

Abstract
X-linked hypophosphatemic rickets (XLH) in humans is caused by mutations in the PHEX gene. Previously, three mutations in the mouse Phex gene have been reported: PhexHyp, Gy, and PhexSka1. Here we report analysis of two new spontaneous mutations in the mouse Phex gene, PhexHyp-2J and PhexHyp-Duk. PhexHyp-2J and PhexHyp-Duk involve intragenic deletions of at least 7.3 kb containing exon 15, and 30 kb containing exons 13 and 14, respectively. Both mutations cause similar phenotypes in males, including shortened hind legs and tail, a shortened square trunk, hypophosphatemia, hypocalcemia, and rachitic bone disease. In addition, mice carrying the PhexHyp-Duk mutation exhibit background-dependent variable expression of deafness, circling behavior, and cranial dysmorphology, demonstrating the influence of modifying genes on Phex-related phenotypes. Cochlear cross-sections from PhexHyp-2J/Y and PhexHyp-Duk/Y males reveal a thickening of the temporal bone surrounding the cochlea with the presence of a precipitate in the scala tympani. Evidence of the degeneration of the organ of Corti and spiral ganglion also are present in the hearing-impaired PhexHyp-Duk/Y mice, but not in the normal-hearing PhexHyp-2J/Y mice. Analysis of the phenotypes noted in PhexHyp-Duk/Y an PhexHyp-2J/Y males, together with those noted in PhexSka1/Y and PhexHyp/Y males, now allow XLH-related phenotypes to be separated from non-XLH-related phenotypes, such as those noted in Gy/Y males. Also, identification of the genetic modifiers of hearing and craniofacial dysmorphology in PhexHyp-Duk/Y mice could provide insight into the phenotypic variation of XLH in humans.
X-linked dominant hypophosphatemia (XLH) is the most frequently occurring familial form of hypophosphatemic rickets in humans with an incidence of 1 per 20,000 individuals (Burnett et al. 1964; Tenenhouse 1999). XLH is caused by loss-of-function mutations in PHEX (phosphate regulating gene with homologies to endopeptidases on the X-chromosome), a gene that encodes an endopeptidase of unknown function (The HYP Consortium 1995). Three X-linked mutations have been identified in the mouse orthologous Phex gene: hypophosphatemia, symbolized PhexHyp, hereafter Hyp (Eicher et al. 1976); skeletal abnormality 1, symbolized PhexSka1, hereafter Ska1 (Carpinelli et al. 2002); and gyro, symbolized Gy (Lyon et al. 1986). Clinical and biochemical abnormalities observed in Hyp/Y, Ska1/Y and Gy/Y mice and in patients with XLH include renal phosphate wasting, hypophosphatemia, impaired mineralization, and growth retardation.
The Hyp mutation is a spontaneous deletion that begins in exon 15 and extends through exons 16–22 (Strom et al. 1997) into downstream intergenic sequences (Sabbagh et al. 2002). The Ska1 mutation is a chemically induced point mutation in a splice donor site immediately following exon 8 (Carpinelli et al. 2002). The Gy mutation is a radiation-induced deletion that removes 160–190 kb, including exons 1–3 and the Sms (spermine synthase) gene (Lorenz et al. 1998; Strom et al. 1997). Given that the gyro deletion includes more than one gene, the previous allelic symbol PhexGy is inadequate; this mutation is here renamed gyro deletion region, symbol Gy (Mouse Genome Database, http://www.informatics.jax.org). Because neither the Hyp nor Gy mutations is restricted to the Phex coding sequence, it has been problematic to assign a specific phenotype solely to the Phex gene.
Here we report the identification of two new spontaneous intragenic deletions in the mouse Phex gene, hypophosphatemia-2 Jackson, symbolized PhexHyp-2J (Hyp-2J) and hypophosphatemia-Duke, symbolized, PhexHyp-Duk (Hyp-Duk). Mice carrying either mutation, together with those carrying Ska1, represent valuable animal models for human XLH because each mutation is restricted solely to the Phex gene. In addition, the resulting abnormal phenotypes produced by these mutations help clarify the specific contribution of a mutated Phex gene to the overall phenotypes observed in Hyp/Y and Gy/Y mice. Finally, the findings reported here may help define the function of Phex in the pathway of bone mineralization and phosphate homeostasis, as well as provide insight into the varying severity noted in humans with XLH.
Materials and methods
Phex alleles
The Hyp-2J allele is a spontaneous mutation that occurred in an inbred C57BL/6J mouse at The Jackson Laboratory and is maintained by mating a Hyp-2J/+ female to a C57BL/6J +/Y male. The Hyp-Duk allele arose at Duke University as a spontaneous mutation in an inbred strain of BALB/cAnBom-Foxn1nu, and mice carrying Hyp-Duk were imported to The Mouse Mutant Resource at The Jackson Laboratory (Davisson 1990). Hyp-Duk is maintained by mating a Hyp-Duk/+ female to a +/Y male or a +/+ female to a Hyp-Duk/Y male (for simplicity, BALB/cAnBomUrd is shortened to BALB/cUrd). The origins of the Hyp and Gy mutations have previously been reported (Eicher et al. 1976; Lyon et al. 1976). Hyp is maintained by mating a Hyp/+ female to a C57BL/6J +/Y male. Gy is maintained by mating a Gy/+ female to a B6EiC3SnF1-a/A +/Y male.
Mice
Mice were housed in polycarbonate cages (51 sq in) on sterilized Northern White Pine shavings bedding and maintained under 14:10 hour light:dark cycles. Acidified water (pH of 2.8–3.2) and sterilized NIH 31 diet (6% fat diet, Ca:P of 1.15:0.85, 19% protein, vitamin and mineral fortified; Purina Mills International) were freely available. All analyses were conducted with mutant males and same-sex littermate controls. (An exception involved craniofacial examinations in which inbred C57BL/6J males were used for comparison with Hyp-2J/Y males.) For each experiment, the numbers of mutant and control males are provided in the Results section.
Serum phosphorus and calcium levels
Blood was obtained from the retro-orbital sinus of male mice at 6 weeks of age and allowed to clot on ice for 2–3 h. Serumwas isolated by centrifugation at 7200 g for 10 min and stored at 4°C. Serum phosphorus and calcium levels were determined with the Beckman SYNCHRON CX5 DELTA System (Beckman Coulter, Inc.). To monitor changes in calibration, along with analytical error and imprecision, Liquid Comprehensive Chemistry Control Serum Levels 1, 2, and 3 were used according to the manufacturer’s instructions for Quality Control. The limits of detection for Ca2+ and PO4 are between 2 and 15 mg/dL and 1.0 and 12.0 mg/dL respectively.
