

Abstract
A series of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamides was synthesized by copper-catalyzed azide–alkyne cycloaddition (CuAAC) and afforded inhibitors of cancer cell growth. For example, compound 13e had an IC50 of 46 nM against MCF-7 human breast tumor cells. Structure–activity relationship (SAR) studies demonstrated that (i) meta-phenoxy substitution of the N-1-benzyl group is important for antiproliferative activity and (ii) a variety of heterocyclic substitutions for the aryl group of the arylamide are tolerated. In silico COMPARE analysis of antiproliferative activity against the NCI-60 human tumor cell line panel revealed a correlation to clinically useful antimicrotubule agents such as paclitaxel and vincristine. This in silico correlation was supported by (i) in vitro inhibition of tubulin polymerization, (ii) G2/M-phase arrest in HeLa cells as assessed by flow cytometry, and (iii) perturbation of normal microtubule activity in HeLa cells as observed by confocal microscopy. The results demonstrate that N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide is a readily accessible small molecule scaffold for compounds that inhibit tubulin polymerization and tumor cell growth.
Keywords: Triazole, Click Chemistry, CuAAC, Antimicrotubule, Antitumor, Antiproliferative, Anticancer, Tubulin, Microtubule
Introduction
Microtubules, dynamic protein polymers composed of α-tubulin and β-tubulin heterodimers, are a well-established cellular target for anticancer drugs.1 Dynamic polymerization of tubulin is a necessary and tightly controlled process during mitosis.2 Perturbing microtubule dynamics with small molecules blocks the cell cycle in the metaphase/anaphase transition and leads to apoptosis.3 Thus, molecules4 that target tubulin halt rapid cell division, a characteristic of cancer cells.5 This therapeutic strategy has been validated by the clinical success of antimicrotubule drugs such as paclitaxel, docetaxel, vincristine, and vinblastine. Nonetheless, neurotoxicity and P-glycoprotein-mediated drug resistance limit the clinical utility of these drugs.6 New-generation taxoids, vinca alkaloids, and other novel chemotypes that modulate microtubule dynamics have been synthesized in efforts to overcome these limitations.7 For example, small molecule modulators of tubulin polymerization that do not elicit neurotoxicity in mice have been identified,8 suggesting that neurotoxicity is not intrinsically linked to antimicrotubule agents. Nonetheless, few of these new antimicrotubule agents have produced useful clinical results. The key limitation to the development of new antimicrotubule drugs is a narrow therapeutic window.9 A new antimicrotubule scaffold amenable to rapid derivatization and combinatorial library synthesis would provide an exceptional opportunity for the discovery of an efficacious antimicrotubule agent with an improved therapeutic window.
Herein we report the synthesis, in vitro antiproliferative activity against select cancer cell lines, and structure–activity relationships of compounds containing the N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide scaffold. We also report mode of action studies based on in silico, in vitro and cell culture experiments, which reveal the potent antimicrotubule activity of this scaffold. The discovery of this scaffold stemmed from our work on Mycobactin S (1),10 a natural product produced by Mycobacterium smegmatis that exhibits anti-tuberculosis activity11 (Chart 1). All synthetic intermediates encountered during our group’s total synthesis of Mycobactin S12 were screened for biological activity. Surprisingly, benzyl ester 2, a small fragment of the natural product, exhibited anti-tuberculosis activity similar to that of Mycobactin S. Furthermore, in contrast to Mycobactin S, compound 2 provided a scaffold that was amenable to rapid structure–activity relationship studies, which led to the discovery of more potent anti-tuberculosis agents.13 While exploring derivatives of 2, we found that 2-phenyl-oxazole-4-carboxamide derivative 3 was also active against M. tuberculosis. 2-Phenyl-oxazole-4-carboxamides are known inhibitors of histone deacetylase,14 Stat3,15 phosphodiesterase,16 phosphatase,17 thromboxane synthase,18 and kinase proteins,19 and known activators of cellular caspase activity.20 We synthesized derivatives of 3 to identify a more potent anti-tuberculosis agent.13 One of the derivatives, compound 4e, had weak anti-tuberculosis activity, but broader biological screening serendipitously revealed that 4e and related derivatives have potent antimicrotubule activity in cancer cells, as disclosed in this report.
Chart 1.

Structures of anti-tuberculosis compounds 2 and 3 and antimicrotubule compound 4e, all derived from a fragment of mycobactin S (1). MIC values indicate in vitro anti-tuberculosis activity against M. tuberculosis H37Rv in GAST medium21, and IC50 values indicate in vitro antiproliferative activity against human breast cancer cell line MCF-7.
Results and Discussion
Chemistry
2-Phenyl-oxazole-4-carboxylic acid 7 was synthesized according to the protocols shown in Scheme 1.11,12 Coupling benzoyl chloride to serine benzyl ester hydrochloride afforded β-hydroxy amide 5. Dehydrative cyclization and oxidation of β-hydroxy amide 5 with diethylaminosulfurtrifluoride (DAST) and DBU/BrCCl3 yielded oxazole 6.22 Catalytic hydrogenolysis of the benzyl ester provided 2-phenyl-oxazole-4-carboxylic acid 7.
Scheme 1.

