensory (afferent) nerves. Airway irritation gives rise to a variety of reflex reactions that tend to be pro-inflammatory. Other reactions to toxins and oxidants in smoke may stimulate inflammation independently of neuropeptide release, including oxidative signaling, transient receptor potential cation channel, subfamily V1 (TRPV1) receptor activation, calcium signaling, activation, and upregulation of nitric oxide synthase and expression of cytokines and chemokines by airway tissues, and secretion of mucus. Host defense mechanisms that normally serve to clear foreign material and degrade toxins and peptides become dysregulated.(Traber et al. 2000; Enkhbaatar and Traber 2004) Activation of the NK-1 receptor by substance P and other neurokinins stimulates vascular leakage in both bronchial and pulmonary circulations, and activation of mast cells releases histamine and TNF-alpha and other cytokines.(Lundberg 1984) The resulting inflammation gives rise to local plasma extravasation and edema. Several mechanisms may amplify the pro-inflammatory actions of toxic smoke, including further release of neuropeptides, activation of nuclear factor kappa-B and poly-adenosly-ribose polymerase, new or increased expression of vascular adhesion molecules, tissue expression of cytokines and chemokines, and activation of platelets by the shear stress resulting from increased microvascular blood flow. By mechanisms that remain to be clarified, the intense acute inflammatory reaction induced in the airways within hours of inhalation injury leads to delayed injury to the lung parenchyma, which in turn leads to protein-rich pulmonary edema, the appearance of myofibroblasts, and pulmonary fibrosis if the host survives the initial reaction to smoke.
Fig. 5. Proposed pathway depicting the role of NK-1 antagonist in lung injury following SB injury in mice.
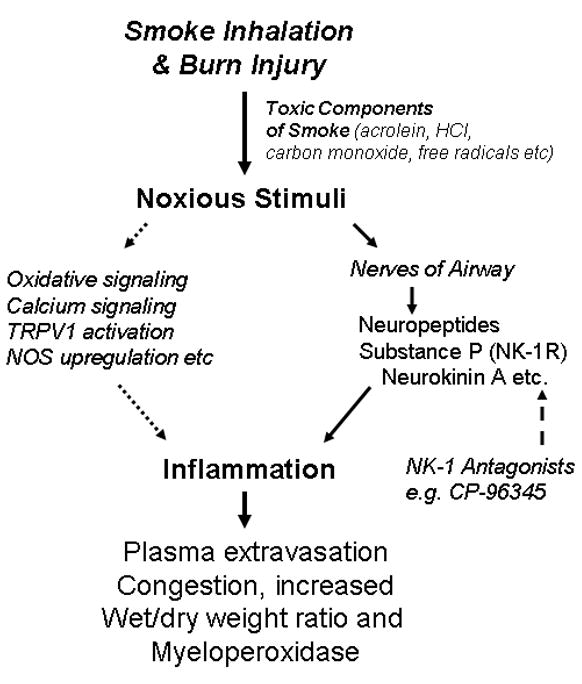
Neurogenic inflammation induced by SB induced noxious stimuli is caused in part by release of neuropeptides from nociceptor nerves, and so treatment with an NK-1 receptor antagonist will reduce plasma leakage, edema, and neutrophil infiltration.
The current study serves to clarify one aspect of this pathogenetic sequence by showing that activation of the NK1 receptor increases plasma leakage and pulmonary edema in a murine model of combined burn and smoke inhalation injury.
Acknowledgments
This work was supported by a program project grant from NIH, # NIGMS PO1-GM66312 and a core facility grant from Shriners Hospitals for Children (Project #8460). The NK-1 antagonist and its inactive stereoisomer were generous gifts of Dr. R. M. Snider of Pfizer, Inc., Groton, CT.
