

Abstract
Anthrax lethal toxin (LeTx) is composed of protective antigen (PA) and lethal factor (LF) – PA is the receptor-binding moiety and LF is a protease that cleaves mitogen-activated protein kinase kinases (MAPKKs). LeTx subverts the immune response to B. anthracis in several ways, such as downregulating interleukin-8 (IL-8) by increasing the rate of IL-8 mRNA degradation. Many transcripts are regulated through cis-acting elements that bind proteins that either impede or promote degradation. Some of these RNA binding proteins are regulated by MAPKs and previous work has demonstrated that interfering with MAPK signaling decreases the half-life of IL-8 mRNA. Here, we have localized a segment within the IL-8 3′ untranslated region responsible for LeTx-induced transcript destabilization and show that this is caused by inhibition of the p38, ERK, and JNK pathways. TTP, an RNA binding protein involved in IL-8 mRNA decay, became hypophosphorylated in LeTx-treated cells and knock-down of TTP prevented LeTx from destabilizing the IL-8 transcript. Cells that were treated with LeTx exhibited increased localization of TTP to Processing-bodies, which are structures that accumulate transcripts targeted for degradation. We furthermore observed that LeTx promoted the formation of Processing-bodies, revealing a link between the toxin and a major mRNA decay pathway.
Keywords: anthrax, lethal toxin, TTP, IL-8, P-body
Introduction
The establishment of a Bacillus anthracis infection relies on the ability of the bacterium to overcome the immune response. One virulence factor that contributes to immune evasion is lethal toxin (LeTx), a protein toxin composed of protective antigen (PA) and lethal factor (LF). PA is the cell binding component that delivers LF to the mammalian cell cytosol, where LF cleaves members of the mitogen activated protein kinase kinase (MAPKK) family (Vitale et al., 2000; Duesbery et al., 1998). LeTx suppresses the immune response through a number of mechanisms, such as impairing dendritic cell maturation (Agrawal et al., 2003), decreasing the proliferation of T and B cells (Fang et al., 2005; Paccani et al., 2005), and blocking the pro-inflammatory cytokine response (Erwin et al., 2001; Pellizzari et al., 1999).
We reported previously that LeTx induces destabilization of IL-8 mRNA (Batty et al., 2006). A molecule that acts both as a chemokine and a cytokine, IL-8 recruits neutrophils to sites of infection and activates them (Rot, 1992). These polymorphonuclear cells can eliminate pathogens by phagocytosis and by production of oxygen radicals and nitric oxide. The importance of neutrophils to the immune response is illustrated by neutropenia, a disorder characterized by severely low neutrophil numbers and by increased patient susceptibility to many pathogens (Janeway, 2001). Thus, IL-8 is an important mediator of inflammation and lowering IL-8 expression through mRNA destabilization is a way that LeTx diminishes this protective host response.
Regulation of mRNA decay mediates rapid changes to gene expression, thereby allowing a short response time to cellular stimuli. The stability of a transcript can be controlled by cis-acting elements within the mRNA molecule, often localized to the 3′ untranslated region (UTR) (Garneau et al., 2007; Wilusz et al., 2001; Chen and Shyu, 1995). AU-rich elements (AREs) are among the best characterized cis-acting elements that influence mRNA stability and are characterized by the pentameric AUUUA motif, although some ARE-containing transcripts lack this motif. The recently constructed ARE database reveals that as many as 8% of human mRNAs contain AREs, including TNF-α, GM-CSF, Il-1β, and IL-8 (Bakheet et al., 2006).
AU-binding proteins (AUBPs) interact with AREs to regulate the stability of ARE-containing transcripts. HuR is an AUBP that stabilizes transcripts (Chen et al., 2002), whereas AUBPs such as tristetraprolin (TTP), K homology-type splicing regulatory protein (KSRP), and T-cell restricted intracellular antigen-1 related protein (TIAR) promote the decay of ARE-containing transcripts (Chou et al., 2006; Briata et al., 2005; Linker et al., 2005; Gherzi et al., 2004; Stoecklin et al., 2004; Lai et al., 2000). Destabilizing AUBPs can recruit transcripts to Processing-bodies (P-bodies), which are sites where mRNA degradation occurs. P-bodies are dynamic cytoplasmic granules that contain translationally silenced transcripts and proteins involved in the breakdown of RNA, such as decapping enzymes and exonucleases (Parker and Sheth, 2007). P-bodies can increase in size and number in response to various stimuli (Kedersha et al., 2005; Sheth and Parker, 2003).
We report here that LeTx activity destabilizes IL-8 mRNA through a 100-nucleotide region in the 3′ UTR. This destabilization can be attributed to the ability of the toxin to inactivate the p38, ERK and JNK pathways, which leads to the dephosphorylation of TTP. Importantly, knocking down TTP using siRNA prevented LeTx from destabilizing the IL-8 transcript. Furthermore, a greater fraction of intoxicated cells, compared to unintoxicated cells, displayed P-bodies and these P-bodies all co-localized with TTP. Our results support a model in which intoxication causes dephosphorylation of TTP that then allows TTP to recruit the transcript to sites of mRNA degradation.
Results
Localization of an element that mediates destabilization of IL-8 mRNA by lethal toxin
The effect of LeTx on IL-8 mRNA stability in HT1080 fibroblasts was assessed in cells pretreated with TNF-α to increase the level of endogenous IL-8 mRNA, and then with actinomycin D, to halt de novo mRNA synthesis. Total RNA was extracted at various times and IL-8 transcript levels were quantified using quantitative real-time PCR (qPCR) and standardized to β-actin mRNA levels. The half-life (t1/2) of IL-8 mRNA in LeTx-treated cells was ∼52 min compared to ∼140 min in unintoxicated cells (Fig. 1A), indicating that LeTx destabilizes IL-8 mRNA in this human fibroblast cell line.
Figure 1.
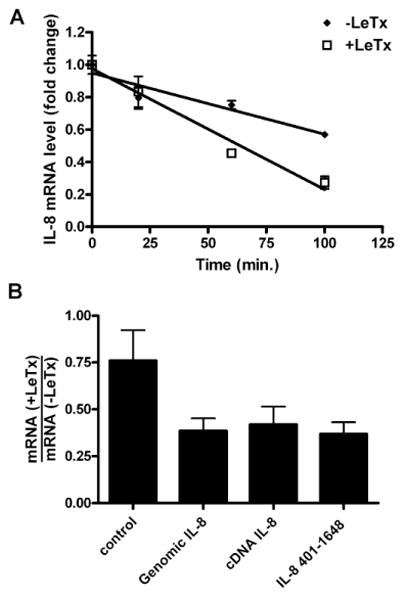
LeTx accelerates IL-8 mRNA decay through the 3′ UTR.