Synthesis of 2-phenyloxazole-4-carboxylic acid.
Our strategy for exploring the chemical space around the 2-phenyl-oxazole-4-carboxamide fragment employed “click chemistry.”23 More specifically, we selected the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction24 because of its wide scope, high efficiency, and recognized utility for drug discovery.25 Following this strategy, N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-2-phenyl-oxazole-4-carboxamides 4a–e were synthesized as shown in Scheme 2. Coupling propargylamine to freshly prepared 2-phenyloxazole-4-carboxyl chloride (derived from the corresponding carboxylic acid 7) provided alkyne 8. With the terminal alkyne precursor in hand, we turned our attention to the syntheses of azides 10a–e. Benzyl bromides 9a–e were treated with NaN3 to afford benzyl azides 10a–e.26 Exposing terminal alkyne 8 to benzyl azides 10a–e in the presence of catalytic Cu(I) produced 1,4-disubstituted triazoles 4a–e in high regioselectivity. Aqueous CuAAC conditions (H2O/t-BuOH, 2:1) facilitated precipitation of the products, which were isolated with high purity.24
Scheme 2.

Synthesis of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-2-phenyl-oxazole-4-carboxamide scaffold.
In order to explore the structure–activity relationships of the aryl amide group, a more general N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide scaffold was synthesized according to the protocols shown in Scheme 3. The simplicity of reaction Scheme 2 and Scheme 3 is anticipated to allow the synthesis of a large library of 1,2,3-triazole-based structures. Moreover, the CuAAC reaction is the convergent synthetic step. Both the arylamide and benzyl groups, attached to opposite sides of the central triazole, are important components of the pharmacophore, as shown by the structure–activity studies described below. The convergence of the synthesis will allow both sides of the scaffold to be systematically varied in future SAR studies.
Scheme 3.

Synthesis of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide scaffold.
In Vitro Antiproliferative Activity
The antiproliferative activity of compounds 4a–e against cancer cells was discovered during broad biological screening of a series of compounds originally anticipated to have anti-tuberculosis activity. Although compounds 4a–e had negligible anti-tuberculosis activity, they inhibited the proliferation of cancer cells in vitro. Therefore, we determined the antiproliferative activities (IC50 values) of hit compounds 4a–e against the breast tumor-derived cell line MCF-7 (Table 1). Because the IC50 of 4e (0.56 µM) was significantly lower than those of 4a–d (IC50 = 7.3–16 µM), we conserved meta-phenoxy benzyl substitution at triazole N-1 in the second series of analogs (Scheme 3).
Table 1.
In vitro antiproliferative activity of compounds 4a–e, colchicine, and 2-methoxyestradiol against human breast cancer cell line MCF-7.
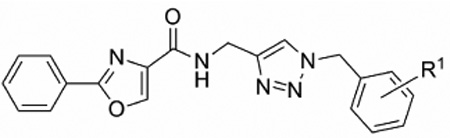 | ||
|---|---|---|
| IC50 (µM) | ||
| Compound | R1 | MCF-7 |
| 4a | H | 15.9 |
| 4b | p-CH3 | 7.59 |
| 4c | p-CF3 | 7.33 |
| 4d | m-OCH3 | 8.35 |
| 4e | m-OPh | 0.56 |
| Colchicine | 0.013 | |
| 2-methoxyestradiol | 0.84 | |
The structure–activity relationships of the triazole C-4 substituent were investigated by changing the carboxamide group (Table 2). In this SAR study, antiproliferative activity was investigated with the MCF-7 cell line and human lymphoma cell line U937. Addition of electron withdrawing or electron donating groups to the para-position of the 2-phenyloxazole group (13a–c) had a significant effect on antiproliferative activity, revealing an avenue for future optimization. Before performing an extensive SAR study via substitution of the 2-phenyloxazole group, we wanted to know if the 2-phenyloxazole was necessary for antiproliferative activity. If simpler aryl groups could replace the 2-phenyloxazole, the three-step synthesis of the 2-phenyloxazole-4-carboxylic acids could be bypassed by using commercial aryl acids. Thus, we conducted a systematic truncation of the 2-phenyloxazole-4-carboxamide group found in 4e. Removing the 2-phenyl group (13d) did not significantly change the activity. Following this lead, which suggested that we could use simpler aryl groups, we synthesized 2-pyridyl derivative 13e. Against the MCF-7 cell line, the IC50 of 13e (46 nM) was significantly lower than that of 4e (560 nM). Likewise, a significant decrease in IC50 was observed with the U937 cell line. Therefore, replacing the 2-phenyloxazole group with simpler aryl groups represents a significant opportunity to improve antiproliferative activity. Phenyl derivative 13f had improved antiproliferative activity compared to 4e, but was less active than 2-pyridyl derivative 13e. Replacing the aryl group with a methyl group (14) gave a meaningful loss of antiproliferative activity and demonstrated the importance of the aryl carboxamide group.
Table 2.
Antiproliferative activities (IC50) of compounds 4e, 13a–f, 14, colchicine, and 2-methoxyestradiol against human breast cancer cell line MCF-7 and human lymphoma cell line U937.