References
- Alarie Y, Stock MF, Matijak-Schaper M, Birky MM. Toxicity of smoke during chair smoldering tests and small scale tests using the same materials. Fundam Appl Toxicol. 1983;3(6):619–26. doi: 10.1016/s0272-0590(83)80112-2. [DOI] [PubMed] [Google Scholar]
- Alarie Y. The toxicity of smoke from polymeric materials during thermal Decomposition. Annu Rev Pharmacol Toxicol. 1985;25:325–47. doi: 10.1146/annurev.pa.25.040185.001545. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Role of neural mechanisms in airway defense. In: Chretien J, Dusser D, editors. Environmental Impact in the Airways. Marcel Dekker; New York: 1996. pp. 93–121. [Google Scholar]
- Barrow RE, Morris SE, Basadre JO, Herndon DN. Selective permeability changes in the lungs and airways of sheep after toxic smoke inhalation. J Appl Physiol. 1990;68:2165–2170. doi: 10.1152/jappl.1990.68.5.2165. [DOI] [PubMed] [Google Scholar]
- Bidani A, Hawkins HK, Wang CZ, Heming TA. Dose dependence and time course of smoke inhalation injury in a rabbit model. Lung. 1999;177:111–122. doi: 10.1007/pl00007630. [DOI] [PubMed] [Google Scholar]
- Borson DB, Brokaw K, Sekizawa DM, Nadel JA. Neutral endopeptidase and neurogenic inflammation in rats with respiratory infections. J Appl Physiol. 1989;66:2653–3658. doi: 10.1152/jappl.1989.66.6.2653. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cox RA, Mlcak RP, Chinkes DL. Upper airway mucus deposition in lung tissue of burn trauma victims. Shock. 2008;29:356–61. doi: 10.1097/shk.0b013e31814541dd. [DOI] [PubMed] [Google Scholar]
- Cox RA, Burke AS, Soejima K, Murakami K, Katahira J, Traber LD, Herndon DN, Schmalstieg FC, Traber DL, Hawkins HK. Airway obstruction in sheep with burn and smoke inhalation injuries. Am J Respir Cell Mol Biol. 2003;29:295–302. doi: 10.1165/rcmb.4860. [DOI] [PubMed] [Google Scholar]
- Cox RA, Burke AS, Traber DL, Herndon DN, Hawkins HK. Production of pro-inflammatory polypeptides by airway mucous glands and its potential significance. Pulm Pharmacol Ther. 2006 doi: 10.1016/j.pupt.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Crapo RO. Smoke-inhalation injuries. JAMA. 1981;246:1694–1696. [PubMed] [Google Scholar]
- Damas V, Bourdon JF, Simmons WH. Influence of several peptidase inhibitors on the pro-inflammatory effects of substance P, capsaicin and collagenase. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1996;354(5):1432–1912. doi: 10.1007/BF00170843. [DOI] [PubMed] [Google Scholar]
- Day AL, Wick E, Jordan TH, Jaffray CE, Bunnett NW, Grady EF, Kirkwood KS. Neutral endopeptidase determines the severity of pancreatitis-associated lung injury. J Surg Res. 2005;128(1):21–7. doi: 10.1016/j.jss.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Demling RH. Smoke inhalation lung injury: an update. Eplasty. 2008;16(8):e27. [PMC free article] [PubMed] [Google Scholar]
- Dost FN. Acute toxicology of components of vegetation smoke. Rev Environ Contam Toxicol. 1991;119:1–46. doi: 10.1007/978-1-4612-3078-6_1. [DOI] [PubMed] [Google Scholar]
- Dusser DJ, Eumeno PD, Graf T, Djokic DB, Borson DB, Nadel JA. Airway neutral endopeptidase-like enzyme modulates tachykinin induced bronchoconstriction in vivo. J Appl Physiol. 1988;65:2585–2591. doi: 10.1152/jappl.1988.65.6.2585. [DOI] [PubMed] [Google Scholar]
- Enkhbaatar P, Murakami K, Traber LD, Cox R, Parkinson JF, Westphal M, Esechie A, Morita N, Maybauer MO, Maybauer DM, Burke AS, Schmalstieg FC, Hawkins HK, Herndon DN, Traber DL. The inhibition of inducible nitric oxide synthase in ovine sepsis model. Shock. 2006;25:522–527. doi: 10.1097/01.shk.0000209525.50990.28. [DOI] [PubMed] [Google Scholar]
- Enkhbaatar P, Traber DL. Pathophysiology of acute lung injury in combined burn and smoke inhalation injury. Clin Sci (Lond) 2004;107:137–143. doi: 10.1042/CS20040135. [DOI] [PubMed] [Google Scholar]
- Hershey AD, Krause JE. Molecular characterization of a functional cDNA encoding the rat substance P receptor. Science. 1990;247:958–962. doi: 10.1126/science.2154852. [DOI] [PubMed] [Google Scholar]
- Hon YL, Madhav B. The effect of CP96, 345 on the expression of tachykinins and neurokinin receptors in acute pancreatitis. J Pathol. 2006;208:364–371. doi: 10.1002/path.1899. [DOI] [PubMed] [Google Scholar]