PIXImus measurements
Areal Bone Mineral Density (aBMD), % lean body mass, and total body weight were determined on 6-week-old males anesthetized by intraperitoneal injection with Avertin (tribromoethanol stabilized in tertiary amyl hydrate), 1 mg per 2 g of body weight. Measurements were determined by using the PIXImus small animal dual-energy X-ray absorptiometry (DEXA) system (Lunar Corp.), with data analysis by software version 1.46. Areal bone mineral density is a two-dimensional measurement comprised of mineral within the area determined to be bone by the preset thresholds in the PIXImus densitometer. Lean body mass measured by the PIXImus includes both lean mass and body water. The combined value is divided by the total body weight and multiplied by 100 to derive % lean body mass. The resolution of the PIXImusis 0.18 ×0.18mm pixels with a usable scanning field of 80 × 65 mm, allowing for measurement of single whole mice and collections of isolated specimens. Calibrations were performed with a phantom of known density, and quality assurance measurements were performed daily with this same phantom. Assessment of accuracy for the PIXImus was done with a set of hydroxyapatite standards (0–2000 mg), yielding a correlation of 0.999 between standards and PIXImus measurement of mineral. The precision for aBMD is >99% for whole body and ~98.5% for specialized regions.
Whole-body X-ray photography
X-ray films were taken of male mice at 6 weeks of age with a Faxitron MX-20 Specimen Radiography System (Wheeling, Illinois) and Kodak MR 2000-1 film (Eastman-Kodak). Measurements were taken at 23KV at 5×magnitude for 3 s.
Craniofacial measurements
Craniofacial measurements on skulls were determined on male mice at 12 weeks of age. Skull measurement landmark descriptions (Fig. 1) and preparation protocols can be found at The Jackson Laboratory Craniofacial Mutant Resource’s website (http://www.jax.org/cranio/characteristics.html).
Fig. 1.

Landmarks for craniofacial hand caliper measurements.
Histology
Tissues were fixed in anesthetized mice by intracardiac perfusion of Bouin’s fixative following a 10 to 20 mL flush with physiological saline. Representative paraffin histological sections of all organs were prepared and stained with hematoxylin and eosin (H&E). Inner ears were dissected, demineralized in Bouin’s fixative, serial sectioned, and stained with H&E. To identify precipitate observed in perilymph and endolymph of the inner ear, we stained sections with periodic acid-Schiff (PAS), phosphatungstic acid hematoxylin (PTAH), and von Kossa stains. For gross examination, whole mounts of inner ears were fixed and cleared for examination as described previously (Ward-Bailey et al. 2000).
Auditory brainstem response (ABR) analysis
Hearing was assessed in mutant and control males between 4 and 42 weeks of age by ABR threshold analysis with equipment from Intelligent Hearing Systems with previously described methods and equipment (Zheng et al. 1999). Evoked responses to broadband click and pure-tone 8 kHz, 16 kHz, and 32 kHz auditory stimuli were recorded; however, for clarity of presentation, only click responses are presented. (The three pure-tone auditory stimuli gave similar results.)
RT-PCR analysis
Total RNA was prepared from brain tissue of Hyp-2J/Y, Hyp-Duk/Y, and respective control mice with Trizol reagent (Life Technologies) as recommended by the manufacturer. First-strand cDNA synthesis was carried out from 1 μg of total RNA with oligo (dT) primer and reverse transcriptase (Amersham Biosciences). As a PCR template, 1–10 ng of first-strand cDNA was used for nested PCR reactions with Phex-specific primers. The amplification products were used for direct DNA sequencing.
DNA sequencing
PCR-amplified cDNA was sequenced with a BigDye terminator cycle sequencing kit (Applied Biosystems) and Phex-specific primers on an ABI 3100 automated sequencer.
Southern blot analysis
Genomic DNA (~5 μg) from Hyp-2J/Y, Hyp-Duk/Y, and +/Y littermate controls was extracted from kidney or tail tip, digested with HindIII, electrophoresed on 0.7% agarose gels, and blotted to Nylon membranes (PALL) by alkaline transfer. Hybridizations were generally performed in a hybridization buffer containing 1.5 × SSPE, 1% SDS and 10% dextran sulfate at 65°C. Probes were labeled by random hexamer priming. Washing was done under stringent conditions (0.1 × SSC, 0.1% SDS at 65°C for 15 min). The mouse genome assembly sequence (http://www.ensemble.org) was used to determine the expected length of the HindIII restriction fragments containing Phex exons 12–18.
Genomic PCR
Primers designed from the mouse genome assembly sequence (http://www.ensemble.org) were used for DNA amplification of all 22 Phex exons (Table 3). After an initial denaturation for 5 min at 95°C, amplification cycles consisted of denaturation at 95°C for 30 s, annealing at the exon-specific temperature for 30 s, and 30 s extension at 72°C for 35 cycles, followed by a final extension for 5 min at 72°C. Reaction volumes (25 μL) contained 50 mM KCl, 10 mM Tris-HCl (pH 8.0), 1.5 mM MgCl2, 200 μM of each dNTP, 0.3 μM of each primer, 1 U AmpliTaq Gold polymerase (Applied Biosystems), and 100 ng of genomic mouse DNA.
Table 3.