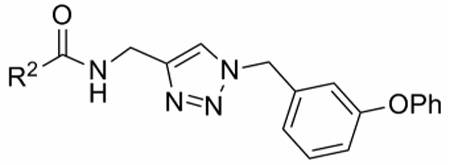 | |||
|---|---|---|---|
| Compound | R2 | IC50 (µM)a | |
| MCF-7 | U937 | ||
| 4e | 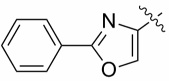 |
0.56 ± 0.11 | 1.40 ± 0.18 |
| 13a | 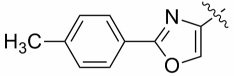 |
0.33 ± 0.045 | 1.13 ± 0.44 |
| 13b | 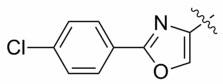 |
1.9 ± 1.4 | nd |
| 13c | 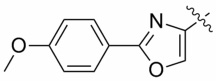 |
0.66 ± 0.31 | 3.75 ± 1.78 |
| 13d | 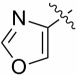 |
0.64 ± 0.35 | 2.19 ± 0.58 |
| 13e | 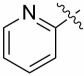 |
0.046 ± 0.022 |
0.58 ± 0.42 |
| 13f | 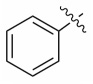 |
0.245 ± 0.007 | nd |
| 14 | 6.4 ± 1.3 | 28 ± 19 | |
| Colchicine | 0.0134 | 0.008 ± 0.003 | |
| 2-methoxyestradiol | 0.842 ± 0.090 | 2.91 ± 1.17 | |
IC50 values represent the concentration at which the cell count was inhibited to 50% of that measured in the vehicle control. Error is S.E.M., n≥3.
Time Dependence of In Vitro Cellular Antiproliferative Activity
In order to distinguish cytotoxic activity from cytostatic activity, the time dependence of the effect of 13e on MCF-7 cells was determined (Figure 1). At a concentration of 39 nM, 13e slowed the cell proliferation rate. At higher concentrations (78 nM and 156 nM), cellular proliferation was halted, but 13e did not decrease the number of cells. Thus, at concentrations moderately higher than the IC50, 13e is cytostatic rather than cytotoxic against MCF-7 cells.
Figure 1.

Time and concentration dependence of the antiproliferative activity of 13e against MCF-7 tumor cells. Time is in terms of time elapsed after addition of the compound. Cell counts are shown relative to the cell count observed in the vehicle control 96 h after addition of the 0.5% DMSO solution.
Broad-Spectrum In Vitro Antiproliferative Activity
The NCI-60 anticancer drug screen is an in vitro assay consisting of 60 different human tumor cell lines.27 Organized by disease type, the NCI-60 panel includes various leukemia cell lines and cell lines derived from solid tumor sources. Cell line selectivity guides further biological evaluation. In the NCI-60 panel, compounds 4b, 4c, 4d, and 4e induced broad-spectrum antiproliferative activity against tumor cell lines derived from leukemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, and breast cancer (see Supplemental Information). Of these four compounds, 4e (NCI-60 mean GI50 = 870 nM) was more than twenty times more potent than the other three compounds (Table 3). This result is consistent with the results from our MCF-7 and U937 assays and further emphasizes the importance of meta-phenoxy benzyl substitution of triazole N-1 for optimal activity within this scaffold. With respect to selectivity among the 60 cell lines in the NCI-60 panel, all four compounds tested in the panel showed greater than average potency against human leukemia cell line HL-60, breast cancer cell line MCF-7, and melanoma28 cell line MDA-MB-435 (Table 3).
Table 3.
Antiproliferative activity of compounds 4b, 4c, 4d, and 4e against selected cell lines in the NCI-60 screen.
| GI50 (µM) | ||||
|---|---|---|---|---|
| compound | mean | HL-60 | MCF-7 | MDA-MB-435 |
| 4b | 21.9 | 1.53 | 8.93 | 3.71 |
| 4c | 16.6 | 2.34 | 3.83 | 1.94 |
| 4d | 17.8 | 2.37 | 4.16 | 2.12 |
| 4e | 0.87 | 0.39 | 0.36 | 0.18 |
COMPARE Analysis Revealed a Correlation to Antimicrotubule Drugs
For a given compound, the antiproliferative activity measured in the NCI-60 differs by cell line. Furthermore, antitumor agents with similar mechanisms of action can produce similar patterns of differential antiproliferative data. We used the matrix COMPARE algorithm29 to measure the correlations between compounds 4b, 4c, 4d, and 4e with respect to differential antiproliferative activity. The matrix produced by the analysis showed that 4c and 4d have highly correlated activities (r = 0.968) (Table 4). On the contrary, 4e (the most potent compound tested in the NCI-60) had low correlations (r < 0.5) with the other three compounds. These matrix COMPARE results suggest that 4c and 4d have the same mechanism of action, but the mechanism of 4e is distinct. Future studies will further investigate the importance of the meta-phenoxy substituent found in 4e for its mechanism action. In the studies reported here, we focused on 4e and related analogs, which also have the meta-phenoxy substituent, because of their superior potency.
Table 4.
