

Abstract
Nutlins, the newly developed small molecule antagonists of MDM2, activate p53 and induce apoptosis in cancer cells, offering a novel strategy of chemotherapy. Recent studies have further suggested synergistic effects of nutlins with other chemotherapeutic drugs. However, it is unclear whether nutlins increase or decrease the side effects of these drugs in normal non-malignant cells or tissues. Cisplatin is a widely used chemotherapy drug, which has a major side effect of kidney injury. Here we show that Nutlin-3 protected kidney cells against cisplatin-induced apoptosis. The cytoprotective effects of Nutlin-3 were not related to its regulation of p53 or consequent gene expression during cisplatin treatment. Moreover, the protective effects were shown in MDM2-, MDM4-, or p53-deficient cells. On the other hand, Nutlin-3 suppressed mitochondrial events of apoptosis during cisplatin incubation, including Bax activation and cytochrome c release. Nutlin-3 attenuated cisplatin-induced oligomerization of Bax and Bak but not their interactions with Bcl-XL. In isolated mitochondria, Nutlin-3 inhibited cytochrome c release induced by Ca2+, Bim peptide, and recombinant tBid. Importantly, it blocked both Bax and Bak oligomerization under these conditions. Together, the results have uncovered a new pharmacological function of nutlins, i.e. suppression of Bax and Bak, two critical mediators of apoptosis.
Nutlins are cis-imidazoline compounds, which were recently identified to be the first potent and selective small molecule antagonists of MDM22 (1). Binding to the same site on MDM2 as p53, nutlins interfere with the molecular interaction between p53 and MDM2. This leads to the inhibition of MDM2-mediated p53 degradation via the proteasomal pathway, resulting in p53 accumulation and activation. The unique targeting ability of nutlins suggests the potential use of these compounds in chemotherapy for cancers with wild-type p53 (2, 3). The chemotherapeutic activity of nutlins has now been demonstrated by recent studies using in vitro and in vivo models (1, 4–13). Nutlins induce p53-dependent cell cycle arrest and apoptosis in cancer cells and suppress the growth of tumor xenografts in nude mice. Importantly, there is evidence that nutlins, while being toxic to cancer cells, do not induce cell death or apoptosis in normal non-malignant cells and tissues (1, 7, 11, 12).
Due to their unique targeting of p53-MDM2 interaction, nutlins have been tested recently for their synergistic effects with existing therapies. It has been shown that nutlins are synergistic with antimitotic agents, genotoxic drugs, and radiation in cancer therapy (4–6, 9, 11, 14). These preclinical studies have further supported the potential use of nutlins in combinational therapy with existing drugs or treatments. Nevertheless, it is unclear whether nutlins may increase or decrease the side effects of the therapies in normal tissues or organs. In this area, a recent study has suggested the possibility of using nutlins to activate temporary cell cycle arrest in normal tissues to protect against the side effects during chemotherapy with mitotic inhibitors such as paclitaxel (14).
Major side effects of chemotherapy are frequently shown in the kidneys and renal tissues, which are the sites for filtration, concentration, and excretion of the drugs. For example, cisplatin, a widely used chemotherapy drug (15–18), induces nephrotoxicity and acute renal failure (19, 20). Although the mechanism underlying cisplatin nephrotoxicity remains unclear, we and others have recently suggested the involvement of p53 signaling (21–24). p53 is phosphorylated and induced by cisplatin in renal tubular cells. Moreover, inhibition of p53 with the pharmacological inhibitor pifithrin-α or a dominant-negative p53 mutant attenuates kidney cell apoptosis during cisplatin treatment (23). p53 may induce apoptosis in these cells by upregulation of apoptotic genes, including PUMA-α (22).
This study sought to: 1) determine the regulation of p53 signaling by nutlins in kidney tubular cells; 2) determine whether nutlins ameliorate or aggravate cisplatin-induced toxicity or apoptosis in these cells; and 3) examine the mechanism underlying the effects of nutlins. We show that Nutlin-3 induced p53 but not PUMA-α and did not induce apoptosis in kidney cells. Importantly, Nutlin-3 suppressed kidney cell apoptosis during cisplatin treatment. Mechanistically, the cytoprotective effects of Nutlin-3 were dissociated from its regulation of MDM-p53 signaling. Nutlin-3 blocked Bax and Bak activation and cytochrome c release in kidney cells and also in isolated mitochondria. The results suggest a new pharmacological function of nutlins, i.e. inhibition of Bax and Bak. By this function, nutlins may protect normal cells and tissues during cancer therapy.
Experimental Procedures
Materials
The rat kidney proximal tubular cell line was from by Dr. U. Hopfer (Case Western Reserve University, Cleveland, OH) and maintained for experiments as described (22, 23). The p53-deficient baby mouse kidney cell line was prepared by E1A transformation of baby mouse kidney epithelial cells that were isolated from p53-deficient mice (25). The MDM2 or MDM4-deficient mouse embryonic fibroblast (MEF) cells were from Dr. G. Lozano (University of Texas M. D. Anderson Cancer Center, Houston, TX) (26, 27). HCT116 cells were obtained from Dr. B. Vogelstein (Howard Hughes Medical Institute and Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, Baltimore, MD). Antibodies used in this study were from the following sources: rabbit polyclonal anti-p53 and anti-phospho-p53 (Ser-15) from Cell Signaling Technology (Beverly, MA); monoclonal anti-MDM2 (SMP14) and monoclonal anti-p21 (F-5) from Santa Cruz Biotechnology (Santa Cruz, CA); monoclonal anti-Bcl-XL (2H12) and monoclonal anti-Bax (1D1) from NeoMarkers (Fremont, CA); polyclonal anti-Bak (NT) and polyclonal anti-Bax (NT) from Upstate Biotechnology; monoclonal anti-cytochrome c from BD Pharmingen; polyclonal anti-PUMA from Dr. J. Yu (University of Pittsburgh, Pittsburgh, PA); all secondary antibodies were from Jackson ImmunoResearch (West Grove, PA). Nutlin-3 was purchased from Cayman Chemical (Ann Arbor, MI). The Bim peptide with a sequence of DNRPEIWIAQELRRIGDEFNAYYAR was commercially synthesized by GenScript Corp. (Scotch Plains, NJ). Chemical cross-linker bismaleimidohexane was from Pierce. Carbobenzoxy-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin (DEVD-AFC) and AFC were purchased from Enzyme Systems Products (Dublin, CA). Annexin V-FITC apoptosis detection kit was from BD Pharmingen. Other reagents, including cisplatin, were from Sigma.
Cisplatin Treatment
Cisplatin treatment of cultured cells was conducted as described in our recent publications (22, 23). Cisplatin was used at 20 μm for kidney cells, 50 μm for HCT116 cells, 100 μm for p53-deficeint baby mouse kidney, MDM2-deficient MEF, and MDM4-deficient MEF cells. Nutlin-3 dissolved in Me2SO was added alone or together with cisplatin at indicated concentrations. One group of cells was added Me2SO as a solvent control. At the end of incubation, cells were monitored morphologically or harvested to collect cell lysates for biochemical analyses. For cell lysis, both floating and adherent cells were collected.