Primers used for amplification of Phex exons 1–22
| Exon | Primer forward | Primer reverse | bp | °C |
|---|---|---|---|---|
| 1 | 5′-AGAGTCTTGAATATCAAACGCC-3′ | 5′-AAGAGTCTCCAGAGATGCCC-3′ | 260 | 62 |
| 2 | 5′-AACTGTCTTGCGTATGTGCC-3′ | 5′-TTTGGTCAACCTCAAGCAAAC-3′ | 266 | 60 |
| 3 | 5′-TCTTGTCAAACAGTGTTCTGG-3′ | 5′-CCAGGGAGACATTTGAGGAG-3′ | 300 | 60 |
| 4 | 5′-TTTTCTGGAGGCTGACCTTC-3′ | 5′-TTCTATTGCCCAAACAACACC-3′ | 296 | 60 |
| 5 | 5′-AGCTTGGTGAACGAGTTTGG-3′ | 5′-CATGTGTGACATCCTAAATAGC-3′ | 506 | 60 |
| 6 | 5′-TGGAGTGAACTTATTTCTTGGG-3′ | 5′-ACCAAACAGTAGCCAGACAAG-3′ | 258 | 60 |
| 7 | 5′-TCCTTCCTACCAGGCTGTTG-3′ | 5′-TTGGATACAAAGGGCAATGGC-3′ | 304 | 62 |
| 8 | 5′-CATGTGAAAGGCAGTCATGC-3′ | 5′-AGGCCAAATTTTATACACAGATG-3′ | 240 | 60 |
| 9 | 5′-CAGAATGGATTATGCTCGGC-3′ | 5′-TTTCAAAGCAATTTTCACCGTAC-3′ | 333 | 60 |
| 10 | 5′-TTGCCAACAGTTTTCCAAAGG-3′ | 5′-AAGCTCCCTACATCCCATCC-3′ | 293 | 60 |
| 11 | 5′-GACTGGTGTGGGATGGAATC-3′ | 5′-TCTTGTCTGTCAGATCTGCC-3′ | 280 | 60 |
| 12 | 5′-TTCAGGTCAAGGAAACCTGG-3′ | 5′-AGCACACACCTACCTCTGGG-3′ | 309 | 60 |
| 13 | 5′-GAAGACTGAAGCCCTATATCC-3′ | 5′-TTAGTTAATAGGCATCAGGCC-3′ | 230 | 60 |
| 14 | 5′-ATAGCGTCTCTTCTGGTTGC-3′ | 5′-GCTGGCTACCCTGAGTTGAG-3′ | 308 | 60 |
| 15 | 5′-AGTCTTGCCAAACTGTGCTC-3′ | 5′-GTCACTTACATTGAAAACACTC-3′ | 387 | 60 |
| 16 | 5′-TTGGGCTATAGTTGGAACCAG-3′ | 5′-AGATATTCTGTCCTTGGCCG-3′ | 217 | 60 |
| 17 | 5′-AAGGATTATGCTGTCAGATTCATATC-3′ | 5′-ACAATTAACACCATACAATCAAC-3′ | 197 | 60 |
| 18 | 5′-TGTGGGAGCTAATCTCAGGG-3′ | 5′-GAAGAAAAATGGCCCACAAGG-3′ | 329 | 62 |
| 19 | 5′-GCTTGGGCTAGTTTGCTATCTC-3′ | 5′-TGAGTTGGTGCTATACACGGAG-3′ | 304 | 62 |
| 20 | 5′-ACCCATGGCAGGAATATGTG-3′ | 5′-TGCCAGTGAGTTTTTCCTCC-3′ | 294 | 60 |
| 21 | 5′-CCAAAATTGTTCTTCAGTACACC-3′ | 5′-ATCTGGCAGCACACTGGTATG-3′ | 258 | 64 |
| 22 | 5′-TTTGTAGTGGAGTATTTCATCC-3′ | 5′-AGGCAGAAAGCAGTAGGTGC-3′ | 324 | 60 |
Statistical analysis
Descriptive statistics are presented as mean ± SEM. All statistical analyses were performed with StatView 4.5 software for Macintosh (Abacus Concepts). Individual genotype means were assessed for significant differences by Fisher’s Protected LSD test. Differences were deemed statistically significant when p ≤ 0.05 unless otherwise noted.
Results
New Phex mutations
The spontaneous X-linked Hyp-2J mutation arose at The Jackson Laboratory in 1998 in the C57BL/6J inbred mouse strain. Hemizygous males (Hyp-2J/Y) and heterozygous females (Hyp-2J/+) resembled Hyp/Y males and Hyp/+ females, respectively, with shortened hind legs and tail, and a squared trunk. Because Phex was an obvious candidate gene for the observed phenotypes noted in Hyp-2J mutant mice, its molecular characterization was pursued (see molecular analysis below).
The Hyp-Duk mutation arose spontaneously in an inbred BALB/cAnBomUrd-Foxn1nu mouse strain at the Department of Urology Research, Duke University. All affected males are mildly growth retarded in overall body size, with a squared shortened body, shortened hind limbs and tail, and some exhibit circling behavior. In contrast, not all Hyp-Duk/+ females show evidence of growth retardation and circling phenotypes, making it difficult to distinguish some heterozygous (Hyp-Duk/+) females from normal (+/+) females. For this reason, only Hyp-Duk/Y males were chosen for examination. The Hyp-Duk mutation was mapped utilizing an F2 intercross with CAST/Ei to the region of the X Chr containing the Phex gene, and serum phosphate analysis indicated that affected males were hypophosphatemic (Table 1). Molecular testing of Phex as a candidate gene then was pursued.
Table 1.
Clinical characterization of Phex mutants and controls
| Genotype | PO4 (mg/dL) | Ca2+ (mg/dL) | ABMD (g/cm2) | Weight (g) | % Lean |
|---|---|---|---|---|---|
| Hyp-Duk/Y | 2.95 ± 0.25* | 8.42 ± 0.12* | 0.027 ± 3.77E-4* | 16.08 ± 0.32* | 90.76* |
| +/Y | 9.55 ± 0.22 | 9.63 ± 0.14 | 0.037 ± 0.001 | 20.25 ± 0.42 | 88.69 |
| Hyp-2J/Y | 3.58 ± 0.19* | 8.38 ± 0.06* | 0.026 ± 2.71E-4* | 16.41 ± 0.11* | 91.75 |
| +/Y | 9.48 ± 0.53 | 9.80 ± 0.38 | 0.037 ± 0.001 | 21.82 ± 0.47 | 90.89 |
| Hyp/Y | 4.45 ± 0.22* | 9.15 ± 0.23* | 0.025 ± 4.93E-4* | 16.85 ± 0.25* | 90.98 |
| +/Y | 10.47 ± 0.81 | 10.93 ± 0.68 | 0.037 ± 0.001 | 21.89 ± 0.61 | 90.61 |
| Gy/Y | 4.23 ± 0.24* | 9.17 ± 0.10* | 0.026 ± 0.001* | 11.31 ± 1.02* | 89.87* |
| +/Y | 8.68 ± 0.37 | 10.55 ± 0.68 | 0.039 ± 0.001 | 25.34 ± 0.88 | 87.35 |
All mutants and controls examined at 6 weeks of age.
Data presented are mean ± SEM.
For all values n = 6.
p ≤ 0.05.
Serum chemistry
Serum PO4 and Ca2+ levels were assessed at 6 weeks of age in Hyp/Y, Hyp-2J/Y, Hyp-Duk/Y, Gy/Y males and same-sex littermate controls by using the Beckman Synchron CX5 DELTA System (Beckman Coulter, Inc.). All mutant males had serum PO4 levels (2.95–4.45 mg/dL) significantly lower than controls (Table 1). Statistically significant hypocalcemia also was present in all mutants within the same range (8.38–9.17 mg/dL) (Table 1). The serum PO4 and Ca2+ levels found in Hyp/Y and Gy/Y mice were consistent with those previously reported (Eicher et al. 1976; Lyon et al. 1986).