Matrix COMPARE analysis of 4b, 4c, 4d, and 4e. Matrix values (r values) are Pearson’s correlation coefficients.30
| compound | 4b | 4c | 4d | 4e |
|---|---|---|---|---|
| 4b | 1.000 | 0.600 | 0.583 | 0.489 |
| 4c | 0.600 | 1.000 | 0.968 | 0.475 |
| 4d | 0.583 | 0.968 | 1.000 | 0.491 |
| 4e | 0.489 | 0.475 | 0.491 | 1.000 |
The COMPARE algorithm can also compare the differential antiproliferative activity of a new compound to those of compounds with known mechanisms of action in the NCI Standard Agent Database.31 Standard COMPARE analysis has been used previously to identify the cellular targets of antitumor agents.32 Thus, the pattern of differential antiproliferative activity of 4e was used to probe the NCI Standard Agent Database for correlations. We used all three measures of activity provided by the NCI-60 screen (GI50, 50% growth inhibition; TGI, total growth inhibition; and LC50, 50% lethal concentration). A standard COMPARE analysis of 4e (Table 5) showed correlations to paclitaxel, maytansine, vincristine, vinblastine, and rhizoxin, all of which affect microtubule polymerization. Given this in silico result, we hypothesized that 4e targets microtubules and directly tested this hypothesis in vitro. In contrast, the first-ranked COMPARE hits for 4b–d did not include antimicrotubule agents (see Supplemental Information).
Table 5.
Standard COMPARE analysis of 4e. The Target Set was the Standard Agent Database and the Target Set Endpoints were set equal to the Seed Endpoints (GI50, TGI, and LC50). Correlation values (r) are Pearson’s correlation coefficients. Some hits appear multiple times because they were tested by the NCI for the Standard Agent Database at multiple concentration ranges.
| rank | compound | r |
|---|---|---|
| Based on GI50 Mean Graph | ||
| 1 | paclitaxel | 0.541 |
| 2 | maytansine | 0.534 |
| 3 | vincristine sulfate | 0.500 |
| 4 | trimetrexate | 0.480 |
| 5 | soluble Baker's Antifol | 0.460 |
| Based on TGI Mean Graph | ||
| 1 | paclitaxel (hiConc = 10−5 M) | 0.654 |
| 2 | paclitaxel (hiConc = 10−6 M) | 0.646 |
| 3 | paclitaxel (hiConc = 10−4.6 M) | 0.642 |
| 4 | maytansine | 0.621 |
| 5 | vinblastine sulfate | 0.606 |
| Based on LG50 Mean Graph | ||
| 1 | rhizoxin (hiConc = 10−9 M) | 0.681 |
| 2 | rhizoxin (hiConc = 10−4 M) | 0.650 |
| 3 | vinblastine sulfate (hiConc = 10−7.6 M) | 0.558 |
| 4 | α-2'-deoxythioguanosine | 0.545 |
| 5 | vinblastine sulfate (hiConc = 10−4 M) | 0.540 |
Inhibition of Tubulin Polymerization In Vitro
The polymerization of microtubules from purified tubulin can be monitored in vitro by measuring an increase in light scattering. This in vitro experiment removes complicating factors, such as microtubule-associated proteins (MAPs), which might be part of a putative target that leads to disruption of microtubules as observed with microscopy. In order to test our hypothesis that the target of the N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide scaffold is tubulin, and not a MAP, we monitored the polymerization of tubulin after treatment with 4e, 13a, and 13e (Figure 2). In this experiment, paclitaxel, a microtubule stabilizer, enhanced the rate of tubulin polymerization, while nocodazole, a microtubule destabilizer, prevented the polymerization of tubulin. Similar to nocodazole, 4e, 13a, and 13e completely inhibited tubulin polymerization at 10 µM. Thus, the triazole-based compounds prevent the formation of microtubules in vitro. We followed this in vitro assay with cell culture experiments to see if microtubules are the primary cellular target.
Figure 2.

Inhibition of tubulin assembly by 4e, 13a, and 13e in vitro. All compounds were tested at a concentration of 10 µM. Effects of compounds on tubulin polymerization were assessed by monitoring the increase in light scattering, measured as optical density (O.D.), at 340 nm. Standards of 10 µM nocodazole (a tubulin assembly inhibitor) and 10 µM paclitaxel (a tubulin assembly promoter) were used for direct comparison.
Cell Cycle Analysis Demonstrated G2/M-Phase Arrest
Antimicrotubule agents induce M-phase arrest. Flow cytometry can quantitatively determine the population of cells in each phase of the cell cycle by measuring the DNA content of individual cells. Cells in G2-phase or M-phase have twice as much DNA as cells in G1-phase. Thus, we conducted flow cytometric cell cycle analysis of HeLa cells treated with 4e, 13a, 13d, 13e, and 14. Consistent with the hypothesized mechanism of action, compounds 4e, 13a, 13d, and 13e significantly increased the population of cells in G2/M-phase (Figure 3). Upon treatment with these four compounds, the population of G2/M-phase cells increased from 13% in the control to over 90%. Compound 14, however, did not induce significant G2/M-phase arrest, which suggested that the arylamide moiety is important for the antimitotic activity of the N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide scaffold.
Figure 3.