A. Endogenous IL-8 mRNA was induced by incubating HT1080 cells for 2 h with TNF-α (10 ng/ml), followed by treatment with LeTx (10-8 M PA and 10-9 M LF) for 1 h. Transcription was inhibited by addition of actinomycin D (1 μg/ml) and total RNA was isolated at the indicated times and transcript levels was measured using qPCR. Error bars indicate SEM of three independent experiments.
B. HT1080 cells were transiently transfected with the indicated plasmids containing IL-8 sequences and treated with LeTx (10-8 M PA and 10-9 M LF). Total RNA was isolated from untreated and intoxicated cells and reporter transcript levels were measured using qPCR. Error bars indicate SEM of three independent experiments.
While we have demonstrated previously that the 3′ UTR of the IL-8 transcript confers LeTx-dependent destabilization to a reporter transcript (Batty et al., 2006), it has been shown that stability of other transcripts can be influenced by regions in the 5′ UTR and coding region and by whether or not the transcript has undergone splicing (Zhao and Hamilton, 2007; Chen et al., 2000). Therefore, to determine if regions outside of the 3′UTR affect LeTx-mediated transcript destabilization, we cloned the IL-8 genomic sequence, the cDNA sequence, and the 3′ UTR into reporter constructs containing tags downstream of the transcription start site that allowed for detection by qPCR. The reporter constructs were transiently transfected into HT1080 cells for 2 h and the cells were either left untreated or were treated with LeTx for 24 h. RNA was isolated from untreated and toxin-treated cells and the ratios of the levels of reporter transcripts were calculated (Fig. 1B). The levels of the transcripts containing IL-8 sequences were reduced by LeTx to similar extents, suggesting that the element(s) responsible for toxin-mediated destabilization are confined to the 3′ UTR and that splicing does not affect the stability of the transcript.
Localization of destabilizing elements in IL-8 mRNA
In order to identify cis-acting elements that destabilize IL-8 mRNA, various truncations of the IL-8 3′ UTR were cloned behind the EYFP coding region (Fig. 2A). The constructs were transiently transfected into HT1080 cells and reporter mRNA was quantified using qPCR and standardized to β-actin mRNA. mRNA containing the entire IL-8 3′ UTR (EYFP-IL-8401-1648) was detected at an ∼10-fold lower level than that of the EYFP control mRNA, confirming the presence of destabilization elements in the 3′ UTR (Fig. 2B). A segment comprising nucleotides 1000-1100 (EYFP-IL-81000-1100), containing 4 clustered AUUUA pentamers, was found to exert significant destabilization to the reporter transcript, whereas other ARE-containing regions did not confer significant destabilization to the EYFP coding region.
Figure 2.
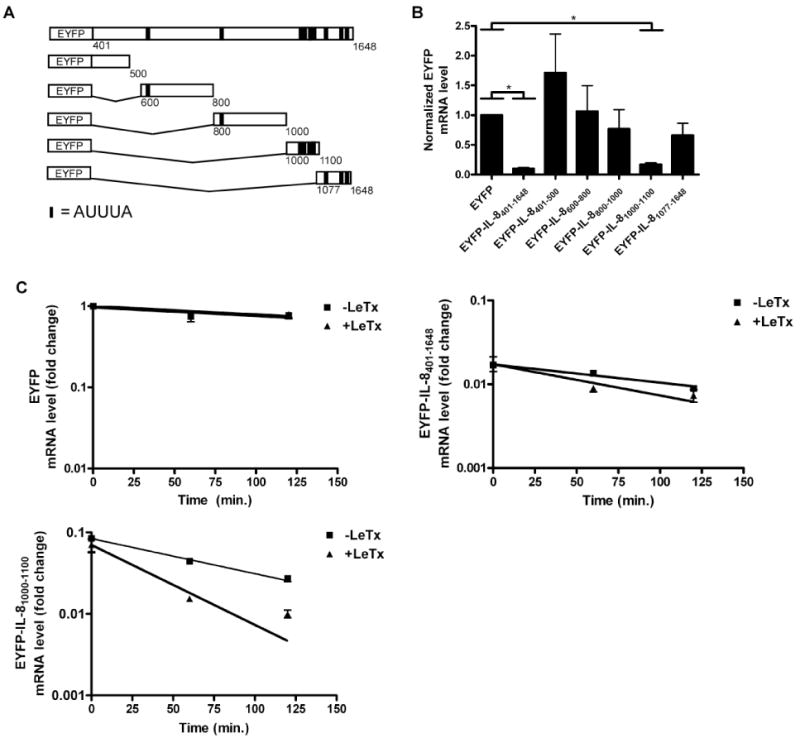
Identification of a region within the IL-8 3′UTR that confers mRNA instability.
A. Scheme depicting the reporter genes containing various truncations of the IL-8 3′UTR. Vertical lines indicate the positions of AUUUA sequences. The thicker vertical lines indicate two adjacent AUUUA sequences.
B. IL-8 3′UTR constructs were transiently expressed in HT1080 cells. Total RNA was isolated and reporter mRNA levels were measured using qPCR. Error bars indicate SEM of three independent experiments and the asterisk indicates a significant difference (p < 0.05).
C-E. Cells stably expressing EYFP, EYFP-IL-8401-1648, and EYFP-IL-81000-1100 were treated with LeTx (10-8 M PA and 10-9 M LF) for 1 h followed by addition of actinomycin D (1 μg/ml). RNA was then isolated at the indicated times and measured using qPCR. Error bars indicate SEM of three independent experiments.
To test if EYFP-IL-81000-1100 mRNA is responsive to LeTx-mediated destabilization, stable transfectants were treated with toxin and half-lives were assessed after the addition of actinomycin D (Fig. 2C). EYFP mRNA that lacks IL-8 sequences had a half-life of more than 200 min and LeTx had no effect on the stability of this transcript. Transcripts containing the EYFP coding region fused to the IL-8 3′ UTR (EYFP-IL-8401-1648) had a half-life of ∼130 min in untreated cells, and ∼95 min in LeTx-treated cells. Similarly, transcripts containing the EYFP coding region fused to nucleotides 1000-1100 of the IL-8 3′ UTR had a half-life of ∼74 min in untreated cells, and ∼42 min in LeTx-treated cells. These results indicate that LeTx causes destabilization of IL-8 mRNA through nucleotides 1000-1100 within the 3′ UTR.
Inhibition of MAPK pathways destabilizes IL-8 mRNA
LeTx downregulates the p38, ERK, and JNK MAPK pathways and we previously reported that disruption of each MAPK pathway using pharmacological inhibitors destabilizes IL-8 mRNA in HUVECs (Batty et al., 2006). Here, we wanted to assess the relative importance of the MAPK pathways on IL-8 mRNA stability in fibroblasts. HT1080 cells were treated with actinomycin D and either SB202190 (p38 inhibitor), SP600125 (JNK inhibitor), U0126 (MEK1/2 inhibitor), or a combination of all three inhibitors. There was no appreciable difference between the decay rates of IL-8 mRNA in cells treated with any one of the inhibitors individually compared to mock treatment, whereas co-treatment with all three inhibitors destabilized IL-8 mRNA by more than two-fold (Fig. 3A).