Histology
Whole-body histopathological examination was conducted on mutant and control males between 6 and 40 weeks of age. In general, under-mineralized bone was present throughout the body of all mutant males. Growth plates of the knee were thickened and irregular (Fig. 2). A whole-body examination found no obvious lesions in any major organs.
Fig. 2.
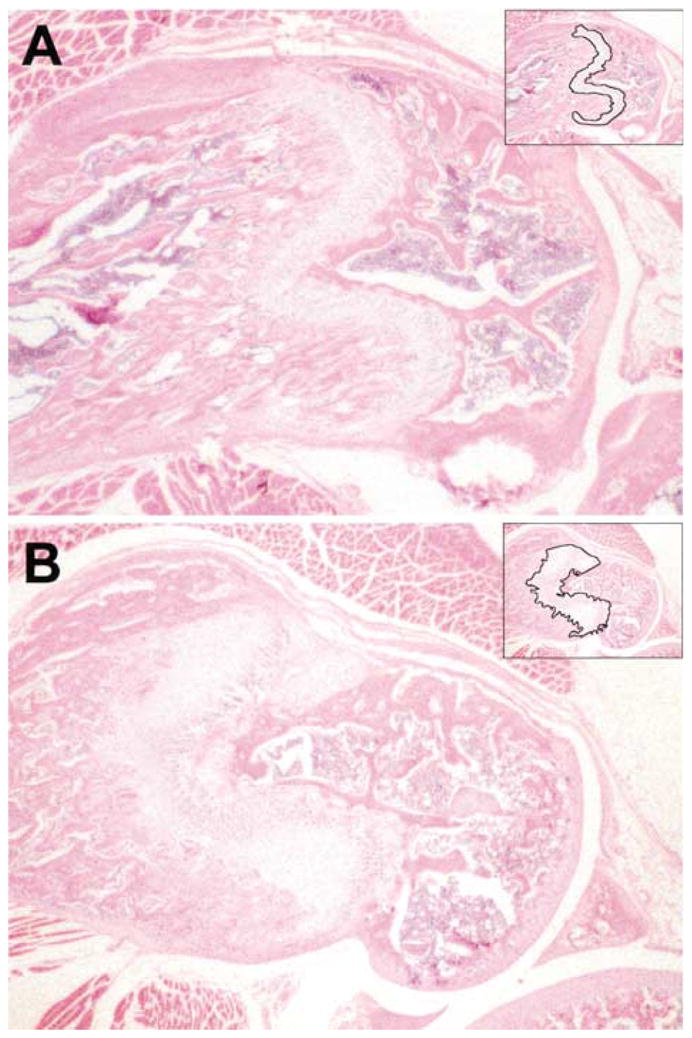
Cross-section of the distal femurs of a Hyp-Duk/Y mutant male (B) compared with a littermate +/Y control (A) at 6 weeks of age. The growth plate (outlined in the upper right hand corner) of the mutant is thicker than that of the control. The proximal and distal sides of the mutant growth plate are not parallel. Similar pathology was found in Hyp-2J/Y mutant males (data not shown).
Body composition, bone density, and X-ray analyses
Mutant and littermate control males were analyzed at 6 weeks of age by dual-energy X-ray absorptiometry (DEXA) (PIXImus, Lunar Corp.) for whole-body aBMD and body composition (Table 1). All four types of mutants had significantly lower whole-body aBMD and a trend toward higher lean body mass than wildtype male controls. Differences in lean body mass were significant only in Gy/Y and Hyp-Duk/Y males when compared with controls. In addition, Gy/Y males weighed significantly less than the other three types of mutant males and ~50% less than their wildtype sibling male controls. In contrast, Hyp/Y, Hyp-2J/Y and Hyp-Duk/Y males weighed ~25% less than their male controls. Subsequent to DEXA analysis, an X-ray at 5×magnification was taken of the body by using a Faxitron MX-20 Specimen Radiography System (Faxitron X-ray Corp.). In all mutant males, shortened and thickened long bones with very disorganized femoral growth plates and splayed tibia and fibula were noted, a finding previously reported in Hyp/Y and Gy/Y mice (Eicher et al. 1976; Lyon et al. 1986).
Craniofacial characterization
Morphological data on skulls were collected from Hyp-Duk/Y and Hyp-2J/Y males and their respective controls at 12 weeks of age (Table 2). Skulls of Hyp-2J/Y mice were significantly shorter in length and width, lower in aBMD, and significantly larger in inner canthal distance than control males. However, skull height, nose length, and lower/upper jaw length were not significantly different.
Table 2.
Craniofacial characterization of Phex mutants
| Hyp-Duk/Y | +/Y | Hyp-2J/Y | +/Y | |
|---|---|---|---|---|
| Skull | ||||
| aBMD (g/cm2) | 0.090 ± 0.003* | 0.107 ± 0.002 | 0.085 ± 0.001* | 0.105 ± 0.001a |
| Height (mm) | 9.69 ± 0.05 | 9.48 ± 0.12 | 9.82 ± 0.11 | 10.34 ± 0.27 |
| Width (mm) | 10.45 ± 0.08 | 10.37 ± 0.09 | 10.48 ± 0.15* | 11.12 ± 0.22 |
| Length (mm) | 21.48 ± 0.31* | 22.63 ± 0.14 | 21.86 ± 0.10* | 22.91 ± 0.22 |
| Nose | ||||
| Length (mm) | 14.12 ± 0.29* | 15.09 ± 0.09 | 14.99 ± 0.16 | 15.34 ± 0.15 |
| Skull/nose length ratio | 1.52 ± 0.02 | 1.50 ± 0.01 | 1.46 ± 0.01* | 1.49 ± 0.01 |
| Inner canthal distance (mm) | 3.79 ± 0.07 | 3.69 ± 0.07 | 5.05 ± 0.09* | 4.20 ± 0.04 |
| Jaw length | ||||
| Lower (mm) | 10.86 ± 0.16 | 10.73 ± 0.17 | 10.99 ± 0.11 | 11.13 ± 0.28 |
| Upper (mm) | 14.62 ± 0.19* | 15.41 ± 0.13 | 15.67 ± 0.28 | 15.40 ± 0.24 |
| Jaw length ratio | 1.35 ± 0.02* | 1.43 ± 0.01 | 1.43 ± 0.03 | 0.39 ± 0.02 |
Mutants and controls were examined at 12 weeks of age.