Morphological Examination of Apoptosis
Apoptotic cells were identified by their morphology as described in our previous studies (23, 28). Briefly, cells were fixed with 4% paraformaldehyde, followed by exposure to 10 μg/ml Hoechst 33342 for 2–5 min to stain the nuclei. Cellular and nuclear morphology was examined by phase-contrast and fluorescence microscopy. Apoptosis was characterized by cellular shrinkage, nuclear condensation and fragmentation, and formation of apoptotic bodies. Four fields with ∼200 cells per field were examined in each condition to estimate the apoptosis percentage. Images of representative fields were also recorded.
Flow Cytometric Analysis of Apoptosis
Annexin V-FITC/PI staining was performed according to the manufacturer's instruction (BD Pharmingen). Briefly, cells were detached by trypsinization and harvested along with the floating cells by centrifugation at 1,000 × g for 5 min. Following washes in phosphate-buffered saline containing 2% bovine serum albumin, the cells were resuspended in binding buffer (10 mm Hepes-NaOH, 140 mm NaCl, 2.5 mm CaCl2) at a final cell density of 1–2 × 106 cells/ml. Single cell suspension of 100 μl (1–2 × 105 cells) was incubated with 5 μl of Annexin V-FITC and 5 μl of PI for 15 min at room temperature in the dark. After adding 400 μl of binding buffer, the samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences). For each sample, 10,000 events were counted.
Measurement of Caspase Activity
Caspase activity was measured enzymatically using the fluorogenic peptide substrate DEVD-AFC (23, 28, 29). Briefly, cells were extracted with 1% Triton X-100. The lysates of 25 μg of protein were incubated with enzymatic reactions containing 50 μm DEVD-AFC at 37 °C for 60 min. Fluorescence at excitation 360 nm/emission 530 nm was then monitored by a GENios plate-reader (Tecan US Inc., Research Triangle Park, NC). A standard curve was constructed using free AFC for each measurement. Based on the standard curve, the fluorescence reading from each enzymatic reaction was converted into a nanomolar amount of liberated AFC per milligram of protein to indicate caspase activity.
Immunoblot Analysis
Protein concentration was determined using the bicinchoninic acid reagent (Pierce). Equal amounts of protein were loaded in each lane for reducing gel electrophoresis. The resolved proteins were then electroblotted to polyvinylidene difluoride membranes. The membranes were sequentially incubated with blocking buffer, primary antibodies, and horseradish peroxidase-conjugated secondary antibodies. Antigens on the blots were revealed by enhanced chemiluminescence (ECL) from Pierce.
Co-immunoprecipitation
Co-immunoprecipitation was conducted according to our recent work (22, 28). Briefly, whole cell lysates were collected with a buffer containing 2% CHAPS. The lysates of 500 μg of protein were precipitated with 20 μl of agarose protein A/G and 1 μg of anti-Bax or anti-Bcl-XL. The resultant immunoprecipitates were dissolved in 2% SDS buffer for electrophoresis and immunoblot analysis to detect the presence of Bax and Bcl-XL.
Dual Immunofluorescence of Cytochrome c and Active Bax
Cells were grown on collagen-coated glass coverslips. After experiments, the cells were fixed with a modified Zamboni fixative containing picric acid and 4% paraformaldehyde. The fixed cells were then permeabilized with 0.1% SDS, followed by blocking in phosphate-buffered saline containing 2% bovine serum albumin, 0.2% milk, and 2% normal goat serum. The cells were subsequently exposed to a mixture of primary antibodies (rabbit anti-active Bax and mouse anti-cytochrome c), followed by incubation with a mixture of FITC-labeled goat-anti-rabbit and Cy3-labeled goat-anti-mouse secondary antibodies. After extensive washes, signals were examined by confocal microscopy under Cy3 and FITC channels.
Cellular Fractionation
Cells were fractionated into cytosolic and membrane-bound organellar fractions by using digitonin, which at low concentrations selectively permeabilizes the plasma membrane without solubilizing mitochondria (22, 28). Briefly, cells were incubated with 0.05% digitonin in an isotonic buffer (in mm: 250 sucrose, 10 Hepes, 10 KCl, 1.5 MgCl2, 1 EDTA, and 1 EGTA (pH 7.1)) for 2 min at room temperature. The soluble extract was collected as cytosolic fraction. The digitonin-insoluble part was washed with isotonic sucrose buffer and further dissolved in 2% SDS buffer as membrane-bound organellar fraction enriched with mitochondria. These two fractions were analyzed for Bax and cytochrome c by immunoblotting.
Isolation of Mitochondria
Mitochondria were isolated as described previously (30) with minor modifications. Cells were rinsed with ice-cold phosphate-buffered saline and harvested by scraping gently. The cells were then homogenized with a 1 ml of Wheaton homogenizer in buffer A (in mm: 250 sucrose, 10 HEPES-NaOH, 10 KCl, 1.5 MgCl2, 1 EDTA, 1 EGTA, and 0.5 phenylmethylsulfonyl fluoride (pH 7.2)). The homogenates were centrifuged at 1,000 × g for 10 min to remove unbroken cells and nuclei. The resultant supernatants were further centrifuged at 10,000 × g for 10 min to sediment the heavy membrane fractions containing mitochondria. All procedures were performed on ice or at 4 °C. The mitochondrial preparation was kept on ice in buffer B (in mm: 250 sucrose, 10 HEPES-NaOH, 10 KCl, 5 MgCl2, 1 EDTA, 1 EGTA, 5 sodium succinate, and 0.1 phenylmethylsulfonyl fluoride (pH 7.4)) and used for experiments within 1 h of isolation.
Cytochrome c Release from Isolated Mitochondria
This assay was modified from our previous work (30). Isolated mitochondria of ∼15 μg of protein were preincubated in 60 μl of buffer B (see above for compositions) at 30 °C for 15 min with Nutlin-3 or vehicle solution, followed by centrifugation at 10,000 × g for 10 min at 4 °C. The preincubated mitochondria were then gently resuspended in 60 μl of buffer B and treated for 20 min at 30 °C with various reagents (1.2 mm CaCl2, 25 μm Bim peptide, 10 ng of wild-type tBid, or mutant tBid) in the absence or presence of Nutlin-3. To analyze cytochrome c release, the incubation mixture was centrifuged at 10,000 × g for 10 min at 4°C. The resultant pellet and supernatant were analyzed for cytochrome c by immunoblotting.
Bax and Bak Oligomerization
Bax and Bak oligomerization in cells and isolated mitochondria was analyzed by a method modified from our previous work (28, 31). Briefly, isolated mitochondria or the digitonin-insoluble membrane fraction of the cells were incubated with 5 mm bismaleimidohexane for 30 min of cross-linking at room temperature under constant mixing. The cross-linked samples were then subjected to immunoblot analysis of Bax and Bak.