Figure 3.
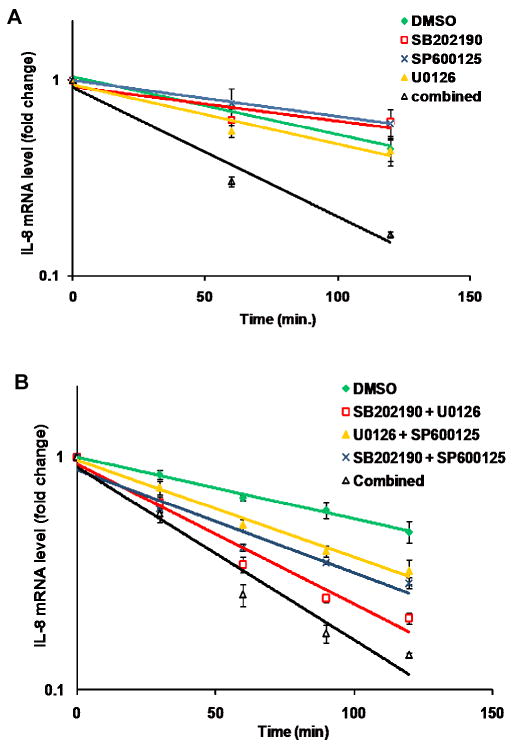
Decay analysis of IL-8 mRNA in response to pharmacological inhibitors.
A. HT1080 cells were treated with 1 μg/ml actinomycin D in combination with either DMSO, 10 μM SB202190, 20 μM SP600125, 10 μM U0126, or all three inhibitors. Total RNA was isolated at the indicated times and IL-8 transcript levels were assessed by qPCR. Error bars indicate SEM of three independent experiments.
B. HT1080 cells were treated with 1 μg/ml actinomycin D and either DMSO, pair-wise combinations of inhibitors, or all three inhibitors as indicated. Total RNA was isolated at the indicated times and IL-8 transcript levels assessed by qPCR. Error bars indicate SEM of three independent experiments.
We next tested pair-wise combinations of the three inhibitors and found that each combination accelerated IL-8 mRNA decay compared to mock treatment (Fig. 3B). Inhibition of the JNK pathway, in combination with either the ERK or the p38 pathways, was least potent at destabilizing IL-8 mRNA, accelerating the decay by ∼1.6 and ∼1.7-fold respectively compared to mock treatment. The combination of inhibiting the p38 and ERK pathway caused a ∼2.3 fold change, nearly as much as inhibiting all three inhibitors together, which caused a ∼2.8-fold change. These results suggest that LeTx-mediated destabilization of IL-8 mRNA is largely due to inhibition of the p38 and ERK pathways.
TTP is involved in LeTx-mediated IL-8 destabilization
Since TTP, TIAR, and KSRP have been demonstrated previously to bind IL-8 mRNA in vitro (Winzen et al., 2007; Suswam et al., 2005a; Suswam et al., 2005b), we assessed whether overexpression of these AUBPs would affect the level of IL-8 mRNA in HT1080 cells. FLAG-tagged forms of these proteins were over-expressed and IL-8 mRNA levels were quantified by qPCR (Fig. 4A, B). IL-8 mRNA expression was lowered in cells over-expressing TTP or TIAR, but not KSRP (Fig. 4A).
Figure 4.
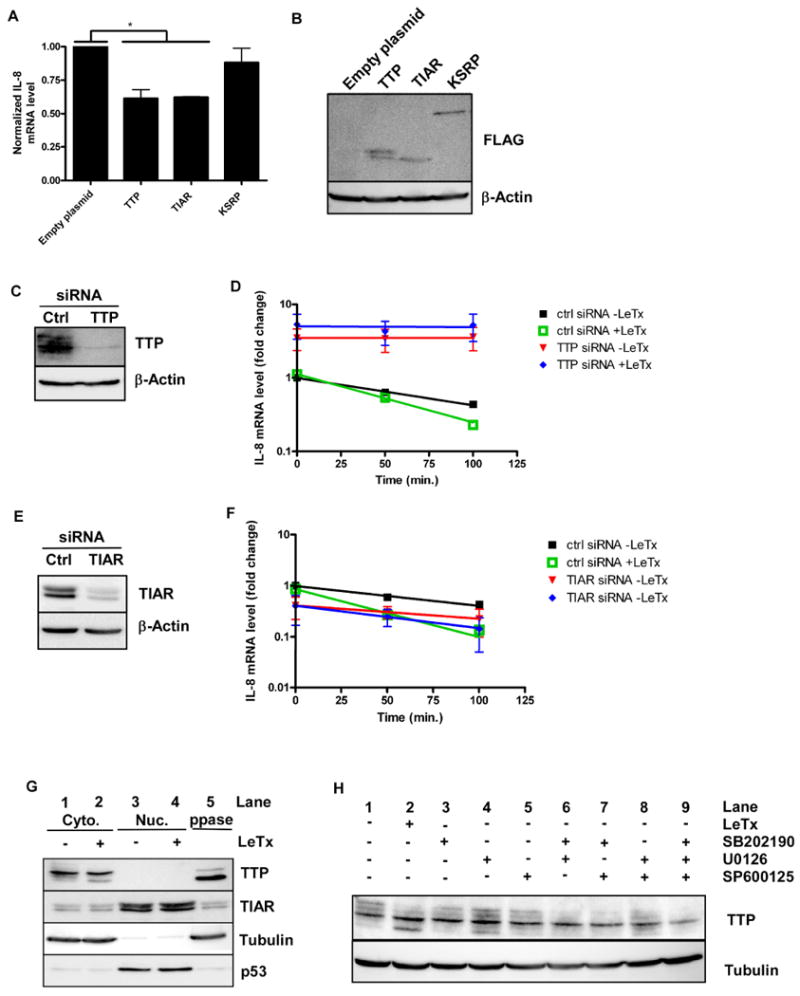
Involvement of AUBPs in IL-8 mRNA stability.
A. Indicated AUBPs were over-expressed in HT1080 cells and total RNA was isolated. Endogenous IL-8 mRNA was measured and normalized to β-actin mRNA levels. Error bars indicate SEM of three independent experiments and asterisks indicate significant differences (p < 0.05).
B. Cytoplasmic extracts were prepared from cells transfected as in (A) and AUBPs were detected by the FLAG-tag. Beta-actin protein levels were measured as a loading control. The blot is representative of three independent experiments.
C. Extracts from cells transfected with negative control siRNA or siRNA directed against TTP were prepared and immunoblotted for TTP. Beta-actin expression was used as loading control. The blot is representative of three independent experiments.
D. HT1080 cells from (C) were treated with 1 μg/ml actinomycin D in the absence or presence of LeTx. Total RNA was isolated at the indicated times and IL-8 transcript levels were assessed by qPCR. Error bars indicate SEM of three independent experiments.