Data presented are mean ± SEM.
For all values n = 6 unless otherwise noted (an = 10).
p ≤ 0.05.
Comparison of Hyp-Duk/Y skulls with +/Y littermate control skulls revealed a reduction in nose, upper jaw, and overall skull length, as well as a reduction in skull aBMD. However, skull width and skull-to-nose length ratio were not different (Table 2).
Auditory brainstem response (ABR) analysis and ear pathology
The average ABR thresholds of Gy/Y, Hyp/Y and Hyp-Duk/Y males were significantly higher than those of +/Y controls, but not for Hyp-2J/Y males (Fig. 3). Mean ABR threshold differences between mutants and controls were 48 dB for Hyp-Duk (t-test p < 0.0001), 20 dB for Hyp (p = 0.002), 21 dB for Gy (p = 0.02), and 5 dB for Hyp-2J (p = 0.4). The average ABR thresholds of Gy/Y and Hyp/Y males were not significantly different from each other (p = 0.38), but were slightly higher than those of Hyp-2J/Y mice (p < 0.01) and much lower than those of Hyp-Duk/Y males (p < 0.0001).
Fig. 3.

Hearing impairment associated with Phex mutations. Histogram depicts click ABR threshold means (with bars indicating two standard errors) for (Gy/Y; 6 mice, +/Y; 3 mice), (Hyp/Y; 4 mice, +/Y; 5 mice), (Hyp-2J/Y; 13 mice, +/Y; 5 mice), (Hyp-Duk/Y; 20 mice, +/Y; 11 mice), and (BALB/cUrd × BALB/cByJ) F1-Hyp-Duk/Y (F1-Hyp-Duk/Y; 5 mice). Increased values are indicative of hearing impairment (* = p ≤ 0.05).
Because the Hyp, Gy, Hyp-2J, and Hyp-Duk mutations cause complete loss of Phex function, the more severe hearing loss exhibited by Hyp-Duk/Y mice is likely the result of strain background genetic differences. To examine this possibility, a BALB/cUrd Hyp-Duk/+ female was mated to a BALBc/ByJ +/+ male. The average ABR thresholds of the mutant male offspring (Fl-Hyp-Duk/Y) were 35 dB lower than those of BALB/cUrd Hyp-Duk/Y males (p < 0.001), but not significantly different from those of Gy/Y males (p = 0.2) or Hyp/Y males (p = 0.7) (Fig. 3). These results suggest that homozygosity for recessive factors (genetic modifiers) present in the BALB/cUrd strain exacerbate the hearing loss in Phex mutant mice.
ABR tests were performed on Hyp-Duk/Y mice at different ages to determine whether hearing loss increases over time (Fig. 4). Regression analysis showed no significant associations of test age with ABR thresholds (p > 0.3), indicating that the hearing loss associated with the Hyp-Duk mutation is not progressive. Of the 20 Hyp-Duk/Y mice tested for ABR thresholds, 11 showed circling behavior indicative of vestibular dysfunction. The 9 non-circling males actually had slightly higher ABR thresholds (mean = 93.8 dB) than the circling males (mean = 79.1 dB SPL; t-test p = 0.04). Thus, the degree of auditory dysfunction is independent of the degree of vestibular dysfunction in Hyp-Duk mutant males.
Fig. 4.
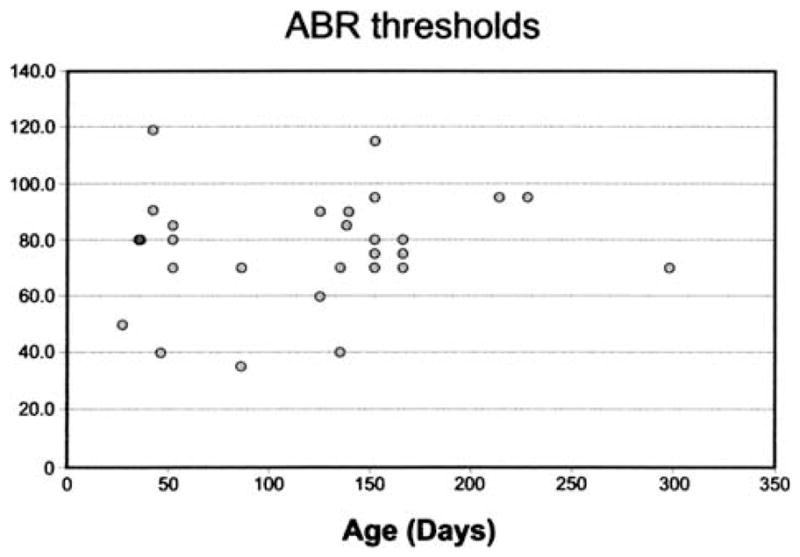
Hearing impairment is non-progressive up to 10 months of age. Scatter plot showing ABR thresholds for click stimulus of 20 Hyp-Duk/Y mice tested at 32 different ages (range 27–298 days).
The whole mounts of inner ears from six Hyp-Duk/Y mutant males were compared with three +/Y controls. No gross morphological abnormalities were noted. Examination of cross-sections of mutant cochleas, however, revealed several pathological abnormalities (Fig. 5). The temporal bone surrounding the cochlea was thickened in Hyp-Duk/Y males, with many interspersed areas of non-mineralization. A precipitate or infiltrate was usually observed in the scala tympani of the cochleas, but also occasionally in the scala vestibuli. This precipitate stained nonspecific for the stains PAS, PTAH, and von Kossa. Degeneration of the organ of Corti and spiral ganglia was obvious in the apical turn of the cochlea, but the severity of degeneration decreased towards the base. Endolymph volume appeared greater in the cochleas, resulting in an increased scala media and a diminished scala vestibule, with a consequent displacement of Reissner’s membrane. Cochlear cross-sections of three Hyp-2J/Y males showed thickening of the temporal bone and cochlear precipitates, but no degeneration of the organ of Corti and the spiral ganglion cells.
Fig. 5.

Mid-modiolar cross-section through cochleas of a 5-month-old Hyp-Duk/Y mutant male (right) compared with an age-matched +/Y control (left). ABR thresholds of the mutant mouse were 50–80 dB above those of the control for all auditory stimuli presented. The temporal bone (TB) surrounding the mutant cochlea is thickened with many areas of nonmineralization (indicated by arrows with stars). A precipitate or infiltrate was often seen in the scala tympani (ST) of mutants. Degeneration of the organ of Corti (OC) and spiral ganglion cells (SG) is apparent in the mutant, especially in the apical turn, as indicated by arrows. Relative to the control, the scala media (SM) of the mutant is increased in size and the scala vestibuli (SV) decreased, with a consequent displacement of Reisner’s membrane (RM).