Statistics
Data were expressed as means ± S.D. (n ≥ 3). Statistical analysis was conducted using GraphPad Prism software. Statistical differences in multiple groups were determined by multiple comparisons with Tukey's post-tests following analysis of variance. Statistical differences between two groups were determined by two-tailed unpaired Student t test. p < 0.05 was considered significantly different.
Results
Apoptotic Effects of Nutlin-3 in Kidney Tubular Cells
Recent studies have shown that nutlins activate p53 by interfering with p53-MDM2 interaction, leading to apoptosis and growth inhibition in cancer cells. Notably, at these concentrations (10–20 μm), nutlins were not toxic to normal cells or tissues (1, 5, 7, 8, 11–13). We confirmed the low toxicity of nutlins in cultured kidney tubular epithelial cells. As shown in Fig. 1A, the basal level of spontaneous apoptosis in these cells was ∼4%, and Nutlin-3 at 1–20 μm did not induce further apoptosis. At 30 μm, Nutlin-3 increased apoptosis to ∼10%. Apoptosis induced by 30 μm of Nutlin-3 showed typical apoptotic morphology with cellular and nuclear condensation and fragmentation (not shown). The morphological examination was consistent with the measurements of caspase activity. As shown in Fig. 1B, Nutlin-3 at 20 μm and lower concentrations did not activate caspases, whereas marginal caspase activation was induced by 30 μm Nutlin-3.
FIGURE 1. Apoptotic effects of Nutlin-3 in normal kidney cells.

Kidney tubular cells were incubated with 0–30 μm Nutlin-3 for 24 h. A, percentage of apoptosis assessed morphologically. B, caspase activity measured by enzymatic assays as described under “Experimental Procedures.” Data are expressed as means ± S.D. (n = 4). *, statistically significantly different from the control group without Nutlin-3 exposure. The results show that Nutlin-3 did not induce apoptosis in kidney cells at 20 μm or lower concentrations; at 30 μm it induced modest apoptosis.
Regulation of p53 Signaling by Nutlin-3
We then determined whether p53 and related signaling were regulated by Nutlin-3 in renal tubular cells. To this end, we treated kidney tubular cells with 20 μm Nutlin-3 for 0–24 h, and collected cell lysates for immunoblot analysis. Representative blots and results of densitometric analysis are shown in Fig. 2. Nutlin-3 treatment for 4 h induced a modest p53 accumulation, which increased markedly at 8 h and was maintained thereafter. p53 was phosphorylated after 4 h of Nutlin-3 treatment, and interestingly the phosphorylation decreased during subsequent incubation. Nutlin-3 also induced a drastic increase of MDM2, and similar to p53 phosphorylation, MDM2 decreased toward basal levels after 16–24 h of Nutlin-3 treatment. For gene expression downstream of p53, we detected p21 induction after 4–8 h of Nutlin-3 treatment, and again the induction decreased thereafter. Interestingly, despite its effects on p53 and p21, Nutlin-3 did not induce PUMA-α at any time points (Fig. 2). The absence of apoptotic gene (e.g. PUMA-α) induction may account for the lack of toxicity of Nutlin-3 in kidney cells.
FIGURE 2. Activation of p53 signaling by Nutlin-3 in kidney cells.

Kidney tubular cells were treated with 20 μm Nutlin-3 for 0–24 h. Whole cell lysates were collected in 2% SDS buffer for immunoblot analysis of total p53, phosphorylated p53, MDM2, p21, and PUMA-α. β-Actin was also probed to monitor sample loading and transfer. A, representative blots. B, results of densitometric analysis of the blots. The signals of control cells were arbitrarily set as 1 in each blot, and the signals of other conditions in the same blot were normalized with the control to indicate the expression level of the proteins. The results show that Nutlin-3 induced a transient p53 phosphorylation, and MDM2 and p21 accumulation, followed by total p53 accumulation. Nutlin-3 did not induce PUMA-α in the kidney cells.
Suppression of Cisplatin-induced Kidney Cell Apoptosis by Nutlin-3
Synergistic effects of nutlins and other chemotherapeutic agents have been reported in cancer cells (4–6, 9, 11, 14). However, whether nutlins ameliorate or exacerbate the side effects of therapy in normal cells or tissues is unclear. This is an important question for potential clinical use of nutlins along with other chemotherapy agents for combinational therapies. With this question, we determined the effects of Nutlin-3 on cisplatin-induced kidney cell apoptosis. Cisplatin is a commonly used cancer therapy drug, which has a major side effect of kidney injury or nephrotoxicity (19, 20). Cellular and nuclear morphology is shown in Fig. 3A. Clearly, massive apoptosis was induced by cisplatin but not by Nutlin-3 at 20 μm. In the cisplatin-treated group, apoptotic cells were shrunken with condensed and fragmented nuclei and formed apoptotic bodies. Importantly, cisplatin-induced apoptosis was suppressed by Nutlin-3. To confirm the results, we analyzed apoptosis by flow cytometric assay of Annexin V-FITC/PI staining. Representative plots are shown in Fig. 3B. Cisplatin treatment for 20 h led to positive Annexin V staining (indication of apoptotic changes in plasma membrane) in 65% cells, which was reduced to 40% by Nutlin-3. The same analysis also showed that Nutlin-3 itself did not induce kidney cell apoptosis at 20 μm, confirming our earlier morphological examination shown in Fig. 1. The cytoprotective effects of Nutlin-3 during cisplatin treatment were dose-dependent, and significant cytoprotection was shown at 10–30 μm (Fig. 3C). Consistently, Nutlin-3 suppressed cisplatin-induced caspase activation in a dose-dependent manner (Fig. 3D).
FIGURE 3. Suppression of cisplatin-induced apoptosis by Nutlin-3.

Kidney tubular cells were treated with 20 μm cisplatin in the absence or presence of Nutlin-3 or purified Nutlin-3b. A, representative images of cell morphology. Cells after cisplatin treatment in the absence or presence of 20 μm Nutlin-3 were fixed for nuclear staining with Hoechst33342. Cell and nuclear morphology was recorded by phase-contrast and fluorescence microscopy, respectively. B, flow cytometric analysis of apoptosis after Annexin V-FITC and PI staining. C and E, percentage of apoptosis assessed morphologically. D and F, caspase activity measured enzymatically by using DEVD-AFC as the substrate. Data are expressed as means ± S.D. (n = 4). *, statistically significantly different from the control group. #, statistically significantly different from the cisplatin-only group. The results show that Nutlin-3 inhibited cisplatin-induced kidney cell apoptosis in a dose-dependent manner, whereas purified Nutlin-3b was not cytoprotective.
Nutlin-3 has two enantiomers, Nutlin-3a and -3b. Interestingly, Nutlin-3b is much less effective in binding and antagonizing MDM2 (1). In the majority of our experiments, we used a mixture of both enantiomers. To determine if the observed cytoprotective effects of Nutlin-3 was specific for an enantiomer, we tested purified Nutlin-3b. The results show that Nutlin-3b was not protective during cisplatin incubation (Fig 3, E and F).