E. Extracts from cells transfected with negative control siRNA or siRNA directed against TIAR were prepared and immunoblotted for TIAR. Beta-actin expression was used as loading control. The blot is representative of three independent experiments.
F. HT1080 cells from (E) were treated with 1 μg/ml actinomycin D in the absence or presence of LeTx. Total RNA was isolated at the indicated times and IL-8 transcript levels were assessed by qPCR. Error bars indicate SEM of three independent experiments.
G. HT1080 cells were left untreated or were treated with LeTx for 2 h. Cytoplasmic and nuclear proteins were isolated and equivalent amounts of extract were subjected to Western blotting using antibodies against TTP, TIAR, tubulin, and p53. A sample of untreated cytoplasmic extract was treated with lambda protein phosphatase (PPase). The blot is representative of three independent experiments.
H. HT1080 cells were treated with LeTx or with indicated pharmacological inhibitors for 2 h. Cytoplasmic proteins were extracted and probed for TTP and tubulin. The blot is representative of seven independent experiments.
To ascertain whether TTP or TIAR is required for LeTx-mediated IL-8 transcript destabilization, RNA interference was used to downregulate TTP and TIAR levels. The TTP protein level in cells transfected with TTP siRNA was reduced to ∼7% of that detected in cells transfected with negative control siRNA (Fig. 4C). Knock-down of TTP increased the stability of IL-8 mRNA and this stability was not diminished by LeTx-treatment (Fig. 4D). In contrast, knock-down of TIAR to ∼10% of the control level increased the half-life of IL-8 mRNA from ∼81 min to ∼116 min, but did not prevent LeTx treatment from increasing the decay rate by 1.6 fold (Fig. 4 E, F). These results indicate that TTP, but not TIAR, mediates LeTx-stimulated IL-8 mRNA decay.
Intoxication causes dephosphorylation of TTP
The possibility that LeTx alters the endogenous expression level or localization of TTP and TIAR was also addressed. Cytoplasmic and nuclear fractions were prepared and probed for tubulin (a cytoplasmic marker) and p53 (a nuclear marker) (Fig. 4G). Most of the TIAR protein was found in the nuclear fraction in untreated cells (compare lane 1 and 3); toxin treatment did not alter its expression level or localization. The doublet observed likely represents 2 isoforms that resulted from alternative splicing (Taupin et al., 1995).
We found that TTP localized predominantly to the cytoplasm in both untreated and intoxicated cells (Fig. 4G). Multiple bands corresponding to TTP were observed in both cell lysates, but the uppermost bands were not apparent in lysates prepared from intoxicated cells and the lower band was more prominent (Fig. 4G, compare lanes 1 and 2). That these bands represented differentially phosphorylated forms of TTP was shown by incubating untreated cell lysates with Lambda protein phosphatase (lane 5), which led to the loss of the upper forms and an increase in the amount of the lower form. Thus, LeTx activity induces the dephosphorylation of TTP.
Pharmacological inhibitors were used to determine which of the MAPK pathways contribute to TTP phosphorylation. Cells were treated for 2 h with LeTx or with pharmacological inhibitors before cytoplasmic proteins were prepared and subjected to Western blotting (Fig. 4H). More of the hypophosphorylated (lower) form of TTP was detected in lysates from cells treated with either LeTx or the MEK1/2 inhibitor (U0126) alone compared to lysates from untreated cells (compare lane 1 with lanes 2 and 4). Only a slight, yet reproducible decrease in the amount of the upper TTP band was observed upon treatment with the p38 or JNK inhibitors alone (compare lane 1 with lanes 3 and 5). Combinations of the inhibitors did not substantially increase the levels of hypophosphorylated TTP compared to the MEK1/2 inhibitor treatment, but the upper bands representing the hyperphosphorylated forms of TTP were clearly less prominent. Together, these results suggest that each of the three MAPK pathways contributes to the phosphorylation of TTP.
LeTx enhances TTP localization to P-bodies
P-bodies are dynamic cytoplasmic loci that are enriched in enzymes involved in 5′ to 3′ mRNA decay. Previous studies have shown that TTP can target ARE-containing transcripts to P-bodies (Fenger-Gron et al., 2005; Kedersha et al., 2005). This finding, together with our data demonstrating that overexpression of TTP lowered IL-8 mRNA levels and that LeTx activity caused dephosphorylation of TTP, led us to investigate the effects of LeTx on P-body formation and TTP localization. We first examined the effects of LeTx on the assembly of P-bodies by subjecting untreated or intoxicated HT1080 cells to immunofluorescence analysis. The helicase DDX6 (p54/RCK) was used as a marker to identify P-bodies (red) and Hoescht dye was used for nuclear staining. The absence (Fig. 5A) or the presence (Fig. 5B) of P-bodies can easily be distinguished using this marker. P-bodies were observed in ∼26% of untreated cells and ∼52% of intoxicated cells (Fig. 5C). Similarly, cells treated with a combination of pharmacological inhibitors against p38, MEK1/2, and JNK also exhibited a significant increase in P-body formation (data not shown). These foci corresponded to P-bodies and not stress granules as they did not colocalize with the stress granule marker TIA-1 (data not shown). We next compared TTP localization in untreated and toxin-treated HT1080 cells and since we could not visualize endogenous TTP in these cells by immunohistochemistry, FLAG-tagged TTP was transiently transfected and cells were stained with anti-FLAG and anti-DDX6 antibodies (Fig. 5D-I). In some cells, FLAG-TTP was diffusely distributed in the cytoplasm (Fig. 5D), whereas in others it concentrated at cytoplasmic foci that always co-localized with the P-body marker DDX6 (Fig. 5H - I). FLAG-TTP colocalized with P-bodies in ∼3% of untreated cells and in ∼15% of LeTx-treated cells (Fig. 5J).
Figure 5.