Effects of 4e, 13a, 13d, 13e, and 14 on the cell cycle distribution of HeLa cells as measured by propidium iodide staining and flow cytometry. HeLa cells were treated with 5 µM compound for 18 h in triplicate.
Confocal Microscopy Showed M-phase Arrest and Disruption of Microtubules
Visual evidence for M-phase arrest can be obtained with confocal microscopy due to DNA condensation resulting in enhanced staining by propidium iodide. This outcome is in contrast to the diffuse staining of DNA in interphase cells. Visual evidence for the disruption of microtubules can be obtained concurrently using a fluorescein isothiocyanate-conjugated anti-tubulin antibody. We therefore used confocal microscopy to examine HeLa cancer cells treated with compounds 4e and 13e. Both DNA condensation and disruption of microtubules were observed at 5 µM of 4e or 13e (Figure 4). Together, these images show that 4e and 13e induce M-phase arrest and interfere with microtubule formation in whole cells.
Figure 4.

Confocal microscopy images of HeLa cells after 18 h incubation in the presence of 5 µM compound. Nocodazole is a known tubulin polymerization inhibitor. Nuclear DNA was stained with propidium iodide (red channel) and tubulin was stained with FITC-conjugated anti-α-tubulin antibody (green channel). Compounds 4e and 13e disrupted normal microtubule structures, caused fragmentation of mitotic spindles, and induced M-phase arrest.
Conclusion
In summary, we identified N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide as a novel and proprietary33 small molecule scaffold for potential antitumor agents. Elucidating structure–activity relationships by subtraction from initial hit compound 4e (MCF-7 IC50 = 560 nM) led to the discovery of 13e (MCF-7 IC50 = 46 nM), a foundational compound for further study. Compound 13e (and related compounds) induced M-phase arrest in HeLa cells at 5 µM and inhibited tubulin polymerization in vitro at 10 µM, providing strong support for antimicrotubule activity as the primary mechanism of action. The NCI-60 screen demonstrated broad-spectrum antitumor activity and prompted further biological evaluation. Compound 13c was recently evaluated by the National Cancer Institute Developmental Therapeutics Program (NCI DTP) for acute toxicity in vivo, and 100, 200 and 400 mg/kg intraperitoneal (IP) doses were well tolerated in non-tumor bearing mice. Ongoing studies in collaboration with the NCI DTP will evaluate in vivo efficacy in hollow fiber assays.34 Extensive SAR studies and the development of a combinatorial library are accessible because compounds based on the N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)arylamide scaffold are readily synthesized with the CuAAC reaction. Our findings will facilitate the design and optimization of potent, cell-permeable antimicrotubule agents.
Experimental Section
2-(4-methoxyphenyl)-N-((1-(3-phenoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)oxazole-4-carboxamide (C27H23N5O4, 13c)
2-(4-Methoxyphenyl)oxazole-4-carboxylic acid (11c, 0.951 g, 4.3 mmol) was suspended in anhydrous CH2Cl2 (12 mL) under argon. Oxalyl chloride (0.45 mL, 5.2 mmol) and N,N-dimethylformamide (20 µL) were added carefully to the mixture because of gas evolution. The reaction slowly turned to a light yellow homogeneous solution over 3 h. The solution was concentrated in vacuo to give 2-(4-methoxyphenyl)oxazole-4-carbonyl chloride (1.0 grams, 97%) as an yellow solid, which was used immediately in the next reaction without characterization.
2-(4-Methoxyphenyl)oxazole-4-carbonyl chloride (1.0 g, 4.0 mmol) was dissolved in anhydrous CH2Cl2 (15 mL) under argon and cooled to 0 °C (ice bath). Propargyl amine hydrochloride (0.443 g, 4.8 mmol) and N,N-diisopropylethylamine (2.1 mL, 12.0 mmol) were added with stirring. The reaction was allowed to warm to room temperature. After stirring for 20 h, TLC analysis indicated completion of the reaction. The mixture was poured into a solution of 10% aqueous NaHCO3 and extracted with CH2Cl2 (2x). The organic layer was separated, washed with 10% aqueous NaHCO3 and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The resultant crude material was purified by column chromatography (SiO2, EtOAc/CH2Cl2 stepwise elution, 1:1 to 10:1) to give 2-(4-Methoxyphenyl)-N-(prop-2-ynyl)oxazole-4-carboxamide (12c) as an off white solid (0.788 g, 77%). mp 151–152 °C; 1H NMR (300 MHz, CDCl3) δ 8.06 (s, 1H), 7.82 (d, J = 8.5 Hz, 2H), 7.13 (bs, NH, 1H), 6.84 (d, J = 8.5 Hz, 2H), 4.15−4.07 (m, 2H), 3.72 (s, 3H), 2.15 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 161.82, 161.57, 160.40, 140.49, 136.44, 128.83, 128.29, 119.13, 114.26, 113.69, 79.17, 71.69, 55.37, 28.66; HRMS–FAB (m/z) [M+H]+ calcd for C14H12N2O3, 257.0921; found, 257.0930.