Nutlin-3 Does Not Diminish Cisplatin-induced p53 Activation
We and others have recently suggested a role of p53 in cisplatin-induced kidney cell apoptosis and nephrotoxicity (21–24). p53 is phosphorylated and induced by cisplatin in kidney cells, triggering apoptosis via apoptotic gene induction (22–24). To investigate the mechanism(s) underlying the cytoprotective effects of Nutlin-3, we first determined whether Nutlin-3 interfered with p53 activation during cisplatin treatment. Representative blots and results of densitometric analysis are shown in Fig. 4. Cisplatin induced a treatment time-dependent p53 accumulation and phosphorylation (lanes 2, 4, 6, and 8), which was not suppressed by Nutlin-3 (lanes 3, 5, 7, and 9). Interestingly, cisplatin also induced MDM2 at 4 and 8 h, which was followed by decreases of MDM2 to undetectable levels. Consistent with the results of Fig. 2, MDM-2 was drastically induced by Nutlin-3 (lanes 3, 5, and 7). The results indicate that Nutlin-3 does not diminish p53 activation during cisplatin treatment, suggesting that the observed cytoprotective effects of Nutlin-3 may be related to downstream apoptotic events.
FIGURE 4. Effects of Nutlin-3 on cisplatin-induced p53 activation.
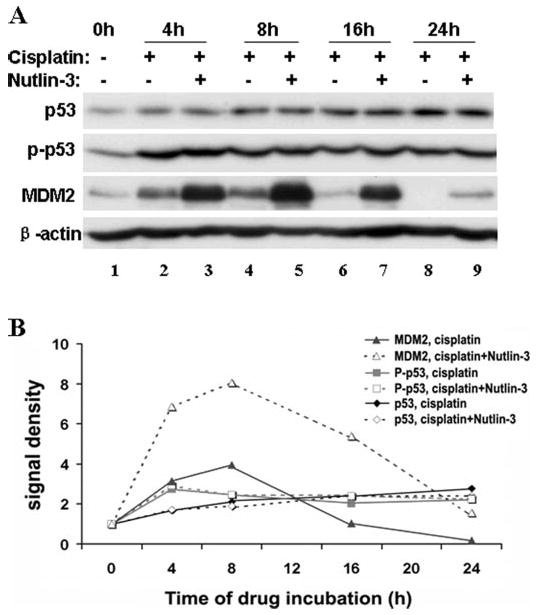
Kidney tubular cells were incubated with 20 μm cisplatin in the absence or presence of 20 μm Nutlin-3 for 0–24 h. Whole cell lysates were collected for immunoblot analysis of total p53, phosphorylated p53, and MDM2. The blots were reprobed for β-actin to monitor protein loading and transferring. A, representative blots. B, densitometric analysis of the blots. The signals of control (0h) cells were arbitrarily set as 1 in each blot, and the signals of other conditions in the same blot were normalized with the control to indicate the expression level of the proteins. The results show that cisplatin induced p53 accumulation and phosphorylation, which was not altered by Nutlin-3, although MDM2 was further induced in the presence of Nutlin-3.
Effects of Nutlin-3 on Bcl-2 Family Protein Expression during Cisplatin Treatment
Bcl-2 family proteins are critical regulators of apoptosis (32–37). Our recent work has suggested the involvement of these proteins in cisplatin-induced kidney cell apoptosis (22). Under these conditions, PUMA-α, a Bcl-2 family protein, is induced via p53 and neutralizes the anti-apoptotic members, including Bcl-XL. This leads to the activation of Bax and Bak, followed by the release of apoptogenic factors from mitochondria, including cytochrome c (22). To determine if Nutlin-3 affected Bcl-2 protein expression, we collected whole cell lysates for immunoblot analysis (Fig. 5). Following cisplatin incubation (lane 2), Bcl-XL and Bcl-2, two anti-apoptotic proteins, decreased. On the contrary, the pro-apoptotic PUMA-α was induced markedly. Bak showed a marginal increase, whereas no obvious changes were observed in Bax. Importantly, Nutlin-3, when added along with cisplatin, did not significantly antagonize these changes. The conclusion is further substantiated by densitometric analysis of blots from four separate experiments (Fig. 5B). It is also noteworthy that Nutlin-3 treatment alone did not change the expression of Bcl-2 family proteins in kidney cells (Fig. 5A, lane 4).
FIGURE 5. Effects of Nutlin-3 on Bcl-2 family protein expression during cisplatin treatment.
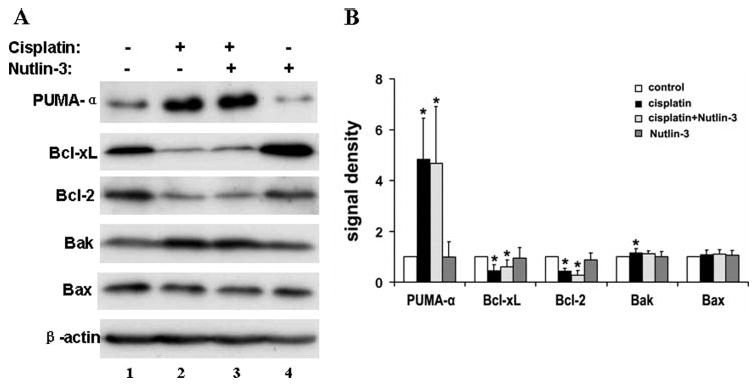
Kidney tubular cells were treated with 20 μm cisplatin in the absence or presence of 20 μm Nutlin-3 for 24 h. Whole cell lysates were harvested for immunoblot analysis of PUMA-α, Bcl-XL, Bcl-2, Bak, and Bax. The blots were also reprobed for β-actin to monitor protein loading and transferring. A, representative blots. B, densitometric analysis of four blots from separate experiments. The signals of the control lysate were arbitrarily set as 1 in each blot, and the signals of other conditions in the same blot were normalized with the control to indicate the expression level of Bcl-2 family proteins. Data are expressed as means ± S.D. (n = 4). *, statistically significantly different from the control group. The results show that cisplatin induced the pro-apoptotic PUMA-α and Bak, accompanied by decreases in the anti-apoptotic Bcl-XL and Bcl-2. Nutlin-3 did not have significant effects on cisplatin-induced changes in Bcl-2 protein expression.
Inhibition of Cisplatin-induced Apoptosis by Nutlin-3 in p53, MDM2, or MDM4-deficient Cells
The results shown above suggest that Nutlin-3 protects kidney cells during cisplatin treatment without altering p53 signaling. To substantiate these findings, we examined the effects of Nutlin-3 in p53-deficient baby mouse kidney cells (25). Cisplatin at 100 μm induced 52% apoptosis in these cells. Under this condition, Nutlin-3 suppressed apoptosis in a dose-dependent manner (Fig. 6A). At 20 μm, Nutlin-3 suppressed apoptosis to 21%. Consistently, Nutlin-3 suppressed caspase activation during cisplatin treatment of p53-deficient cells (data not shown). Nutlins antagonize MDMs (3, 8, 38, 39) and, as a result, lead to p53 induction. Thus we further examined the cytoprotective effects of Nutlin-3 in MDM2- or MDM4-deficient MEFs (Fig. 6, B and C). Clearly, Nutlin-3 dose dependently protected these gene-deficient cells during cisplatin treatment. Together the results indicate that cytoprotection by Nutlin-3 does not depend on its effects on MDM-p53 signaling.