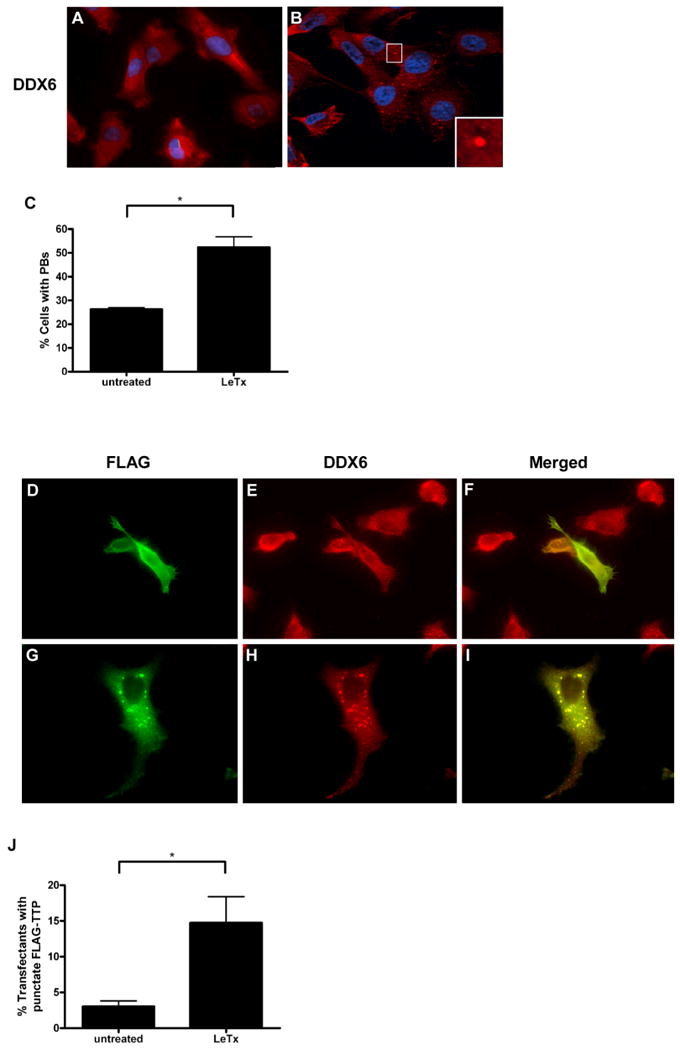
Effect of LeTx on P-body formation and TTP localization.
A-C. HT1080 cells were left untreated or treated with LeTx for 1 h. DDX6 was used to visualize formation of P-bodies. Representative immunofluorescence micrographs of HT1080 cells exhibiting diffuse (A) or punctate (B) staining of endogenous DDX6 (red). Hoechst dye (blue) was used for nuclear staining. A P-body at higher magnification is shown in the insert. The fraction of cells exhibiting P-bodies from untreated or intoxicated cells was quantified from a minimum of 100 cells per sample (C). Values indicate mean and SEM of three independent experiments. Asterisk indicates significant difference (P < 0.05).
D-J. HT1080 cells transiently transfected with FLAG-TTP were left untreated or were treated with LeTx for 1 h. Representative immunofluorescence micrographs show cells exhibiting diffuse (D) or punctate (G) staining of FLAG-TTP. Localization of FLAG-TTP to P-bodies was examined by co-staining with DDX6 (E, H). Merged images are shown (F, I). The fraction of cells exhibiting punctate FLAG-TTP staining was quantified (J). Values indicate mean and SEM of three independent experiments. Asterisk indicates significant difference (P < 0.05).
We next wanted to examine the effect of LeTx on TTP localization in HeLa cells, which constitutively display P-bodies and are used by numerous groups to study mRNA decay (Franks and Lykke-Andersen, 2007; Fenger-Gron et al., 2005; Stoecklin et al., 2004). HeLa cells were transfected with FLAG-TTP and stained for FLAG and DDX6. In contrast to HT1080 cells, P-bodies were visible in almost all of the HeLa cells, and treatment with LeTx did not affect P-body size or number in these cells. Diffuse staining of FLAG-TTP (Fig 6A) that does not colocalize with P-bodies (Fig. 6B and C) is largely seen in untreated cells. A majority of cells treated with LeTx exhibited punctate staining of FLAG-TTP in the cytoplasm (Fig. 6D) that colocalized to P-bodies (Fig. 6E and F). FLAG-TTP accumulated at P-bodies in ∼22% of the untreated cells and in ∼54% of the toxin-treated cells (Fig. 6G). These data suggest, therefore, that LeTx causes TTP to be recruited to P-bodies in both HT1080 and HeLa cells.
Figure 6.
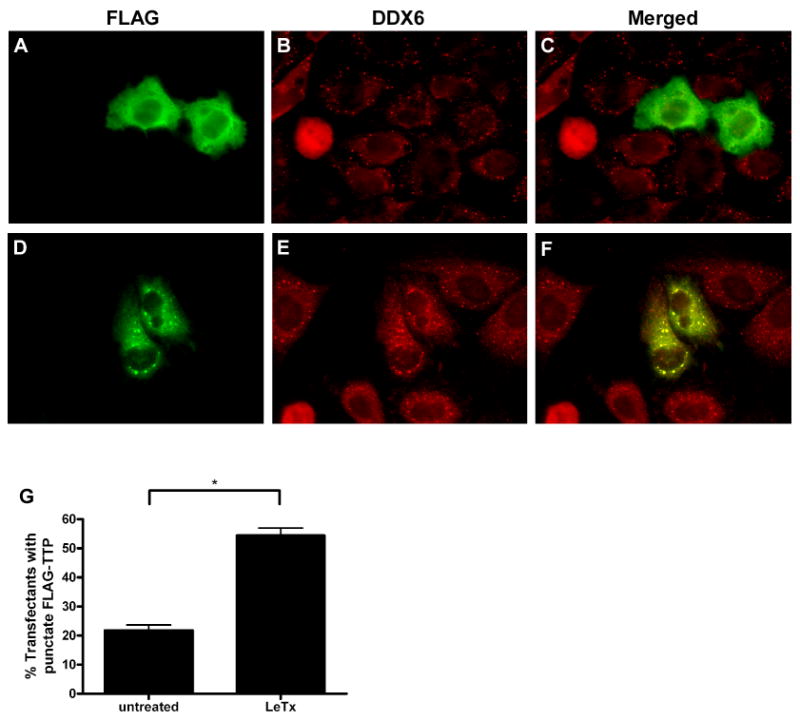
LeTx increases recruitment of TTP to P-bodies in HeLa cells.
HeLa cells transiently transfected with FLAG-TTP were left untreated or were treated with LeTx for 1 h. Representative immunofluorescence micrographs show cells exhibiting diffuse (A) or punctate (D) staining of FLAG-TTP. Localization of FLAG-TTP to P-bodies was examined by co-staining with DDX6 (B, E). Merged images are shown (C, F). The fraction of cells exhibiting punctate FLAG-TTP staining was quantified (G). Values indicate mean and SEM of three independent experiments. Asterisk indicates significant difference (P < 0.05).
Discussion
Interfering with host gene expression is an effective means for a bacterial pathogen to evade the immune response. Not surprisingly then, bacteria and their toxins have developed various ways to downregulate gene expression. Anthrax lethal toxin downregulates expression of the neutrophil attractant IL-8 both transcriptionally and post-transcriptionally. A recent study demonstrated that through the inhibition of histone phosphorylation, LeTx decreased chromatin accessibility to NF-κB, leading to lowered IL-8 transcription. This group further implicated this mechanism in reducing neutrophil recruitment during a B. anthracis infection (Raymond et al., 2009). We previously demonstrated that LeTx post-transcriptionally regulates IL-8 expression by increasing the rate of IL-8 transcript decay (Batty et al., 2006). In the current study, we have characterized cis-acting and trans-acting elements involved in this process.
