

Abstract
Motivation: Functional similarity between proteins is evident at both the sequence and structure levels. SeSAW is a web-based program for identifying functionally or evolutionarily conserved motifs in protein structures by locating sequence and structural similarities, and quantifying these at the level of individual residues. Results can be visualized in 2D, as annotated alignments, or in 3D, as structural superpositions. An example is given for both an experimentally determined query structure and a homology model.
Availability and Implementation: The web server is located at http://www.pdbj.org/SeSAW/
Contact: standley@ifrec.osaka-u.ac.jp
1 INTRODUCTION
Sequence alignment and structural alignment are widely used techniques for inferring functional or evolutionary relationships between proteins. However, most alignment methods do not integrate sequence and structural information into one measure of similarity or describe the similarity at the level of individual residues. We recently introduced a sequence and structure-based scoring method that employs sequence profile–profile comparisons, but is anchored by structural alignments and showed that the functional information associated with the top-scoring hits found by the method agreed well with expert annotations published in the literature (Standley et al., 2008b). Subsequently, we have shown that this approach can be used to identify functional sites in remote (e.g.10–20% sequence identity) homology models, even when the structural template used to build the model is itself un-annotated (Standley et al., 2008a). That is, a structure without a known function (e.g. a structural genomics target) can be used as an intermediate template to subsequently locate a functionally characterized structure, and thus map putative functional sites onto a distantly related query sequence. Here, we describe a web-based implementation of the method called SeSAW (sequence-derived structural alignment weights) that can automatically perform putative functional residue mapping. We emphasize that such mapping is intended to guide subsequent experiments rather than to serve as a substitute for experimental annotations.
2 ALGORITHM
SeSAW takes as input a PDB-formatted query file, chain ID, and, in the case of a template-based model, the PDB ID and chain ID of the template. As illustrated in Figure 1, A PSI-BLAST position specific scoring matrix (PSSM) for the query is retrieved or computed, as necessary (we maintain a database of PSSMs for every unique PDB chain). The query is partitioned into unique structural domains using SCOP (Murzin et al., 1995), CATH (Pearl et al., 2005) and Protein Domain Parser (Alexandrov and Shindyalov, 2003). For each domain, SeSAW attempts to construct a list of representative structure neighbors by mapping from a pre-computed list of pairwise structural alignments using PSI-BLAST; if this fails, SeSAW performs direct structural alignment on the representative list using ASH (Standley et al., 2007). The representative neighbors are then expanded to include their sequence homologs. The resulting hits are structurally aligned to the query and ranked by the SeSAW score. The score is given by adding the ASH structure alignment score to the sum of a per-residue similarity score:
| (1) |
where per-residue similarity score Si is defined as:
| (2) |
Here, d is the distance between Cα atoms in the two aligned residues (after superposition of the query and template), dmax is a reference distance (4 Å used on all calculations), wB is a scalar weight (0.8 used in all calculations), SB is the 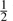 bit Blosum62 matrix, aQ and aT are the amino acid types of the query and template, respectively, wP is a scalar weight (1.5 used in all calculations), and ST and SQ are the odds column vectors of the query and template PSSMs, respectively. The SeSAW score is reported, along with a P-value computed by numerically integrating the known distribution of scores. Functional annotations, extracted regularly at the Protein Data Bank Japan, are then mapped onto the query–template alignment.
bit Blosum62 matrix, aQ and aT are the amino acid types of the query and template, respectively, wP is a scalar weight (1.5 used in all calculations), and ST and SQ are the odds column vectors of the query and template PSSMs, respectively. The SeSAW score is reported, along with a P-value computed by numerically integrating the known distribution of scores. Functional annotations, extracted regularly at the Protein Data Bank Japan, are then mapped onto the query–template alignment.
Fig. 1.

Outline of the server. The rectangles on the left indicate major steps that are performed in real-time. Those on the right indicate steps that are done offline. Ovals in the center represent external software used for both types of calculations. Colored lines indicate their interconnection.
3 VISUALIZATION
Query–template alignments, with residue-level functional descriptions, when available, are displayed with Jalview (Waterhouse et al., 2009). Superpositions can be downloaded or visualized in 3D with an interactive table of residues pairs that score highly according to the per-residue similarity score.