FIGURE 6. Inhibition of cisplatin-induced apoptosis by Nutlin-3 in p53-, MDM2-, or MDM4 -deficient cells.

A, p53-deficient baby mouse kidney cells were treated with 100 μm cisplatin for 8 h in the presence of 0–30 μm Nutlin-3. B, MDM2-deficient MEF cells were treated with 100 μm cisplatin for 36 h in the presence of 0–30 μm Nutlin-3. C, MDM4-deficient MEF cells were treated with 100 μm cisplatin for 36 h in the presence of 0–30 μm Nutlin-3. Apoptosis was assessed morphologically. Data are expressed as mean ± S.D. (n = 3). *, statistically significantly different from the control group. #, statistically significantly different from the cisplatin-only group. The results show the cytoprotective effects of Nutlin-3 in all three kinds of deficient cells.
Suppression of Cisplatin-induced Bax Activation and Cytochrome c Release by Nutlin-3
To further investigate the mechanism of Nutlin-3-mediated cytoprotection, we examined its effects on Bax activation and cytochrome c release, two critical events in cisplatin-induced kidney cell apoptosis (22, 40). We first examined Bax activation in cisplatin-treated cells by immunofluorescence using an antibody specific for active (and not inactive) Bax. Control cells without cisplatin exposure did not show significant immunofluorescence of active Bax (not shown). Following cisplatin treatment, many cells showed punctate organellar/mitochondrial staining of active Bax (Fig. 7A, Active Bax, arrowed). The same cells released cytochrome c into the cytosol (Cytochrome c, arrowed). The conclusion that Bax activation in mitochondria correlated with cytochrome c release was further indicated by overlapping of the Bax and cytochrome c images (Fig. 7A, Overlap). The cells that did not show active Bax maintained cytochrome c in mitochondria (Fig. 7A, asterisks). We further detected Bax accumulation in mitochondria and cytochrome c release by immunoblotting (Fig. 7, B and C). In this assay, cells were fractionated into cytosolic and organellar fractions. As shown in Fig. 7B, in control cells the majority of Bax was detected in the cytosolic fraction (lane 1), and, after cisplatin treatment, a significant amount of Bax moved to the organellar fraction containing mitochondria (lane 2). Importantly, Nutlin-3 effectively suppressed cisplatin-induced Bax accumulation in mitochondria (lane 3). Immunoblots of cytochrome c are shown in Fig. 7C. Cisplatin treatment for 24 h induced a nearly complete release of cytochrome c into the cytosolic fraction (lane 6), which was partially suppressed by Nutlin-3 (lane 7). Nutlin-3 alone did not induce Bax accumulation or cytochrome c release (lanes 4 and 8), an observation that was consistent with the low toxicity of Nutlin-3 in kidney cells (Fig. 1). Together, these results suggest that Nutlin-3 may protect against cisplatin-induced apoptosis by inhibiting Bax activation and subsequent cytochrome c release.
FIGURE 7. Suppression of cisplatin-induced Bax activation and cytochrome c release by Nutlin-3.
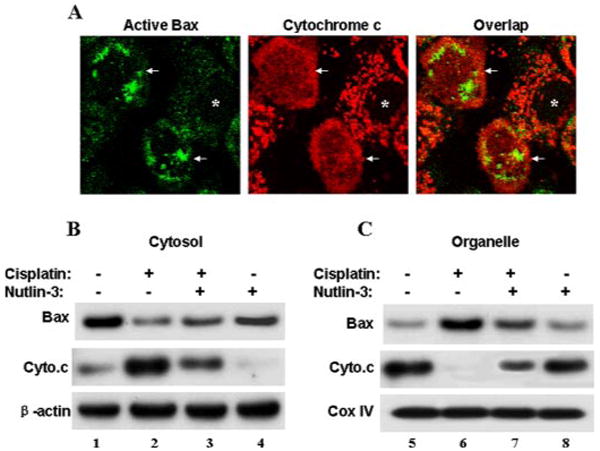
A, dual immunofluorescence of cytochrome c and active Bax in cisplatin-treated cells. Kidney tubular cells were incubated with 20 μm cisplatin and fixed for dual immunofluorescence of cytochrome c and active Bax. Arrows: cells with active Bax and cytochrome c release. Asterisks: cells without active Bax and cytochrome c release. B, immunoblot analysis of Bax translocation to mitochondria. C, immunoblot analysis of cytochrome c release. Kidney tubular cells were incubated with 20 μm cisplatin in the absence or presence of 20 μm Nutlin-3 for 24 h. The cells were fractionated into cytosolic (Cytosol) and membrane-bound organellar fractions containing mitochondria (Organelle) for immunoblot analysis of Bax and cytochrome c. β-Actin and cytochrome oxidase IV (CoxIV) were used as internal protein loading controls for cytosolic and membrane fractions, respectively. The results show Bax activation and translocation to mitochondria during cisplatin treatment, accompanied by cytochrome c release. Both Bax activation and cytochrome c release were suppressed by Nutlin-3.
Inhibition of Bax/Bak Oligomerization by Nutlin-3 during Cisplatin Treatment of Kidney Cells
Upon activation, Bax and Bak form oligomers in mitochondria, which are critical to the development of porous defects in mitochondrial membrane and consequent cytochrome c release (32–37). Thus we determined the effects of Nutlin-3 on Bax/Bak oligomerization during cisplatin incubation. As shown in Fig. 8 (lanes 2 and 5), cisplatin treatment led to the formation of Bax and Bak oligomers in kidney cells. Nutlin-3, added along with cisplatin, partially inhibited Bax/Bak oligomerization.
FIGURE 8. Inhibition of Bax and Bak oligomerization during cisplatin treatment of kidney cells by Nutlin-3.

Kidney tubular cells were treated with 20 μm cisplatin in the absence or presence of 20 μm Nutlin-3 for 20 h. After treatment, the membrane-bound organellar fraction was collected and cross-linked with bismaleimidohexane, followed by immunoblot analysis of Bax (A) and Bak (B). The results show that Nutlin-3 inhibited Bax and Bak oligomerization in kidney cells during cisplatin incubation.
No Effects of Nutlin-3 on the Interaction between Bax/Bak and Bcl-XL
We further determined whether Nutlin-3 affected the interactions between Bax/Bak and the anti-apoptotic proteins. In the kidney cells used in this study, Bcl-XL was expressed at a much higher level than Bcl-2. Thus we analyzed the interactions between Bax/Bak and Bcl-XL by co-immunoprecipitation. As shown in Fig. 9, there was a detectable Bax interaction with Bcl-XL in control cells, which was enhanced following cisplatin treatment. Importantly, Nutlin-3 did not significantly affect the molecular interaction. Nutlin-3 did not affect the interaction between Bak and Bcl-XL either (not shown). Thus Nutlin-3 protected the kidney cells during cisplatin treatment without affecting the interactions between Bax/Bak and Bcl-XL.