The element in the IL-8 transcript that confers sensitivity to LeTx is confined to the 3′ UTR. This region, encompassing nucleotides 1000-1100, has an AU content of 82% and contains 4 AUUUA motifs; this ARE has been identified previously as a potent destabilization element (Winzen et al., 2004). Surprisingly, the EYFP-IL-81000-1100 reporter transcript had a shorter half-life than that of the EYFP-IL-8401-1648 transcript. Thus, there may be a stabilizing element in the 3′ UTR located outside of this ARE, or alternatively, the distance between the stop codon and destabilizing element might affect the efficiency of decay. Inhibition of the MAPK pathways using pharmacological inhibitors was found to have similar destabilizing effects as LeTx on EYFP-IL-81000-1100 mRNA (data not shown), suggesting that it is the inactivation of these pathways by LeTx that causes IL-8 mRNA decay.
Investigation into the possible involvement of the ARE-binding proteins TTP, TIAR, and KSRP in LeTx-mediated IL-8 mRNA destabilization was motivated by past studies that demonstrated their participation in IL-8 mRNA decay (Suswam et al., 2008; Winzen et al., 2007; Suswam et al., 2005b). We found that overexpression of KSRP did not alter the level of IL-8 mRNA in HT1080 cells, whereas overexpression of either TTP or TIAR lowered the level of IL-8 mRNA. When TIAR expression was knocked-down, the half-life of IL-8 mRNA increased from ∼81 min in control cells to ∼116 minutes. However, as was observed in control cells, LeTx accelerated IL-8 mRNA decay by 1.6-fold in TIAR knock-down cells. This observation suggests that while TIAR influences IL-8 transcript stability, its activity is not regulated by LeTx or by the MAPK pathways. Not surprisingly then, treatment with LeTx did not cause the redistribution of TIAR from the nucleus to the cytoplasm or affect its expression level.
Treatment of cells with LeTx decreased the level of phosphorylation of TTP. Knocking down TTP using siRNA caused increased stability of IL-8 mRNA – no appreciable decay was measured in these cells and importantly, LeTx-treatment did not destabilize the transcript (Fig. 4D). These results correlated with the observation that IL-8 mRNA decay increased upon pharmacological inhibition of p38, ERK and JNK, and that these inhibitors also caused dephosphorylation of TTP. Past studies identified TTP as a substrate of the p38/MAPKAP2 pathway (Chrestensen et al., 2004; Stoecklin et al., 2004), although inhibition of MEK1/2 was found to have minimal effect on TTP phosphorylation (Suswam et al., 2008), suggesting that the involvement of the ERK pathway may differ between cell types. Phosphorylation of TTP through the JNK pathway has not been reported previously.
TTP activity is regulated by phosphoryation in several ways. It has been reported that phosphorylated TTP has a lower affinity for mRNA, which would reduce its ability to mediate transcript degradation (Carballo et al., 2001). Furthermore, phosphorylated TTP binds 14-3-3, a ubiquitously expressed phospho-serine and phospho-threonine binding protein that exerts a variety of effects on its binding partners (Bridges and Moorhead, 2005). The binding of 14-3-3 to TTP affects its localization and potentially its ability to interact with components of the mRNA degradation machinery.
We found that hypophosphorylation of TTP in intoxicated cells was associated with an increase in the number of cells exhibiting P-bodies and an increase in the localization of TTP to P-bodies. P-bodies are cytoplasmic granules that contain translationally repressed mRNA. The protein composition of P bodies has not been fully defined, but the known components include decapping enzymes, activators of decapping enzymes, and exonucleases (Parker and Sheth, 2007; Eystathioy et al., 2002). P-bodies are dynamic structures that vary in size and number depending on the availability of nontranslating mRNA pools. TTP can nucleate the formation of P-bodies or deliver ARE-containing transcripts to pre-existing P-bodies for storage or degradation. Our work suggests that LeTx might affect mRNA turnover by altering the number and mRNA content of P-bodies.
Experimental procedures
Cell culture
The human cell lines HT1080 and HeLa were maintained under an atmostphere of 5% CO2 at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin (10,000 IU) and streptomycin (10,000 μg/ml) (Wisent Inc.). Stable cell lines were generated using the Flp-In System following manufacturer's protocol (Invitrogen). For decay assays, cells were seeded at 7.2 × 105 cells per well in a 6-well dish and treated the next day as indicated. For transfections, cells were seeded at 2.0 × 106 per 10 cm dish and transfected the next day with SuperFect (Qiagen). RNA extraction was performed 16 h post transfection.
Plasmids
pcDNA3-FLAG-TTP and pcDNA3-FLAG-HuR were described previously (Lykke-Andersen and Wagner, 2005). The coding region of TIAR was amplified from HT1080 cDNA using primers 5′-GCGGAATTCATGATGGAAGACGACGGG-3′ and 5′-GCGGCGGCCGCTCACTGTGTTTGGTAACTTG-3′. The coding region of HuR in pcDNA3-FLAG-HuR was excised using EcoRI and NotI and the coding region of TIAR was inserted into these sites to create pcDNA3-FLAG-TIAR.
The coding region of KSRP was excised with EcoRI and XhoI from pOTB7-KSRP (ATCC) and inserted into pcDNA3.1. Nucleotides between the EcoRI site and the ATG start codon of KSRP were excised by digestion with EcoRI and SgrAI and replaced with annealed oligonucleotides 5′-AATTCATGTCGGACTACAGCACGGGAGGACCCCCGCCCGGGCCGCCGCCGCCCG-3′ and 5′-CCGGCGGGCGGCGGCGGCCCGGGCGGGGGTCCTCCCGTGCTGTAGTCCGACATG-3′. Then, EcoRI and XbaI was used to excise the KSRP cDNA and inserted into these sites in pcDNA3-FLAG.