Molecular analysis of the Hyp-2J mutation
Overlapping RT-PCR products corresponding to Phex exons 1–9 and 7–22 were sequenced by using total RNA isolated from two Hyp-2J/Y and one +/Y male mice. No DNA sequence variation was found in exons 1–9. Within the amplified transcript comprising exons 7–22, exon 15 was absent.
To confirm that the Hyp-2J mutation involves the deletion of exon 15, we performed Southern blot analysis on HindIII-digested genomic DNA from mutant and control males. The probe consisted of an RT-PCR product corresponding to Phex exons 12–18. A~7.3 kb fragment corresponding to Phex exon 15 was deleted in Hyp-2J/Y DNA (Fig. 6), whereas fragments corresponding to Phex exon 12 (4.8 kb), exon 13 (6.4 kb), exon 14 (2.8 kb), exon 16 (4.5 kb), and exons 17, 18 (10.8 kb) were present. PCR primers were designed to confirm the deletion of exon 15 in genomic DNA. The combined RT-PCR, genomic PCR, and Southern blot analysis indicated that the Hyp-2J deletion involves at least 7.3 kb of DNA containing Phex exon 15.
Fig. 6.

Southern blot of HindIII digests of mouse genomic DNA. Hybridization with an RT-PCR product corresponding to Phex exons 12–18. A 6.4-kb fragment corresponding to exon 13 (underlined) and a 2.8-kb fragment corresponding to exon 14 (underlined) were absent in Hyp-Duk/Y DNA but present in the control DNA (+/Y). Instead of a 7.3-kb genomic HindIII fragment containing Phex exon 15, a 3.8-kb junction fragment was found in Hyp-Duk/Y DNA. In Hyp-2J/Y DNA, the 7.3-kb fragment corresponding to Phex exon 15 (bold) is absent, while it is present in the control DNA (+/Y).
Molecular analysis of the Hyp-Duk mutation
The public mouse genome assembly sequence was used to design intronic primers for the PCR amplification of all 22 Phex exons (Table 3). Genomic DNA was isolated from two Hyp-Duk/Y and two +/Y male mice. While all exons were amplified in +/Y DNA, Phex exons 13 and 14 were missing in Hyp-Duk/Y DNA.
To confirm the deletion of the two exons, an RTPCR product corresponding to Phex exons 12–18 was hybridized to a Southern blot of genomic DNA. The two HindIII fragments corresponding to exon 13 (6.4 kb) and exon 14 (2.8 kb) were deleted in Hyp-Duk/Y DNA (Fig. 6). Furthermore, instead of the 7.3-kb HindIII fragment containing Phex exon 15 present in +/Y DNA, a 3.8 kb junction fragment was detected in the Hyp-Duk/Y DNA. The HindIII fragments corresponding to Phex exon 12 (4.8 kb), exon 16 (4.5 kb), and exons 17, 18 (10.8 kb) were present in mutant and control DNA. In summary, genomic PCR and Southern blot analysis indicated that the Hyp-Duk deletion involves at least 30 kb containing Phex exons 13 and 14.
Discussion
Male mice hemizygous for the Phex mutant alleles Hyp, Hyp-2J, Hyp-Duk, Gy, or Ska1 exhibit similar clinical phenotypes characterized by shortened hind legs and tail, shortened squared trunk, hypophosphatemia due to impaired renal absorption of phosphate, hypocalcemia, and rachitic bone disease. In addition, male mice hemizygous for Hyp, Hyp-2J, Hyp-Duk, or Gy mutant alleles have decreased aBMD, which is attributed to the under-mineralization of the skeleton (no aBMD data have been reported for Ska/Y males). Hyp-Duk/Y males exhibit a circling phenotype similar to that described in the Gy/Y males, which is rare in Hyp/Y and Hyp-2J/Y males and not reported for Skal/Y males (Carpinelli et al. 2002).
The original Hyp mutation involves a deletion of the Phex gene starting in intron 15 and extending through the 3′ region and ~10 kb beyond, but does not include the next downstream gene Sat (Sabbagh et al. 2002). We found that the deletion breakpoints for Hyp-2J are in introns 14 and 15 of the Phex gene and that those for Hyp-Duk are in intron 12 and intron 14 (Fig. 7). The deletion of Phex exon 15 in the Hyp-2J allele and exons 13 and 14 in the Hyp-Duk allele lead to a premature stop codon in the open reading frame after 94 nucleotides and 156 nucleotides, respectively. The mutations are likely to cause loss of a functional Phex protein. Because Hyp and Hyp-2J mutations arose and are maintained on the same genetic background (C57BL/6J) and both cause a similar phenotype, it is unlikely that either affects expression of a closely linked gene. This suggestion is strengthened by results reported for the mutation, skeletal abnormality 1, symbolized PhexSka1(Ska1). Ska1 was recovered from a C57BL/6J inbred mouse that was the first generation of a random mutagenesis screen with the chemical mutagen N-ethyl-N-nitrosourea (Carpinelli et al. 2002). The Ska1 mutation involves a point mutation in a splice donor site immediately following exon 8 of the Phex gene and causes skipping of exon 8 in the Phex mRNA, creating a null allele. We conclude that Hyp-2J, Hyp-Duk, and Ska1 are the first mouse mutations restricted to the Phex gene, and males carrying these mutations provide better models for the analysis of the pathophysiology of XLH in humans.
Fig. 7.

Overview of the five mouse Phex mutations. The Gy mutation deletes the first three Phex exons and the complete spermine synthase gene. The Hyp mutation deletes the last seven exons and additional undetermined downstream sequence. The Skal mutation involves a point mutation in a splice donor site immediately following exon 8 of the Phex gene. The Hyp-Duk mutation deletes exons 13 and 14, while Hyp-2J deletes exon 15.
In contrast, the Gy mutation deletes the first three Phex exons together with the upstream spermine synthase (Sms) gene (Lorenz et al. 1998). The compromised fertility of Gy/Y males is likely the result of a deletion of Sms. Gy/Y mice are smaller at birth than their normal male siblings, a condition that persists throughout life (Table 1) and may be attributed to the deletion of the Sms gene rather than the strain background.