FIGURE 9. Effects of Nutlin-3 on the interaction between Bax and Bcl-XL during cisplatin treatment of kidney cells.

Kidney tubular cells untreated (control) or treated with 20 μm cisplatin for 20 h in the absence or presence of 20 μm Nutlin-3. Whole cell lysates were collected for immunoprecipitation using an antibody to Bax (A) or Bcl-XL (B). The resultant precipitates were analyzed by immunoblots of Bax and Bcl-XL The results show that Bax interacted with Bcl-XL following cisplatin incubation and Nutlin-3 did not prevent the molecular interaction.
Inhibition of Cytochrome c Release from Isolated Mitochondria by Nutlin-3
Nutlin-3 did not alter p53 signaling or Bcl-2 protein expression during cisplatin treatment of kidney cells (Figs. 4 and 5), but it interfered with Bax/Bak activation and cytochrome c release (Figs. 7 and 8). The results are particularly interesting, because they have suggested a new pharmacological function of nutlins, i.e. antagonizing mitochondrial events of apoptosis. To further pursue these findings, we determined the effects of Nutlin-3 on cytochrome c release in isolated mitochondria. Mitochondria were isolated from kidney cells and then treated with high Ca2+, Bim peptide, or tBid. These treatments are known to induce mitochondrial membrane permeabilization and cytochrome c release in isolated mitochondria (30, 41). Representative results are shown in Fig. 10. The majority of cytochrome c was preserved in mitochondria during control incubation (Fig. 10A, lane 1). Ca2+ at 1.2 mm induced a marked release of cytochrome c into the incubation buffer (Fig. 10A, lane 2), which was suppressed by Nutlin-3 (Fig. 10A, lane 3). Similarly, a Bim peptide released cytochrome c from isolated mitochondria (Fig. 10B, lane 2), and Nutlin-3 inhibited this release in a dose-dependent manner (Fig. 10B, lanes 3–5). Nutlin-3 also prevented cytochrome c release induced by recombinant tBid (Fig. 10C), a potent inducer of mitochondrial permeabilization (30, 41). The inhibition was detected at 20 μm Nutlin-3 and became complete at 50–100 μm (Fig. 10C, lanes 4–6). As a negative control, mutant tBid did not release cytochrome c (Fig. 10C, lane 7). These results, together with earlier observations of this study, suggest that Nutlin-3 may directly inhibit mitochondrial damage and, as a result, prevent cisplatin-induced apoptosis.
FIGURE 10. Inhibition of cytochrome c release from isolated mitochondria by Nutlin-3.

Mitochondria isolated from kidney tubular cells were treated with 1.2 mm Ca2+ (A), 25 μm Bim peptide (B), or 10 ng of recombinant tBid (C) in the presence of 0–100 μm Nutlin-3. The incubation mixtures were centrifuged to collect pellets and supernatants for immunoblot analysis of cytochrome c. The mitochondrial fraction blot was reprobed for cytochrome oxidase IV (CoxIV) to monitor protein loading and transferring. The results show that Nutlin-3 dose dependently inhibited cytochrome c release from isolated mitochondria.
Inhibition of tBid-induced Bax/Bak Oligomerization in Isolated Mitochondria by Nutlin-3
We hypothesized that Nutlin-3 might prevent cytochrome c release from isolated mitochondria by blocking Bax and Bak oligomerization in the organelles. To test this possibility, we determined the effects of Nutlin-3 on tBid-induced Bax and Bak oligomerization in isolated mitochondria. Isolated mitochondria were incubated with tBid in the presence of 0–100 μm Nutlin-3. The samples were then cross-linked with bismaleimidohexane to preserve the oligomers for immunoblot analysis. As shown in Fig. 11 A, tBid induced Bax oligomerization with the dimer, trimer, and tetramer (lane 2). Nutlin-3 ameliorated Bax oligomerization in a dose-dependent manner. At 50–100 μm, Nutlin-3 blocked Bax oligomerization completely (Fig. 11 A, lanes 3–6). Similarly, Nutlin-3 inhibited Bak oligomerization during tBid treatment (Fig. 11B, lanes 2–6). Thus Nutlin-3 may attenuate Bax and Bak oligomerization in mitochondria and, as a result, prevent the formation of cytochrome c releasing pore and subsequent apoptosis.
FIGURE 11. Inhibition of tBid-induced Bax and Bak oligomerization in isolated mitochondria by Nutlin-3.

Mitochondria isolated from kidney tubular cells were treated with recombinant tBid in the presence of 0–100 μm Nutlin-3. After treatment, the mitochondrial pellets were collected for cross-linking with bismaleimidohexane. The cross-linked samples were analyzed for Bax (A) and Bak (B) by immunoblotting. Asterisk: a nonspecific band shown in all lanes. The results show that Nutlin-3 inhibited tBid-induced Bax and Bak oligomerization in isolated mitochondria.
Effects of Nutlin-3 on Cisplatin-induced Apoptosis of HCT116 Colon Cancer Cells
To determine if Nutlin-3 also protects cancer cells during cisplatin treatment, we examined HCT116, a well characterized colon cancer cell line. As shown in Fig. 12A, cisplatin at 50 μm induced >60% apoptosis in 24 h. Nutlin-3 suppressed apoptosis in these cells in a dose-dependent manner. Statistically significant cytoprotective effects of Nutlin-3 were shown at 5 μm, and at 20 μm Nutlin-3 inhibited apoptosis to ∼20%. Consistently, Nutlin-3 ameliorated caspase activation during cisplatin treatment of HCT116 cells (Fig. 12B). In addition, cytoprotective effects of Nutlin-3 were also shown in A2780 ovarian cancer cells (not shown).
FIGURE 12. Effects of Nutlin-3 on cisplatin-induced apoptosis and caspase activation in HCT116 cells.

HCT116 cells were treated with 50 μm cisplatin for 24 h in the presence of 0–20 μm Nutlin-3. A, apoptosis assessed by morphology. B, caspase activity analyzed by enzymatic assays. Data are expressed as means ± S.D. (n =4). *, statistically significantly different from the control group. #, statistically significantly different from the cisplatin-only group. The results show that Nutlin-3 inhibited cisplatin-induced apoptosis in the HCT116 colon cancer cell line.
Discussion
The synergistic effects of nutlins and existing chemotherapeutic drugs in cancer cells have laid a foundation for future combinational therapy. A major issue of cancer therapy is the potential side effects or toxicity in normal tissues. Nutlins, when used alone, do not show obvious toxicity in normal non-malignant cells and tissues. However, it is unclear whether nutlins may increase or decrease the side effects of existing drugs in combinational therapy. Using kidney cells, we have shown in the current study that Nutlin-3 can protect normal cells or tissues during cisplatin therapy. Notably, the cytoprotective effects of Nutlin-3 are not related to its regulation of MDM-p53 signaling during cisplatin treatment. Instead, Nutlin-3 blocks Bax and Bak activation in mitochondria and, as a result, prevents the release of apoptogenic factors (e.g. cytochrome c) and apoptosis. Thus, this study has uncovered a new pharmacological function of nutlins, i.e. antagonizing Bax and Bak. These findings may have important implications in the use of nutlins in combinational therapy.