4 EXAMPLES
The SeSAW method was used to find templates related to the hypothetical protein TTHA1568 from Thermus thermophilus HB8 (PDB identifier 2czlA), a structural genomics target with unknown function (Standley et al., 2008b). The biochemical function of TTHA1568 has subsequently been determined (Hiratsuka et al., 2008). In our original work, while we were unable to pinpoint the exact biochemical function, our analysis indicated a likely active site near residues S57, T105 and T106, as well as a highly significant glycine (G82) that we proposed would act as a hinge, allowing substrate access. These predictions are supported by recent experimental evidence (Arai et al., 2009). This result is significant since the closest sequence homolog with known function at the time of our prediction, a glutamate transport protein, had a sequence identity of only 15%.
The second example illustrates the use of SeSAW in annotating a homology model. Zc3h12a from Mus musculus is a protein that was found to be required for mRNA stability of inflammatory cytokines. Because of the very low sequence homology to known folds, a number of models were built and submitted to SeSAW, and the model with the highest raw score retained for further analysis. This model was built on a structural genomics target of unknown function (PDB ID 2qipA). The top two SeSAW hits to this model were to a Mg-dependent hydrolase (2ho4B) and a Mg-dependent phosphatase (1k1e). From these hits, a cluster of conserved aspartic acids that bind Mg could be identified in the query. The second highest hit was to the nuclease domain of the Taq DNA polymerase (1tauA). These three hits are consistent with a possible Mg-dependent ribonuclease function, and this prediction was subsequently demonstrated in vitro and in vivo; moreover, when we mutated one of the predicted Mg-binding aspartic acids to asperagine, the nuclease activity was abolished, confirming the predicted active site location (Matsushita et al., 2009).
These two examples are typical of SeSAW results when only very low sequence homologs exist. More recently, SeSAW was used to correctly identify the dual (Ser/Thr or Tyr) specificity of the kinase ROP16 from Toxoplasma gondii using a more modest (20%) homology model, while sequence analysis alone indicated greater similarity to Ser/Thr kinases (Yamamoto et al., 2009). The structural alignment step allows very distantly related templates to be recognized, while the use of profile–profile sequence comparison highlights the residue pairs that are mutually conserved. However, not all such residues are expected to be part of the active site; structurally important amino acids such as proline and some large hydrophobic groups often score highly as well. Another limitation of SeSAW is that, in difficult cases such as these, the exact biochemical function is not automatically revealed, although residues that make up the active site can often be located. Prediction of the biochemical role of the protein requires some investigation and, ultimately, biochemical experimentation. Nevertheless, SeSAW is a significant improvement over running structural and sequence analysis separately (Standley et al., 2008b), and can thus play an important role in automated functional annotation of structural genomics targets or homology models.
ACKNOWLEDGEMENTS
The authors would like to thank A. Yoshihara for technical assistance.
Funding: This work was supported by a kakenhi grant 21570169: Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS).
Conflict of Interest: none declared.
REFERENCES
- Alexandrov N, Shindyalov I. PDP: protein domain parser. Bioinformatics. 2003;19:429–430. doi: 10.1093/bioinformatics/btg006. [DOI] [PubMed] [Google Scholar]
- Arai R, et al. Crystal structure of MqnD (TTHA1568), a menaquinone biosynthetic enzyme from Thermus thermophilus HB8. J. Struct. Biol. 2009;168:575–581. doi: 10.1016/j.jsb.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Hiratsuka T, et al. An alternative menaquinone biosynthetic pathway operating in microorganisms. Science. 2008;321:1670–1673. doi: 10.1126/science.1160446. [DOI] [PubMed] [Google Scholar]
- Matsushita K, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185–1190. doi: 10.1038/nature07924. [DOI] [PubMed] [Google Scholar]
- Murzin AG, et al. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Pearl F, et al. The CATH Domain Structure Database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucleic Acids Res. 2005;33:D247–D251. doi: 10.1093/nar/gki024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley DM, et al. ASH structure alignment package: sensitivity and selectivity in domain classification. BMC Bioinformatics. 2007;8:116. doi: 10.1186/1471-2105-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley DM, et al. Structure-based functional annotation of protein sequences guided by comparative models. In: Zhang XS, Chen L, Wu LY, Wang Y, editors. The Second International Symposium on Optimization and Systems Biology. China: Lijiang; 2008a. pp. 395–403. [Google Scholar]
- Standley DM, et al. Functional annotation by sequence-weighted structure alignments: statistical analysis and case studies from the Protein 3000 structural genomics project in Japan. Proteins. 2008b;72:1333–1351. doi: 10.1002/prot.22015. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, et al. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 2009;206:2747–2760. doi: 10.1084/jem.20091703. [DOI] [PMC free article] [PubMed] [Google Scholar]