Differences in lean body mass were significant only in Gy/Y and Hyp-Duk/Y males compared with their normal male siblings, possibly because of the circling behavior associated with both mutations. Furthermore, the hearing impairment and circling behavior of Hyp-Duk/Y males suggest that the disruption of Phex is responsible for the circling behavior in Gy/Y mice and not the deletion of a contiguous 5′ gene. There are indications that genetic background impacts the circling phenotype. For example, when the Hyp mutation is present on the B6EiC3SnF1-a/A background (on which Gy has been characterized), a subset of Hyp/Y mice circled (Meyer et al. 1995).
Hearing impairment was much greater in Hyp-Duk/Y males than in Hyp/Y, Hyp-2J/Y and Gy/Y males (Fig. 3). Because all four mutations appear to cause complete loss of Phex function, the more severe hearing loss exhibited by Hyp-Duk/Y mice is likely the result of strain background effects. Specific factors (genetic modifiers) present in the BALB/cUrd genome that exacerbate the hearing impairment associated with Phex mutations are probably recessive because inter-strain F1 Hyp-Duk/Y males did not show this effect. The hearing impairment caused by the Phex mutation in Hyp-Duk/Y males is non-progressive, at least by 10 months of age (Fig. 4). At older ages, other genetic factors affecting age-related hearing loss may complicate interpretations (Johnson et al 2000).
Inner ear abnormalities were reported previously for Gy/Y and Hyp/Y males. Similar to our observations in Hyp-Duk/Y males, thickening of the temporal bone with bands of incomplete calcification was reported in inner ears of Gy/Y mutants (Sela et al. 1982), and a perilymphatic precipitate was reported in cochleas of Gy/Y and Hyp/Y mutants (Barkway et al. 1988, 1989). These abnormalities, however, cannot cause the hearing impairment observed in Hyp-Duk/Y males because the identical temporal bone pathology and cochlear precipitates were noted in normal-hearing Hyp-2J/Y mutants. The most likely cause of hearing impairment in Hyp-Duk/Y males is a degeneration of the organ of Corti and spiral ganglion cells (Fig. 5). Although there is variability in severity, this cochlear pathology always was noted in Hyp-Duk/Y mutants with impaired hearing, but never in normal-hearing Hyp- 2J/Y mutants. The organ of Corti is the sensorineural epithelium present along the length of the cochlear duct, consisting of outer and inner hair cells and supporting cells, and spiral ganglia are the afferent neurons of inner hair cells. This sensorineural degeneration, however, may be a secondary consequence of an increased endolymph volume, as evidenced by an enlarged scala media, reduced scala tympani, and displaced Reissner’s membrane.
Similar hearing impairment and inner ear pathologies have been reported in humans with X-linked hypophosphatemia, including a thickening of the petrous temporal bone and cochlear dysfunction (Boneh et al. 1987; Davies et al. 1984; O’Malley et al. 1985, 1988; Weir 1977). Transtympanic electrocochleography results suggest that the cochlear abnormality is endolymphatic hydrops (Davies et al. 1984; O’Malley et al. 1985), which is similar to our observation of increased endolymph volume in hearing-impaired Hyp-Duk/Y males. The molecular mechanisms and pathways involving the Phex gene that underlie hearing impairment and inner ear pathology await further investigation. The Hyp-Duk/Y mouse may provide an opportunity to genetically identify modifier genes that interact with Phex mutations to cause cochlear dysfunction.
Skull measurements inHyp/Y mice were reported previously by Gonzalez et al., who found that at 10 weeks of age C57BL/6J Hyp/Y and B6EiC3Sn-a/A Hyp/Y males had significantly decreased skull and nose length compared with controls (Gonzalez et al. 1992). This is consistent with our data for Hyp-Duk/Y males, indicating that skull and nose length are significantly shorter than in male controls (Table 2). Skull, but not nose length, also was significantly reduced in Hyp-2J/Y males. At 10 weeks of age, B6EiC3Sn-a/A Gy/Y males had significantly smaller skull, nose, and lower jaw lengths than controls. Upper jaw length was not affected, indicating that reduced skull size is not the result of overall smaller skeleton morphology (Shetty and Meyer 1991). However, we did not find similar lower jaw length reduction in Hyp-Duk/Y or Hyp-2J/Y mutants. The variability in skull morphology of these four mutants suggests that genetic background affects these craniofacial characteristics.
Craniofacial and dental malformations associated with human XLH include retardation of maxillary and mandibular growth (Tracy and Campbell 1968), bossing of frontal and occipital regions, oxycephaly (Marks et al. 1965), and craniosynostosis (Reilly et al. 1964). The retardation of maxillary and mandibular growth in humans is an example of a craniofacial dysmorphology that can be powerfully modeled by mice. Using Ts65Dn mice, Richtsmeier et al. demonstrated that generally the reduction in nose length, upper jaw length, and/or lower jaw length observed in mice could be considered comparable to human craniofacial phenotypes (Richtsmeier et al. 2000). Hyp-2J/Y skulls showed no reduction in lower/upper jaw or nose length, whereas Hyp-Duk/Y skulls are smaller in nose, skull and upper jaw length, making Hyp-Duk/Y mice better models to study craniofacial abnormalities as a result of XLH in humans.
In their mutational analysis study, Holm et al. found no correlation between the severity of XLH and the type or location of the mutation in the PHEX gene in humans (Holm et al. 2001). However, they did suggest that other genes and environmental factors affect the severity of the disease, a hypothesis supported by our results presented here for mutations in the Phex gene in mice. Although all four Phex mutations examined in our study cause loss of function, changes in genetic background effects (i.e., modifier genes) can alleviate or exacerbate the mutant phenotype. Identification of these genetic modifiers in mice will lend insight into the varying severity of the disease in humans.
In summary, these two new mouse models of human XLH reported here offer outstanding resources for pharmacological investigations. In addition, both may help define the function and role of Phex in the pathway of bone mineralization and phosphate homeostasis.
Acknowledgments
We thank Wesley G. Beamer and Tali Shalom-Barak for their careful review of this manuscript, as well as H. Hellebrand, Heping Yu, and Sandra Gray for their technical assistance and contribution to this study. We are indebted to Susan Poulton for originally discovering the Hyp-Duk mutation at The Urology Research Department at Duke University. We also thank Roderick Bronson for pathohistological evaluation and Coleen Marden for colony maintenance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (STR304/2-1), by the National Institutes of Health grants RR01183, CF-DE13078-03, contract DC62108 and by the Cancer Core grant CA34196 from the National Cancer Institute.