As promising chemotherapeutic drugs, nutlins potently induce apoptosis in p53-proficient cancer cells but not in normal cells or tissues (1, 5, 7, 8, 11–13). This inference is supported by our observations in kidney cells. Nutlin-3 did not induce significant apoptosis in these cells at 20 μm, a concentration that can effectively kill cancer cells (Fig. 1). At this concentration, Nutlin-3 did induce p53 and MDM2 in kidney cells, although the induction was transient and was not maintained after 4–8 h (Fig. 2). The results suggest that nutlins antagonized MDM2 in these cells. However, the cells did not undergo apoptosis even though p53 signaling was activated (Fig. 1). Our results show that the transient p53 activation by Nutlin-3 induced p21 expression but not the expression of PUMA-α (Fig. 2), suggesting that apoptotic gene expression is required for p53-mediated apoptosis. The results are consistent with our recent work that the induction of PUMA-α, a pro-apoptotic Bcl-2 family protein, is critical to kidney cell apoptosis during cisplatin treatment (22). Compared with cancer cells, normal cells show higher tolerance of p53 activation; however, the underlying mechanism is not known. Our results suggest that the tolerance may be related to the transient nature of p53 activation and the absence of apoptotic gene induction in these non-malignant cells.
Nutlin-3 induced p53 phosphorylation in kidney cells in our experiments (Fig. 2). This was surprising, because it has been shown that nutlins induce p53 by specifically antagonizing MDM2 and preventing consequent p53 degradation (3). In two colon cancer cell lines (HCT116 and RKO), nutlins did not induce p53 phosphorylation (42). However, p53 phosphorylation upon nutlin treatment was recently shown in B-cell chronic lymphocytic leukemia cells (6). In our study, Nutlin-3-induced p53 phosphorylation was transient in kidney cells, which returned to basal levels after 16–24 h. Thus, whether p53 is phosphorylated during nutlin treatment may depend on cell types. In addition, the concentrations of nutlins used and the duration of nutlin treatment may also affect the results, because p53 phosphorylation is time-dependent and can be transient.
A major finding of this study is that Nutlin-3 protects against cisplatin-induced apoptosis in kidney cells. This finding is important, because it suggests that nutlins may enhance the therapeutic efficacy during combinational therapies with existing drugs by alleviating the side effects of these drugs in normal tissues. A recent study has proposed a strategy of using nutlins to protect normal tissues during cancer therapy (14). This strategy proposes pretreatment with nutlins to induce mitotic arrest in normal non-malignant cells and tissues, followed by the use of mitotic toxins (e.g. paclitaxel) to kill cancer cells that have mutant p53 and do not undergo mitotic arrest during nutlin pretreatment. This is a clever design, which takes advantage of the different p53 status in normal and cancer cells. Of note, the experimental condition of the current study is different from the previous one. In our experiments, cells were not pre-treated; instead Nutlin-3 was provided during cisplatin incubation. Cisplatin treatment alone induces p53 and p21 in kidney cells, which effectively trigger cell cycle and mitotic arrest (22, 43). Nutlin-3 added during cisplatin incubation did not induce further p53 activation in our study (Fig. 4). These observations suggest that the cytoprotective effects of Nutlin-3 shown in our study may not be a result of mitotic arrest by this compound. This conclusion is further supported by the observation that Nutlin-3 protected kidney cells in the absence of p53, MDM2, or MDM4 (Fig. 6).
In kidney cells, Nutlin-3 suppressed cisplatin-induced Bax translocation and cytochrome c release from mitochondria (Fig. 7). In isolated mitochondria, Nutlin-3 blocked cytochrome c release induced by three types of insults, including Ca2+, a Bim peptide, and recombinant tBid (Fig. 10). Moreover, Nutlin-3 blocked Bid-induced Bax and Bak oligomerization in isolated mitochondria (Fig. 11). These results suggest that, in addition to MDM-p53, nutlins may antagonize Bax and Bak, two molecules that provide the requisite gateway to mitochondrial injury and apoptosis (44). In normal unchallenged cells, Bax is in the cytosol, whereas Bak is in mitochondria. Upon apoptotic stimulation, Bax inserts into mitochondria. Subsequently, Bax and Bak form oligomers, which may form porous defects for cytochrome c release. Alternatively, the oligomers may disturb the lipid bilayer to promote leakage of the proteins from the intermembrane space (32–37). Our results show that Nutlin-3 inhibits Bax translocation to mitochondria and Bax/Bak oligomerization in mitochondria. This pharmacological function of Nutlin-3 may underlie its cytoprotective effects in kidney cells. Of note, Nutlin-3 does not affect the interaction of Bax and Bak with Bcl-XL (Fig. 9). This observation suggests that in mitochondria there are two pools of Bax and Bak; one is bound to and probably neutralized by Bcl-XL/Bcl-2, and the other is free to form oligomers. Nutlin-3 may specifically interfere with free Bax/Bak molecules and, as a result, prevent their oligomerization. Nevertheless it is currently unclear whether Nutlin-3 directly binds Bax and Bak.
Identification of the new pharmacological function of nutlins may have important implications for future combinational cancer therapy. On one hand, by inhibiting Bax and Bak nutlins may suppress apoptosis in normal cells and tissues. This would increase the tolerance of normal tissues to therapy-associated toxicity or side effects and, as a result, enhance the therapeutic efficacy. On the other hand, nutlins may also suppress the toxicity in cancer cells and reduce the therapeutic effects. We showed that Nutlin-3 partially inhibited cisplatin-induced apoptosis in colon cancer HCT116 and ovarian cancer A2780 cells (Fig. 12 and data not shown). Of note, the concentrations of Nutlin-3 used in the current study are within the high range of cancer therapeutics. During therapy, drugs are excreted through the urinary system and the kidney tubular cells are frequently exposed to high concentrations due to their function of reabsorption. Thus it is important and makes it possible to determine the optimal conditions (e.g. dosage, treatment time, etc.) to use nutlins to protect normal tissues without diminishing the therapeutic effects in cancer cells.
Acknowledgments
We thank Dr. L. Vassilev (Roche Research Center, Hoffmann-La Roche Inc.) for purified Nutlin-3b, Dr. U. Hopfer (Case Western Reserve University) for the kidney tubular rat kidney proximal tubular cell line, Dr. G. Lozano (University of Texas M. D. Anderson Cancer Center) for the MDM2 and MDM4-deficient mouse embryonic fibroblast cells, Dr. B. Vogelstein (Howard Hughes Medical Institute and Johns Hopkins Sidney Kimmel Comprehensive Cancer Center) for HCT116 cells, and Dr. J. Yu (University of Pittsburgh) for the PUMA antibody. We also thank Dr. F. Balis (NCI, National Institutes of Health) for support of cisplatin measurement.