2-(4-Methoxyphenyl)-N-(prop-2-ynyl)oxazole-4-carboxamide (12c, 300 mg, 1.17 mmol) and 1-(azidomethyl)-3-phenoxybenzene (290 mg, 1.29 mmol) were suspended in a 2:1 mixture of water and tert-butyl alcohol (4.7 mL total volume). Sodium ascorbate (0.12 mmol, 0.12 mL, 1 M) and copper(II) sulfate (0.012 mmol, 0.12 mL, 0.1 M) were added sequentially. After stirring for 4 days at room temperature, TLC analysis indicated complete consumption of the reactants. The reaction mixture was diluted with water (5 mL) and cooled on ice. The white precipitate was isolated by vacuum filtration and washed with cold water (3 × 5 mL) and cold diethyl ether (3 × 3 mL) to afford 506 mg (90%) of pure product (13c) as a white powder. TLC Rf = 0.42 (EtOAc); HPLC tr = 6.82 min (9:1 hexanes/2-propanol); m.p. 119.1 – 119.4 °C; 1H NMR (300 MHz, CDCl3) δ 8.18 (s, 1 H), 7.99 − 7.93 (m, 2 H), 7.60 (bs, NH, 1 H), 7.55 (s, 1 H), 7.39 − 7.28 (m, 3 H), 7.16 − 7.09 (m, 1 H), 7.03 − 6.90 (m, 7 H), 5.47 (s, 2 H), 4.72 (d, J=6.1 Hz, 2 H), 3.88 (s, 3 H); 13C NMR (151 MHz, CDCl3) δ 161.83, 161.56, 160.84, 158.04, 156.36, 145.05, 140.26, 136.70, 136.29, 130.46, 129.85, 128.31, 123.79, 122.46, 122.27, 119.26, 119.18, 118.56, 118.04, 114.28, 55.40, 53.84, 34.46. HRMS–FAB (m/z) [M+H]+ calcd for C27H23N5O4, 482.1823; found, 482.1803.
Supplementary Material
Acknowledgment
Initial support by the NIH (R01 AI 05419) of the antituberculosis research program that generated the lead compounds is gratefully acknowledged. We would like to thank the University of Notre Dame especially the Mass Spectrometry & Proteomics Facility (Bill Boggess, Nonka Sevova, Michelle Joyce) which is supported by the by Grant CHE-0741793 from the National Science Foundation and Dr. Jaroslav Zajicek for assistance in obtaining mass spectral and NMR data, respectively. We thank Dr. Kamlesh Gupta and Prof. Holly Goodson for advice regarding the tubulin experiments and Dr. Timothy Mitchison for the gift of purified tubulin. We also thank Dr. Jed Fisher for helpful discussions. We acknowledge financial support from the University of Notre Dame (J.A.S. was supported by a Notre Dame College of Science Summer Undergraduate Research Fellowship). We would also like to acknowledge the assistance of the Cell Flow Cytometry facility of the Biotechnology Center at the University of Illinois at Urbana-Champaign.
Abbreviations
- CuAAC
copper-catalyzed azide–alkyne cycloaddition
- MIC
minimum inhibition concentration to kill 90% of the bacterium
- GI50
50% growth inhibition
- TGI
total growth inhibition
- LC50
50% lethal concentration, IC50, half maximal inhibitory concentration
- TB
Mycobacterium tuberculosis
- GAST
medium of glycerol-alanine-salts-Tween 80 without added iron
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- S.E.M.
standard error of the mean
Footnotes
Supporting Information Available. Materials and methods, characterization data for all compounds, cell proliferation curves, cell cycle arrest profiles, and COMPARE analysis results.
References
- 1.Jordan MA, Wilson L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.(a) Mitchison TJ. Microtubule Dynamics and Kinetochore Function in Mitosis. Annu. Rev. Cell Biol. 1988;4:527–549. doi: 10.1146/annurev.cb.04.110188.002523. [DOI] [PubMed] [Google Scholar]; (b) Rusan NM, Fagerstrom CJ, Yvon AC, Wadsworth P. Cell Cycle-Dependent Changes in Microtubule Dynamics in Living Cells Expressing Green Fluorescent Protein-α Tubulin. Mol. Biol. Cell. 2001;12:971–980. doi: 10.1091/mbc.12.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jordan MA. Mechanism of Action of Antitumor Drugs that Interact with Microtubules and Tubulin. Curr. Med. Chem. – Anti-Cancer Agents. 2002;2:1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- 4.(a) Islam MN, Iskander MN. Microtubulin Binding Sites as Target for Developing Anticancer Agents. Mini-Reviews in Medicinal Chemistry. 2004;4:1077–1104. doi: 10.2174/1389557043402946. [DOI] [PubMed] [Google Scholar]; (b) Hadfield JA, Ducki S, Hirst N, McGown AT. Tubulin and Microtubules as Targets for Anticancer Drugs. Progress in Cell Cycle Research. 2003;5:309–325. [PubMed] [Google Scholar]; (c) Hamel E. Antimitotic Natural Products and Their Interactions with Tubulin. Med. Res. Rev. 1996;16:207–231. doi: 10.1002/(SICI)1098-1128(199603)16:2<207::AID-MED4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Kuppens IELM. Current State of the Art of New Tubulin Inhibitors in the Clinic. Curr. Clin. Pharmacol. 2006;1:57–70. doi: 10.2174/157488406775268200. [DOI] [PubMed] [Google Scholar]
- 7.(a) Jordan A, Hadfield JA, Lawrence NJ, McGown AT. Tubulin as a Target for Anticancer Drugs: Agents Which Interact with the Mitotic Spindle. Med. Res. Rev. 1998;18:259–296. doi: 10.1002/(sici)1098-1128(199807)18:4<259::aid-med3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (b) Kiselyov A, Balakin KV, Tkachenko SE, Savchuk N, Ivachtchenko AV. Recent Progress in Discovery and Development of Antimitotic Agents. Anti-Cancer Agents in Med. Chem. 2007;7:189–208. doi: 10.2174/187152007780058650. [DOI] [PubMed] [Google Scholar]