A GFP tag for qPCR detection was cloned between the BamHI and EcoRI restriction sites of the MCS of pcDNA 3.1 (+) (Invitrogen) by insertion of an oligonucleotide created by annealing 5′-GATCCAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCGTGACCGCCGCCG-3′ and 5′-AATTCGGCGGCGGTCACGAACTCCAGCAGGACCATGTGATCGCGCTTCTCGTTGGGGTCTTTCTG-3′. The genomic sequence and cDNA sequence of IL-8 was then cloned 3′ of the GFP tag using the NotI and XbaI restriction sites. The IL-8 genomic sequence was amplified from Human Genomic DNA (roche) with primers 5′- CGC GCG GCC GCC TCC ATA AGG CAC AAA CTT TC-3′ and 5′-CGC TCT AGA TTG ACA ACA AAT TAT ATT TTA AAT G-3′. The IL-8 cDNA sequence was amplified from HUVEC cDNA using the same primers. The IL-8 3′UTR was cloned into pd2EYFP-N1 (Clontech) as described previously (Batty et al., 2006) to make EYFP-401-1648. This construct was then used as template to amplify for all subsequent truncations of IL-8 3′UTR, which were then digested with NotI and XbaI and ligated into pd2EYFP-N1. Primers 5′-CGCGCGGCCGCTAAAAAAATTCATTCTCTGTGG-3′ and 5′-GCGTCTAGAACAACAGACCCACACAATAC-3′ was used to amplify nucleotides 401-500. Primers 5′-CGCGCGGCCGCTAAAAAAATTCATTCTCTGTGG-3′ and 5′-GCGTCTAGATCCCATCATTTTTATGTGATG-3′ was used to amplify nucleotides 401-800. Primers 5′- GCGGCGGCCGCACA ATAAATTTTGCCATAAAGTCA-3′ and 5′-GCGTCTAGAAAAGTGCTTCCACATGTC C-3′ was used to amplify nucleotides 800-1000. Primers 5′-GCGGCGGCCGCTAAGTTTTTTCATCATAACATAAATT-3′ and 5′-GCGTCTAGAAAATTCTTGCACAAATATTTGATG-3′ was used to amplify nucleotides 1000-1100. Primers 5′-GCGGCGGCCGCCATCAAATATTT GTG CAA GAA TT-3′ and 5′-CGCTCTAGATTGACAACAAATTATATTTTAAATG-3′ was used to amplify nucleotides 1077-1648. Cloning into the pcDNA5/FRT expression vector for creation of stable cell lines is as follows: the pd2EYFP-N1 vectors were used as templates and amplified products were digested with either NotI and XhoI (EYFP), or XhoI and ApaI (EYFP-401-1648 and EYFP-1000-1100) and ligated into pcDNA5/FRT. Primers 5′- GCG GCG GCC GC GCC ACC ATG GTG AGC AAG-3′ and 5′- GCGCTCGAGCTACAC ATTGATCCTAGCAG-3′ were used to amplify the EYFP gene from the pd2EYFP vector. Primers 5′-GCGCTCGAGCGCCACCATGGTGAGCAAGG-3′ and 5′-GCGGGGCCCTTGACAACAAATTATATTTTAAATGTTTC-3′ was used to amplify EYFP-401-1648 from the EYFP-401-1648 vector. Primers 5′-GCGCTCGAGCGCCACCATGGTGAGCAAGG-3′ and 5′-GCGGGGCCCAAATTCTTGCACAAATATTTGATG C-3′ was used to amplify EYFP-1000-1100 from the EYFP-1000-1100 vector.
Western blotting
Cytoplasmic and nuclear lysates were prepared with the NE-PER Nuclear and Cytoplasmic Extract Reagents (Pierce) according to manufacturer's protocol. Protein concentration was determined with a protein assay reagent (Bio-Rad). SDS-polyacrylamide gel electrophoresis was performed with 10% polyacrylamide gel and transferred onto nitrocellulose blotting membrane (Pall). Membranes were blocked in 0.1% Tween-20 TBS containing 5% skim milk powder and probed with the primary antibody for 1 h at room temperature or overnight at 4°C. Dilutions of primary antibodies were used as follows: 1:5000 TTP (Abcam), 1:300 TIAR (Santa Cruz), 1:1000 tubulin (Sigma), 1:1000 p53 (Calbiochem).
Expression and purification of PA and LF
Lethal factor was purified as previously described (Kassam et al., 2005). PA was purified as previously described (Miller et al., 1999). Endotoxin was removed from purified LF and PA using Detoxi-Gel AffinityPak columns (Pierce). Endotoxin contamination was assessed using Limulus Amoebocyte Lysate (Cambrex) with a detection limit of less than 0.03 endotoxin units ml-1.
RNA purification and quantification
Total RNA was isolated and quantified as previously described (Batty et al., 2006). Briefly, total RNA was isolated using Rneasy Mini kit (Qiagen) and treated with Dnase (RNase-free Dnase kit, Qiagen). RNA was reverse transcribed using SuperScript II Reverse Transcriptase (Invitrogen) and qPCR was performed using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems). To detect IL-8, the following primers were used: 5′-AATCTGGCAACCCTAGTTGCTA-3′ and 5′-AAACCAAGGCACAGTGGAACA-3′. Primers 5′-AGCAAAGACCCCAACGAGAAG-3′ and 5′-GGCGGCGGTCACGAA-3′ were used to detect EYFP mRNA levels. Primers 5′-AAAGCCACCCCACTTCTCTCTAA-3′ and 5′- ACCTCCCCTGTGTGGACTTG-3′ were used to detect β-actin mRNA levels. To determine mRNA fold change in decay assays, a previously described mathematical model was employed (Pfaffl, 2001). Values were normalized to β -actin levels and expressed relative to untreated samples at 0 min.
siRNA transfection
HT1080 cells were transfected using Lipofectamine RNAiMAX (Invitrogen). The negative control siRNA was obtained from Ambion and siRNA directed against TTP and TIAR with dTdT 3′ overhangs were from Dharmacon Research. For TTP knock-down, cells were transfected with 17 nM of the siRNA duplex with the sense sequence 5′ -CGCUGCCACUUCAUCCACAAC-3′, which can also target Tis11B and Tis11D. IL-8 mRNA decay was then assayed 24 h post-transfection. For TIAR knock-down, 167 nM of siRNA with the sense sequence 5′ –AAGGGCUAUUCAUUUGUCAGA-3′ was used for each transfection. Cells were re-transfected 2 days after the first transfection and IL-8 mRNA decay assayed 24 h later. The cell density on the day of siRNA transfection was ∼30% confluence.
Fluorescence microscopy
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton-X. Blocking was done in 5% BSA for 30 min, and then in Image-iT® FX signal enhancer (Invitrogen) for 30 min. This treatment was followed by 1 h incubation with primary antibodies which were diluted in the blocking agent as follows: 1:1000 DDX6 (Bethyl) and 1:1000 FLAG (Sigma). Then, cells were incubated for 45 min with 1:800 Alexa Fluor® 488 or Alexa Fluor® 594 (Invitrogen). Cells were then incubated with 1:1000 Hoescht dye if applicable and then mounted on Mowiol solution. For quantification of P-bodies or colocalization of FLAG-TTP with P-bodies, at least 100 cells were analyzed from three or more independent experiments. Conventional fluorescence microscopy was performed on Zeiss Axioplan 2 and the images compiled using AxioVision LE software.
Acknowledgments
We thank Jens Lykke-Andersen for reagents. This research was supported by NIH grant RO1 AI067683. J.M. holds the Canada Research Chair in Bacterial Pathogenesis.