Footnotes
This work was supported by grants from the National Institutes of Health and the U. S. Department of Veterans Affairs.
The abbreviations used are: MDM2, murine double minute 2; MEF, mouse embryonic fibroblast; AFC, 7-amino-4-trifluoromethyl coumarin; FITC, fluorescein isothiocyanate; PI, propidium iodide;CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
References
- 1.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 2.Fischer PM, Lane DP. Trends Pharmacol Sci. 2004;25:343–346. doi: 10.1016/j.tips.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Vassilev LT. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 4.Cao C, Shinohara ET, Subhawong TK, Geng L, Woon Kim K, Albert JM, Hallahan DE, Lu B. Mol Cancer Ther. 2006;5:411–417. doi: 10.1158/1535-7163.MCT-05-0356. [DOI] [PubMed] [Google Scholar]
- 5.Coll-Mulet L, Iglesias-Serret D, Santidrian AF, Cosialls AM, de Frias M, Castano E, Campas C, Barragan M, de Sevilla AF, Domingo A, Vassilev LT, Pons G, Gil J. Blood. 2006;107:4109–4114. doi: 10.1182/blood-2005-08-3273. [DOI] [PubMed] [Google Scholar]
- 6.Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Blood. 2006;108:993–1000. doi: 10.1182/blood-2005-12-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, Andreeff M. Blood. 2005;106:3150–3159. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Cancer Res. 2006;66:3169–3176. doi: 10.1158/0008-5472.CAN-05-3832. [DOI] [PubMed] [Google Scholar]
- 9.Ribas J, Boix J, Meijer L. Exp Cell Res. 2006;312:2394–2400. doi: 10.1016/j.yexcr.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 10.Scoumanne A, Chen X. Cancer Res. 2006;66:6271–6279. doi: 10.1158/0008-5472.CAN-06-0121. [DOI] [PubMed] [Google Scholar]
- 11.Secchiero P, Barbarotto E, Tiribelli M, Zerbinati C, di Iasio MG, Gonelli A, Cavazzini F, Campioni D, Fanin R, Cuneo A, Zauli G. Blood. 2006;107:4122–4129. doi: 10.1182/blood-2005-11-4465. [DOI] [PubMed] [Google Scholar]
- 12.Stuhmer T, Chatterjee M, Hildebrandt M, Herrmann P, Gollasch H, Gerecke C, Theurich S, Cigliano L, Manz RA, Daniel PT, Bommert K, Vassilev LT, Bargou RC. Blood. 2005;106:3609–3617. doi: 10.1182/blood-2005-04-1489. [DOI] [PubMed] [Google Scholar]
- 13.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Proc Natl Acad Sci U S A. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Cancer Res. 2005;65:1918–1924. doi: 10.1158/0008-5472.CAN-04-3576. [DOI] [PubMed] [Google Scholar]
- 15.Siddik ZH. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 16.Jordan P, Carmo-Fonseca M. Cell Mol Life Sci. 2000;57:1229–1235. doi: 10.1007/PL00000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niedner H, Christen R, Lin X, Kondo A, Howell SB. Mol Pharmacol. 2001;60:1153–1160. [PubMed] [Google Scholar]
- 18.Perez JM, Fuertes MA, Alonso C, Navarro-Ranninger C. Crit Rev Oncol Hematol. 2000;35:109–120. doi: 10.1016/s1040-8428(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 19.Arany I, Safirstein RL. Semin Nephrol. 2003;23:460–464. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 20.Taguchi T, Nazneen A, Abid MR, Razzaque MS. Contrib Nephrol. 2005;148:107–121. doi: 10.1159/000086055. [DOI] [PubMed] [Google Scholar]
- 21.Cummings BS, Schnellmann RG. J Pharmacol Exp Ther. 2002;302:8–17. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]
- 22.Jiang M, Wei Q, Wang J, Du C, Yu J, Zhang L, Dong Z. Oncogene. 2006;25:4056–4066. doi: 10.1038/sj.onc.1209440. [DOI] [PubMed] [Google Scholar]
- 23.Jiang M, Yi X, Hsu S, Wang CY, Dong Z. Am J Physiol. 2004;287:F1140–F1147. doi: 10.1152/ajprenal.00262.2004. [DOI] [PubMed] [Google Scholar]
- 24.Seth R, Yang C, Kaushal V, Shah SV, Kaushal GP. J Biol Chem. 2005;280:31230–31239. doi: 10.1074/jbc.M503305200. [DOI] [PubMed] [Google Scholar]
- 25.Degenhardt K, Sundararajan R, Lindsten T, Thompson C, White E. J Biol Chem. 2002;277:14127–14134. doi: 10.1074/jbc.M109939200. [DOI] [PubMed] [Google Scholar]
- 26.Montes de Oca Luna R, Wagner DS, Lozano G. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 27.Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 28.Dong Z, Wang J. J Biol Chem. 2004;279:9215–9221. doi: 10.1074/jbc.M312225200. [DOI] [PubMed] [Google Scholar]
- 29.Dong Z, Saikumar P, Patel Y, Weinberg JM, Venkatachalam MA. Biochem J. 2000;347:669–677. [PMC free article] [PubMed] [Google Scholar]
- 30.Kim TH, Zhao Y, Barber MJ, Kuharsky DK, Yin XM. J Biol Chem. 2000;275:39474–39481. doi: 10.1074/jbc.M003370200. [DOI] [PubMed] [Google Scholar]
- 31.Yi X, Yin XM, Dong Z. J Biol Chem. 2003;278:16992–16999. doi: 10.1074/jbc.M300039200. [DOI] [PubMed] [Google Scholar]
- 32.Adams JM, Cory S. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 33.Bernardi P, Petronilli V, Di Lisa F, Forte M. Trends Biochem Sci. 2001;26:112–117. doi: 10.1016/s0968-0004(00)01745-x. [DOI] [PubMed] [Google Scholar]
- 34.Danial NN, Korsmeyer SJ. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 35.Green DR, Kroemer G. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 36.Wang X. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 37.Tsujimoto Y. J Cell Physiol. 2003;195:158–167. doi: 10.1002/jcp.10254. [DOI] [PubMed] [Google Scholar]
- 38.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. J Biol Chem. 2006;281:33030–33035. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 39.Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. J Biol Chem. 2006;281:33036–33044. doi: 10.1074/jbc.M605405200. [DOI] [PubMed] [Google Scholar]
- 40.Park MS, De Leon M, Devarajan P. J Am Soc Nephrol. 2002;13:858–865. doi: 10.1681/ASN.V134858. [DOI] [PubMed] [Google Scholar]
- 41.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT. J Biol Chem. 2004;279:53015–53022. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- 43.Megyesi J, Safirstein RL, Price PM. J Clin Invest. 1998;101:777–782. doi: 10.1172/JCI1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]