References
- 1.Barkway C, Glenn N, Harvey D, Moorjani P, Palmer A, et al. Hearing impairment associated with hypophosphatemia: the Gyro mutant mouse. Hered Deafness Newslett. 1988;1:20–21. [Google Scholar]
- 2.Barkway C, Glenn N, Moorjani P, Palmer A, Stabler S, et al. Hearing impairment in two mouse mutants with hypophosphatemia. Hered Deafness Newslett. 1989;3:20–21. [Google Scholar]
- 3.Boneh A, Reade TM, Scriver CR, Rishikof E. Audiometric evidence for two forms of X-linked hypophosphatemia in humans, apparent counterparts of Hyp and Gy mutations in mouse. Am J Med Genet. 1987;27:997–1003. doi: 10.1002/ajmg.1320270434. [DOI] [PubMed] [Google Scholar]
- 4.Burnett CH, Dent CE, Harper C, Warland BJ. Vitamin-D resistant rickets. Am J Med. 1964;36:222–232. doi: 10.1016/0002-9343(64)90085-3. [DOI] [PubMed] [Google Scholar]
- 5.Carpinelli MR, Wicks IP, Sims NA, O’Donnell K, Hanzinikolas K, et al. An ethyl-nitrosourea-induced point mutation in Phex causes exon skipping, X-linked hypophosphatemia, and rickets. Am J Pathol. 2002;161:1925–1933. doi: 10.1016/S0002-9440(10)64468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies M, Kane R, Valentine J. Impaired hearing in X-linked hypophosphataemic (vitamin-D-resistant) osteomalacia. Ann Intern Med. 1984;100:230–232. doi: 10.7326/0003-4819-100-2-230. [DOI] [PubMed] [Google Scholar]
- 7.Davisson MT. The Jackson Laboratory mouse mutant resource. Lab Anim. 1990;19:23–29. [Google Scholar]
- 8.Eicher EM, Southard JL, Scriver CR, Glorieux FH. Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc Natl Acad Sci USA. 1976;73:4667–4671. doi: 10.1073/pnas.73.12.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez CD, Meyer RA, Jr, Iorio RJ. Craniometric measurements of craniofacial malformations in the X-linked hypophosphatemic (Hyp) mouse on two different genetic backgrounds: C57BL/6J and B6C3H. Teratology. 1992;46:605–613. doi: 10.1002/tera.1420460610. [DOI] [PubMed] [Google Scholar]
- 10.Holm IA, Nelson AE, Robinson BG, Mason RS, Marsh DJ, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86:3889–3899. doi: 10.1210/jcem.86.8.7761. [DOI] [PubMed] [Google Scholar]
- 11.Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70:171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz B, Francis F, Gempel K, Boddrich A, Josten M, et al. Spermine deficiency in Gy mice caused by deletion of the spermine synthase gene. Hum Mol Genet. 1998;7:541–547. doi: 10.1093/hmg/7.3.541. [DOI] [PubMed] [Google Scholar]
- 13.Lyon MF, Scriver CR, Baker LR, Tenenhouse HS, Kronick J, et al. The Gy mutation: another cause of X-linked hypophosphatemia in mouse. Proc NatI Acad Sci USA. 1986;83:4899–4903. doi: 10.1073/pnas.83.13.4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marks SC, Lindahl RL, Bawden JW. Dental and cephalometric findings in vitamin D resistant rickets. J Dent Child. 1965;32:259–265. [PubMed] [Google Scholar]
- 15.Meyer RA, Jr, Meyer MH, Gray RW, Bruns ME. Femoral abnormalities and vitamin D metabolism in X-linked hypophosphatemic (Hyp and Gy) mice. J Orthop Res. 1995;13:30–40. doi: 10.1002/jor.1100130107. [DOI] [PubMed] [Google Scholar]
- 16.O’Malley S, Ramsden RT, Latif A, Kane R, Davies M. Electrocochleographic changes in the hearing loss associated with X-linked hypophosphataemic osteomalacia. Acta Otolaryngol. 1985;100:13–18. doi: 10.3109/00016488509108581. [DOI] [PubMed] [Google Scholar]
- 17.O’Malley SP, Adams JE, Davies M, Ramsden RT. The petrous temporal bone and deafness in X-linked hypophosphataemic osteomalacia. Clin Radiol. 1988;39:528–530. doi: 10.1016/s0009-9260(88)80224-1. [DOI] [PubMed] [Google Scholar]
- 18.Reilly B, Leeming J, Fraser M. Craniosynostosis in the rachitic spectrum. J Pediatr. 1964;64:396–401. doi: 10.1016/s0022-3476(64)80192-x. [DOI] [PubMed] [Google Scholar]
- 19.Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217:137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 20.Sabbagh Y, Gauthier C, Tenenhouse HS. The X chromosome deletion in HYP mice extends into the intergenic region but does not include the SAT gene downstream from Phex. Cytogenet Genome Res. 2002;99:344–349. doi: 10.1159/000071613. [DOI] [PubMed] [Google Scholar]
- 21.Sela J, Bab I, Deol MS. Patterns of matrix vesicle calcification in osteomalacia of Gyro mice. Metab Bone Dis Relat Res. 1982;4:129–134. doi: 10.1016/0221-8747(82)90026-1. [DOI] [PubMed] [Google Scholar]
- 22.Shetty NS, Meyer RA., Jr Craniofacial abnormalities in mice with X-linked hypophosphatemic genes (Hyp or Gy) Teratology. 1991;44:463–472. doi: 10.1002/tera.1420440412. [DOI] [PubMed] [Google Scholar]
- 23.Strom TM, Francis F, Lorenz B, Boddrich A, Econs MJ, et al. Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum Mol Genet. 1997;6:165–171. doi: 10.1093/hmg/6.2.165. [DOI] [PubMed] [Google Scholar]
- 24.Tenenhouse HS. X-linked hypophosphataemia: a homologous disorder in humans and mice. Nephrol Dial Transplant. 1999;14:333–341. doi: 10.1093/ndt/14.2.333. [DOI] [PubMed] [Google Scholar]
- 25.The HYP Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 26.Tracy WE, Campbell RA. Dentofacial development in children with vitamin D resistant rickets. J Am Dent Assoc. 1968;76:1026–1031. doi: 10.14219/jada.archive.1968.0172. [DOI] [PubMed] [Google Scholar]
- 27.Ward-Bailey PF, Wood B, Johnson KR, Bronson RT, Donahue LR, et al. Neuromuscular ataxia: a new spontaneous mutation in the mouse. Mamm Genome. 2000;11:820–823. doi: 10.1007/s003350010167. [DOI] [PubMed] [Google Scholar]
- 28.Weir N. Sensorineural deafness associated with recessive hypophosphataemic rickets. J Laryngol Otol. 1977;91:717–722. doi: 10.1017/s0022215100084255. [DOI] [PubMed] [Google Scholar]
- 29.Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]