- 8.Backer G, Beckers T, Emig P, Klenner T, Kutscher B, Nickel B. New small-molecule tubulin inhibitors. Pure Appl. Chem. 2001;73:1459–1464. [Google Scholar]
- 9.Wood KW, Cornwell WD, Jackson JR. Past and future of the mitotic spindle as an oncology target. Curr. Opin. Pharmacol. 2001;1:370–377. doi: 10.1016/s1471-4892(01)00064-9. [DOI] [PubMed] [Google Scholar]
- 10.(a) Snow GA. Mycobactins: Iron-Chelating Growth Factors from Mycobacteria. Bacteriol. Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) White AJ, Snow GA. Isolation of Mycobactins form Various Mycobacteria: The Properties of Mycobactins S and H. Biochem. J. 1969;111:785–792. doi: 10.1042/bj1110785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Maurer PJ, Miller MJ. Total Synthesis of a Mycobactin: Mycobactin S2. J. Am. Chem. Soc. 1983;105:240–245. [Google Scholar]; (b) Hu J, Miller MJ. Total Synthesis of a Mycobactin S, a Siderophore and Growth Promoter of Mycobacterium Smegmatis, and Determination of its Growth Inhibitory Activity against Mycobacterium tuberculosis. J. Am. Chem. Soc. 1997;119:3462–3468. [Google Scholar]
- 12.Vergne AF, Walz AJ, Miller MJ. Iron chelators from mycobacteria (1954–1999) and potential therapeutic applications. Nat. Prod. Rep. 2000;17:99–116. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 13.(a) Moraski GC, Chang M, Villegas-Estrada A, Franzblau S, Möllmann U, Miller MJ. Structure-Activity Relationship of New Antituberculosis Agents Derived from Oxazoline and Oxazole Benzyl Esters. Eur. J. Med. Chem. 2010 doi: 10.1016/j.ejmech.2009.12.074. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moraski GC, Franzblau SG, Miller M. Utilization of the Suzuki Coupling to Enhance the Antituberculosis Activity of Aryl Oxazoles. Heterocyles. 2010;80:977–988. doi: 10.3987/COM-09-S(S)69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fennell KA, Miller MJ. Synthesis of Amamistatin Fragments and Determination of Their HDAC and Antitumor Activity. Org. Lett. 2007;9:1683–1685. doi: 10.1021/ol070382e. [DOI] [PubMed] [Google Scholar]
- 15.Siddiquee KAZ, Gunning PT, Glenn M, Katt WP, Zhang S, Schroeck C, Sebti SM, Jove R, Hamilton AD, Turkson J. An Oxazole-Based Small-Molecule Stat3 Inhibitor Modulates Stat3 Stability and Processing and Induces Antitumor Cell Effects. ACS Chem. Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- 16.Kuang R, Shue HJ, Blythin DJ, Shih NY, Gu D, Chen X, Schwerdt J, Lin L, Ting PC, Zhu X, et al. Discovery of a highly potent series of oxazole-based phosphodiesterase 4 inhibitors. Bioorg. Med. Chem. Lett. 2007;17:5150–5154. doi: 10.1016/j.bmcl.2007.06.092. [DOI] [PubMed] [Google Scholar]
- 17.(a) Rice RL, Rusnak JM, Yokokawa F, Yokokawa S, Messner DJ, Boynton AL, Wipf P, Lazo JS. A Targeted Library of Small-Molecule, Tyrosine, and Dual-Specificity Phosphatase Inhibitors Derived from a Rational Core Design and Random Side Chain Variation. Biochemistry. 1997;36:15965–15974. doi: 10.1021/bi971338h. [DOI] [PubMed] [Google Scholar]; (b) Ducruet AP, Rice RL, Tamura K, Yokokawa F, Yokokawa S, Wipf P, Lazo JS. Identification of New Cdc25 Dual Specificity Phosphatase Inhibitors in a Targeted Small Molecule Array. Bioorg. Med. Chem. 2000;8:1451–1466. doi: 10.1016/s0968-0896(00)00069-9. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi K, Kohn TJ, True TA, Mais DE, Wikel JH, Utterback BG, Wyss VL, Jakubowski JA. Development of Dual-Acting Agents for Thromboxane Receptor Antagonism and Thromboxane Synthase Inhibition. 3. Synthesis and Biological Activities of Oxazolecarboxamide-Substituted ω-Phenyl-ω-(3-pyridyl)alkenoic Acid Derivatives and Related Compounds. J. Med. Chem. 1998;41:5362–5374. doi: 10.1021/jm980173n. [DOI] [PubMed] [Google Scholar]
- 19.Morwick T, Berry A, Brickwood J, Cardozo M, Catron K, DeTuri M, Emeigh J, Homon C, Hrapchak M, Jacober S. Evolution of the Thienopyridine Class of Inhibitors of IκB Kinase-β: Part I: Hit-to-Lead Strategies. J. Med. Chem. 2006;49:2898–2908. doi: 10.1021/jm0510979. [DOI] [PubMed] [Google Scholar]
- 20.Tai VWF, Sperandio D, Shelton EJ, Litvak J, Pararajasingham K, Cebon B, Lohman J, Eksterowicz J, Kantak S, Sabbatini P, et al. Discovery and structure-activity relationship of 2-phenyl-oxazole-4-carboxamide derivatives as potent apotosis inducers. Bioorg. Med. Chem. Lett. 2006;16:4554–4558. doi: 10.1016/j.bmcl.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 21.GAST medium is glycerol-alanine-salts-Tween 80 medium without added iron. See, Voss JJD, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Low-Oxygen-Recovery Assay for High-Throughput Screening of Compounds against Nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06.