- Jacob S, Deyo DJ, Cox RA, Traber DL, Hawkins HK. Assessment of LungInflammation in a Mouse Model of Smoke Inhalation and Burn Injury: Strain-Specific Differences. Toxicol Mech Meth. 2008;18:1–9. doi: 10.1080/15376510802251993. [DOI] [PubMed] [Google Scholar]
- Jacob S, Deyo DJ, Cox RA, Traber DL, Herndon DN, Hawkins HK. Mechanisms of toxic smoke inhalation and burn injury: role of neutral endopeptidase and vascular leakage in mice. Toxicol Mech Methods. 2009 Mar;19(3):191–6. doi: 10.1080/15376510902725649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AR, Ashthon J, Scypinski L, Dusser D, Nadel J, Borson D. Toluene diisothiocynate increases airway responsiveness to substance P and decreases airway neutral endopeptidase in human lung tissue and cultured cells. Am Rev Resp Dis. 1985;132:564–568. [Google Scholar]
- Kuo H-PJA, Rohde K, Tokuyama PJ, Barnes PJ, Rodgers DF. Capsaicin and sensory neuropeptides stimulation of goblet cell secretion in guinea pig trachea. J Appl Physiol. 1990;431:629–641. doi: 10.1113/jphysiol.1990.sp018351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde C, Nayak U, Hennigan J, Demling R. Antioxidants prevent the cellular deficit produced in response to burn injury. J Burn Care Rehabil. 1996;17:379–383. doi: 10.1097/00004630-199609000-00002. [DOI] [PubMed] [Google Scholar]
- Lau HY, Bhatia M. The effect of CP96345 on the expression of tachykinins and neurokinin receptors in acute pancreatitis. J Pathol. 2006 Feb;208(3):364–71. doi: 10.1002/path.1899. [DOI] [PubMed] [Google Scholar]
- Linares HA. A report of 115 consecutive autopsies in burned children: 1966–80. Burns. 1981;8:263–270. doi: 10.1016/0305-4179(82)90007-9. [DOI] [PubMed] [Google Scholar]
- Lowe JA, III, Drozda SE, Snider RM, Longo KP, Zorn SH, Jackson ER, Morrone J, McLean S, Bryce DK, Bordner J. Discovery of CP-96345 and its characterization in disease models involving substance P. Regul Pept. 1993;46:20–23. doi: 10.1016/0167-0115(93)90006-t. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Brodin E, Hua X, Saria A. Vascular permeability changes and smooth muscle contraction in relation to substance P neurons with special reference to the trachea and lungs. Acta Physiol Scand. 1984;119:243–252. [Google Scholar]
- Martins MA, Shore SA, Gerard NP, Gerard C, Drazen JM. Peptidase modulation of the pulmonary effects of tachykinins in tracheal superfused guinea pig lungs. J Clin Invest. 1990;85:170–176. doi: 10.1172/JCI114408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DM, Bowden JJ, Baluk P, Bunnett NW. Neurogenic inflammation. A model for studying efferent actions of sensory nerves. Adv Exp Med Biol. 1996;410:453–62. [PubMed] [Google Scholar]
- Nadel JA. Neutral endopeptidase modulates neurogeic inflammation. Euro Respir J. 1991;4:745–754. [PubMed] [Google Scholar]
- Nicolau M, Dovichi SS, Cuttle G. Pro-inflammatory Effect of Quercetin by Dual Blockade of Angiotensin Converting-enzyme and Neutral Endopeptidase In Vivo. Nutritional Neuroscience. 2003;6(5):309–316. doi: 10.1080/10284150310001595609. [DOI] [PubMed] [Google Scholar]
- Pham D, Jeng AY, Escher E, Sirois P, Battistini B. Effects of a selective neutral endopeptidase and a nonselective neutral endopeptidase/endothelin-converting enzyme inhibitor on lipopolysaccharide-induced endotoxaemia in anaesthetized Sprague-Dawley rats. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S362–6. doi: 10.1097/00005344-200036051-00105. [DOI] [PubMed] [Google Scholar]
- Prien T, Traber DL. Toxic smoke compounds and inhalation injury--a review. Burns Incl Therm Inj. 1988;14(6):451–60. doi: 10.1016/s0305-4179(88)80005-6. [DOI] [PubMed] [Google Scholar]
- Pryor WA. Biological effects of cigarette smoke, wood smoke, and the smoke from plastics: the use of electron spin resonance. Free Radic Biol Med. 1992;13(6):659–76. doi: 10.1016/0891-5849(92)90040-n. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular Mechanisms of Neurogenic Inflammation. J Pharmacol Exp Ther. 2002;302(3):839–45. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Roques BP, Noble F, Dauge V, Fournie-Zaluski MC, Beaumont A. Neurtral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmaco l Rev. 1993;45:87–146. [PubMed] [Google Scholar]