References
- Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, Pulendran B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature. 2003;424:329–334. doi: 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- Bakheet T, Williams BRG, Khabar KSA. ARED 3.0: the large and diverse AU-rich transcriptome. Nucl Acids Res. 2006;34:D111–114. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty S, Chow EMC, Kassam A, Der SD, Mogridge J. Inhibition of mitogen-activated protein kinase signalling by Bacillus anthracis lethal toxin causes destabilization of interleukin-8 mRNA. Cellular Microbiology. 2006;8:130–138. doi: 10.1111/j.1462-5822.2005.00606.x. [DOI] [PubMed] [Google Scholar]
- Briata P, Forcales SV, Ponassi M, Corte G, Chen CY, Karin M, et al. p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol Cell. 2005;20:891–903. doi: 10.1016/j.molcel.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Bridges D, Moorhead GB. 14-3-3 proteins: a number of functions for a numbered protein. Sci STKE. 2005;2005:re10. doi: 10.1126/stke.2962005re10. [DOI] [PubMed] [Google Scholar]
- Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem. 2001;276:42580–42587. doi: 10.1074/jbc.M104953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Chen CY, Xu N, Shyu AB. Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol Cell Biol. 2002;22:7268–7278. doi: 10.1128/MCB.22.20.7268-7278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Gherzi R, Andersen JS, Gaietta G, Jurchott K, Royer HD, et al. Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation. Genes Dev. 2000;14:1236–1248. [PMC free article] [PubMed] [Google Scholar]
- Chou CF, Mulky A, Maitra S, Lin WJ, Gherzi R, Kappes J, Chen CY. Tethering KSRP, a Decay-Promoting AU-Rich Element-Binding Protein, to mRNAs Elicits mRNA Decay. Mol Cell Biol. 2006;26:3695–3706. doi: 10.1128/MCB.26.10.3695-3706.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. MAPKAP Kinase 2 Phosphorylates Tristetraprolin on in Vivo Sites Including Ser178, a Site Required for 14-3-3 Binding. J Biol Chem. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- Erwin JL, DaSilva LM, Bavari S, Little SF, Friedlander AM, Chanh TC. Macrophage-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect Immun. 2001;69:1175–1177. doi: 10.1128/IAI.69.2.1175-1177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy T, Chan EKL, Tenenbaum SA, Keene JD, Griffith K, Fritzler MJ. A Phosphorylated Cytoplasmic Autoantigen, GW182, Associates with a Unique Population of Human mRNAs within Novel Cytoplasmic Speckles. Mol Biol Cell. 2002;13:1338–1351. doi: 10.1091/mbc.01-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Cordoba-Rodriguez R, Lankford CSR, Frucht DM. Anthrax Lethal Toxin Blocks MAPK Kinase-Dependent IL-2 Production in CD4+ T Cells. J Immunol. 2005;174:4966–4971. doi: 10.4049/jimmunol.174.8.4966. [DOI] [PubMed] [Google Scholar]
- Fenger-Gron M, Fillman C, Norrild B, Lykke-Andersen J. Multiple Processing Body Factors and the ARE Binding Protein TTP Activate mRNA Decapping. Molecular Cell. 2005;20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- Franks TM, Lykke-Andersen J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 2007;21:719–735. doi: 10.1101/gad.1494707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- Gherzi R, Lee KY, Briata P, Wegmuller D, Moroni C, Karin M, Chen CY. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Janeway C. Immunobiology : the immune system in health and disease. New York: Garland Pub.; 2001. [Google Scholar]
- Kassam A, Der SD, Mogridge J. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell Microbiol. 2005;7:281–292. doi: 10.1111/j.1462-5822.2004.00458.x. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fitzler MJ, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. Interactions of CCCH Zinc Finger Proteins with mRNA. BINDING OF TRISTETRAPROLIN-RELATED ZINC FINGER PROTEINS TO AU-RICH ELEMENTS AND DESTABILIZATION OF mRNA. J Biol Chem. 2000;275:17827–17837. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- Linker K, Pautz A, Fechir M, Hubrich T, Greeve J, Kleinert H. Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucl Acids Res. 2005;33:4813–4827. doi: 10.1093/nar/gki797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, Wagner E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005;19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D'Elios MM, et al. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J Exp Med. 2005;201:325–331. doi: 10.1084/jem.20041557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U. P Bodies and the Control of mRNA Translation and Degradation. Molecular Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999;462:199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond B, Batsche E, Boutillon F, Wu YZ, Leduc D, Balloy V, et al. Anthrax Lethal Toxin Impairs IL-8 Expression in Epithelial Cells through Inhibition of Histone H3 Modification. PLoS Pathog. 2009;5:e1000359. doi: 10.1371/journal.ppat.1000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot A. Endothelial cell binding of NAP-1/IL-8: role in neutrophil emigration. Immunol Today. 1992;13:291–294. doi: 10.1016/0167-5699(92)90039-A. [DOI] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and Decay of Messenger RNA Occur in Cytoplasmic Processing Bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. Embo J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suswam E, Li Y, Zhang X, Gillespie GY, Li X, Shacka JJ, et al. Tristetraprolin Down-regulates Interleukin-8 and Vascular Endothelial Growth Factor in Malignant Glioma Cells. Cancer Res. 2008;68:674–682. doi: 10.1158/0008-5472.CAN-07-2751. [DOI] [PubMed] [Google Scholar]
- Suswam EA, Li YY, Mahtani H, King PH. Novel DNA-binding properties of the RNA-binding protein TIAR. Nucl Acids Res. 2005a;33:4507–4518. doi: 10.1093/nar/gki763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suswam EA, Nabors LB, Huang Y, Yang X, King PH. IL-1beta induces stabilization of IL-8 mRNA in malignant breast cancer cells via the 3′ untranslated region: Involvement of divergent RNA-binding factors HuR, KSRP and TIAR. Int J Cancer. 2005b;113:911–919. doi: 10.1002/ijc.20675. [DOI] [PubMed] [Google Scholar]
- Taupin JL, Tian Q, Kedersha N, Robertson M, Anderson P. The RNA-binding protein TIAR is translocated from the nucleus to the cytoplasm during Fas-mediated apoptotic cell death. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1629–1633. doi: 10.1073/pnas.92.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352(Pt 3):739–745. [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wormington M, Peltz SW. The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol. 2001;2:237–246. doi: 10.1038/35067025. [DOI] [PubMed] [Google Scholar]
- Winzen R, Gowrishankar G, Bollig F, Redich N, Resch K, Holtmann H. Distinct domains of AU-rich elements exert different functions in mRNA destabilization and stabilization by p38 mitogen-activated protein kinase or HuR. Mol Cell Biol. 2004;24:4835–4847. doi: 10.1128/MCB.24.11.4835-4847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzen R, Thakur BK, Dittrich-Breiholz O, Shah M, Redich N, Dhamija S, et al. Functional Analysis of KSRP Interaction with the AU-Rich Element of Interleukin-8 and Identification of Inflammatory mRNA Targets. Mol Cell Biol. 2007;27:8388–8400. doi: 10.1128/MCB.01493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Hamilton T. Introns Regulate the Rate of Unstable mRNA Decay. J Biol Chem. 2007;282:20230–20237. doi: 10.1074/jbc.M700180200. [DOI] [PubMed] [Google Scholar]