- 22.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Synthesis of Functionalized Oxazolines and Oxazoles with DAST and Deoxo-Fluor. Org. Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- 23.(a) Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (b) Moses JE, Moorhouse AD. The Growing Applications of Click Chemistry. Chem. Soc. Rev. 2007;36:1249–1262. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 24.(a) Rostovtsev VV, Green LG, Fokin VV, Sharpless KBA. Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (b) Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 25.Kolb HC, Sharpless KB. The Growing Impact of Click Chemistry on Drug Discovery. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez SG, Alvarez MT. A Practical Procedure for the Synthesis of Alkyl Azides at Ambient Temperature in Dimethyl Sulfoxide in High Purity and Yield. Synthesis. 1997;4:413–414. [Google Scholar]
- 27.(a) Boyd MR, Paull KD. Some Practical Considerations and Applications of the National Cancer Institute In Vitro Anticancer Drug Discovery Screen. Drug Develop. Res. 1995;34:91–109. [Google Scholar]; (b) Shoemaker RH. The NCI60 Human Tumor Cell Line Anticancer Drug Screen. Nature Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 28.(a) Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JCF, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. Systematic variation of gene expression patterns in human cancer cell lines. Nature Genetics. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]; (b) Ellison G, Klinowska T, Westwood RFR, Docter E, French T, Fox JC. Further evidence to support the melanocytic origin of MDA-MB-435. J. Clin. Pathol: Mol. Pathol. 2002;55:294–299. doi: 10.1136/mp.55.5.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]; (b) Zaharevitz DW, Holbeck SL, Bowerman C, Svetlik PA. COMPARE: a web accessible tool for investigating mechanisms of cell growth inhibition. J. Mol. Graphics Modell. 2002;20:297–303. doi: 10.1016/s1093-3263(01)00126-7. [DOI] [PubMed] [Google Scholar]
- 30.(a) Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J. Natl. Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]; (b) Zaharevitz DW, Holbeck SL, Bowerman C, Svetlik PA. COMPARE: a web accessible tool for investigating mechanisms of cell growth inhibition. J. Mol. Graphics Modell. 2002;20:297–303. doi: 10.1016/s1093-3263(01)00126-7. [DOI] [PubMed] [Google Scholar]
- 31.DTP Human Tumor Cell Line Screen, Standard Agent Database. Online: http://dtp.nci.nih.gov/docs/cancer/searches/standard_agent.html.
- 32.(a) Bai R, Paull KD, Herald CL, Malspeis L, Pettit GR, Hamel E, Halichondrin B, Homohalichondrin B. Marine Natural Products Binding in the Vinca Domain of Tubulin. J. Biol. Chem. 1991;266:15882–15889. [PubMed] [Google Scholar]; (b) Paull KD, Lin CM, Malspeis L, Hamel E. Identification of Novel Antimitotic Agents Acting at the Tubulin Level by Computer-assisted Evaluation of Differential Cytotoxicity Data. Cancer Res. 1992;52:3892–3900. [PubMed] [Google Scholar]; (c) Kuo SC, Lee HZ, Juang JP, Lin YT, Wu TS, Chang JJ, Lednicer D, Paull KD, Lin CM, Hamel E, Lee KH. Synthesis and Cytotoxicity of 1,6,7,8-Substituted 2-(4’-Substituted phenyl)-4-quinolones and Related Compounds: Identification as Antimitotic Agents Interacting with Tubulin. J. Med. Chem. 1993;36:1146–1156. doi: 10.1021/jm00061a005. [DOI] [PubMed] [Google Scholar]
- 33.Stefely JA, Moraski GA, Miller MJ. Anti-cancer Compounds, Synthesis Thereof, And Methods Of Using Same. WO/2009/111502. [Google Scholar]
- 34.Hollingshead MG, Alley MC, Camalier RF, Abbott BJ, Mayo JG, Malspeis L, Grever MR. In vivo cultivation of tumor cells in hollow fibers. Life Sci. 1995;57:131–141. doi: 10.1016/0024-3205(95)00254-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.