- Sakurai H, Johnigan R, Kikuchi Y, Harada M, Traber LD, Traber DL. Effectof reduced bronchial circulation on lung fluid flux after smoke inhalation in sheep. J Appl Physiol. 1998;84:980–986. doi: 10.1152/jappl.1998.84.3.980. [DOI] [PubMed] [Google Scholar]
- Shirani KZ, Moylan JA, Pruitt BA., Jr The influence of inhalation injury and pneumonia on burn mortality. Ann Surg. 1987;205(1):82–7. doi: 10.1097/00000658-198701000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore SA, Drazen JM. Relative bronchoconstrictor activity of neurokinin A and neurokinin A fragments in the guinea pig. J Appl Physiol. 1991;71:452–457. doi: 10.1152/jappl.1991.71.2.452. [DOI] [PubMed] [Google Scholar]
- Sio SW, Puthia MK, Lu J, Moochhala S, Bhatia M. The neuropeptide substance P is a critical mediator of burn-induced acute lung injury. J Immunol. 2008;180:8333–8341. doi: 10.4049/jimmunol.180.12.8333. [DOI] [PubMed] [Google Scholar]
- Snider RM, Constantine JW, Lowe JA, III, Longo KP, Lebel WS, Woody HA, Drozda SE, Desai MC, Vinick FJ, Spencer RW, Hess HJ. Science. 1991;251:435–437. doi: 10.1126/science.1703323. [DOI] [PubMed] [Google Scholar]
- Stephenson SF, Esrig BC, Polk HC, Fulton RL. The pathophysiology of smoke inhalation injury. Ann Surg. 1975;182:652–660. doi: 10.1097/00000658-197511000-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimler-Gerard NP. Neutral endopeptidase-like enzyme controls the contractile activity of substance P in guinea pig lung. J Clin Invest. 1987;79:1819–1825. doi: 10.1172/JCI113023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka DT, Grunstein MM. Effect of substance P on neurally mediated contraction of rabbit airway smooth muscle. J Appl Physio! 1986;60:458–463. doi: 10.1152/jappl.1986.60.2.458. [DOI] [PubMed] [Google Scholar]
- Traber DL, Herndon DN. Pathophysiology of Smoke Inhalation. In: Haponik EF, Munster AM, editors. Respiratory Injury. New York: McGraw-Hill; 1990. pp. 61–71. [Google Scholar]
- Traber DL, Hawkins HK, Enkhbaatar P, Cox RA, Schmalstieg FC, Zwischenberger JB, Traber LD. Pulm Pharmacol Ther. 2007;20(2):163–6. doi: 10.1016/j.pupt.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Traber DL, Herndon DN. Pathophysiology of smoke inhalation. In: Haponik EF, Munster AM, editors. Respiratory Injury: Smoke Inhalation and Burns. McGraw-Hill; New York: 1991. pp. 61–71. [Google Scholar]
- Vinggaard AM, Nielsen GD, Fries AS. Sensory and pulmonary irritation of inhaled n-butylamine in CF-1 and NMRI mice. Lab Anim. 1989;23(1):1–6. doi: 10.1258/002367789780886957. [DOI] [PubMed] [Google Scholar]
- Wang LF, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, Mehta S. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med. 2002;165:1634–1639. doi: 10.1164/rccm.2110017. [DOI] [PubMed] [Google Scholar]
- Westphal M, Cox RA, Traber LD, Morita N, Enkhbaatar P, Schmalstieg FC, Hawkins HK, Maybauer DM, Maybauer MO, Murakami K, Burke AS, Westphal-Varghese BB, Rudloff HE, Salsbury JR, Jodoin JM, Lee S, Traber DL. Combined burn and smoke inhalation injury impairs ovine hypoxic pulmonary vasoconstriction. Crit Care Med. 2006;34:1428–1436. doi: 10.1097/01.CCM.0000215828.00289.B9. [DOI] [PubMed] [Google Scholar]
- Wong SS, Sun NN, Keith I, Kweon CB, Foster DE, Schauer JJ, Witten ML. Tachykinin substance P signaling involved in diesel exhaust-induced bronchopulmonary neurogenic inflammation in rats. Arch. Toxicol. 2003;77:638–650. doi: 10.1007/s00204-003-0485-4. [DOI] [PubMed] [Google Scholar]
- Wong SS, Sun NN, Lantz RC, Witten ML. Substance P and neutral endopeptidase in development of acute respiratory distress syndrome following fire smoke inhalation. Am J Physiol Lung Cell Mol Physiol. 2004;287:L859–L866. doi: 10.1152/ajplung.00388.2003. [DOI] [PubMed] [Google Scholar]
- Zhao M, Fernandez LG, Doctor A, Sharma AK, Zarbock A, Tribble CG, Kron IL, Laubach VE. Alveolar Macrophage Activation is a Key Initiation Signal for Acute Lung Ischemia-Reperfusion Injury. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L1018–26. doi: 10.1152/ajplung.00086.2006. [DOI] [PubMed] [Google Scholar]