

Abstract
Autosomal recessive woolly hair (ARWH)/hypotrichosis is a hereditary hair disorder which is characterized by tightly curled hair, and is occasionally associated with sparse hair. ARWH can be caused by mutations in the P2RY5 or lipase H (LIPH) gene. Disruption of both genes results in phenotypes with features of both WH and hypotrichosis. In this study, we identified two Guyanese families with ARWH. Both families are of recent Indian descent. Mutation analysis resulted in the identification of mutations in the LIPH gene in both families. Affected individuals in the first family carry compound heterozygous mutations Ex7_8del and 1303_1309dupGAAAACG in the LIPH gene, and those in the second family have a homozygous mutation 659_660delTA in LIPH. The mutations Ex7_8del and 659_660delTA were previously identified in several Pakistani families with ARWH. Haplotype analysis using microsatellite markers close to the LIPH gene defined a founder haplotype shared in families from Pakistan and Guyana. Proteomic analysis of hair shaft samples from one of the families revealed no substantial changes among the proteins identified, indicating that the syndrome does not involve global alterations in protein expression. Our results further suggest a crucial role of lipase H in hair growth.
Keywords: LIPH, woolly hair, hypotrichosis, P2RY5, proteomics
INTRODUCTION
The hair shaft is a highly keratinized structure that is produced by the hair follicle. The growth of the hair shaft originates in the matrix region located in the bulb portion of the HF, which gives rise to a highly cohesive structure with close interactions among hair keratins and their associated proteins (Langbein and Schweizer, 2005). In addition, HF compartments which are composed of several distinct layers surround and support the hair shaft. Recent advances in molecular genetics enabled the identification of numerous genes that are expressed in the HF. Furthermore, mutations in some of these genes have been shown to underlie hereditary hair disorders, such as T cell immunodeficiency, congenital alopecia, and nail dystrophy (OMIM 601705), localized autosomal recessive hypotrichosis (OMIM 607903), and autosomal dominant hypotrichosis simplex of the scalp (OMIM 146520), which are caused by mutations in FOXN1 (Frank et al., 1999), DSG4 (Kljuic et al., 2003), and CDSN (Levy-Nissenbaum et al., 2003), respectively. In addition, it has been shown that mutations in CDH3 gene underlie both hypotrichosis with juvenile macular dystrophy (OMIM 601553) (Sprecher et al., 2001; Shimomura et al., 2008a) and ectodermal dysplasia, ectrodactyly, and macular dystrophy (OMIM 225280) (Kjaer et al., 2005; Shimomura et al., 2008a). Most recently, we have identified mutations in the P2RY5 gene in several consanguineous Pakistani families affected with autosomal recessive woolly hair (ARWH; OMIM 278150) (Shimomura et al., 2008b). The P2RY5 gene encodes a G protein-coupled receptor P2Y5 which is expressed abundantly in the inner root sheath of the hair follicle.
WH is a hair shaft anomaly characterized by tightly curled hair (Chien et al., 2006). WH can appear as a part of some systemic diseases, such as Naxos disease (OMIM 601214) (McKoy et al., 2000) and Carvajal syndrome (OMIM 605676) (Norgett et al., 2000). In addition, an isolated form of WH without associated findings also exists and can display either autosomal dominant (ADWH; OMIM 194300) (Hutchinson et al., 1974) or recessive inheritance (ARWH) (Salamon, 1963; Hutchinson et al., 1974). Before our recent findings of P2RY5 mutations, only two families with ARWH had been reported in the literature (Salamon, 1963; Hutchinson et al., 1974). Importantly, affected individuals in both families showed not only WH, but also sparse and depigmented hairs (Salamon, 1963; Hutchinson et al., 1974). Consistent with these reports, affected individuals in Pakistani families with P2RY5 mutations also exhibited various degrees of sparse hair, even though WH was the only common trait among all affected individuals (Shimomura et al., 2008b). In addition, mutations in the P2RY5 gene were recently reported in Saudi Arabian families with autosomal recessive hypotrichosis simplex (OMIM 146520) which is characterized by early-onset paucity of scalp and body hair (Pasternack et al., 2008).
More recently, we analyzed additional Pakistani families with ARWH, and identified mutations in the lipase H (LIPH) gene (Shimomura et al., 2008c). It was originally reported that a common founder mutation in the LIPH gene caused an autosomal recessively inherited hypotrichosis (OMIM 604379) in isolated Russian populations (Kazantseva et al., 2006). Later, additional mutations in LIPH were identified in families affected with an autosomal recessive hypotrichosis (Ali et al., 2007; Jelani et al., 2008; Kamran-Ul-Hassan Naqvi et al., 2009; Nahum et al., 2009). Notably, the WH phenotype was not mentioned in these reports. Although some of the Pakistani families with LIPH mutations showed hypotrichosis, all affected individuals had WH during their early childhood (Shimomura et al., 2008c). Collectively, we have shown that, similar to P2RY5 mutations, individuals with LIPH mutations can also show overlapping phenotypes between ARWH and hypotrichosis.
In this report, we studied two families of Guyanese origin with ARWH, and identified three distinct pathogenic mutations in the LIPH gene in both families. In addition, we performed proteomic analysis with hairs from members of one family to analyze how LIPH mutations affect the expression of proteins in the hair shaft. Since two of three mutations were previously identified in Pakistani families with ARWH, we performed haplotype analysis with microsatellite markers close to the LIPH gene, and defined common founder alleles among these geographically diverse populations.
RESULTS
Clinical features
Clinical features of both families were examined and evaluated by dermatologists who are experts in hair diseases (Y.S. and A.Z.). Family A is a two-generation pedigree with two affected siblings (Figure 1a). There is no consanguinity between the parents, who are unaffected and have straight scalp hair with normal hair density. Both affected individuals had tightly curled hair during their childhood (Figure 1b). Their hair grew slowly and stopped growing after a few inches. Individual II-1 has gradually lost her hair, and at present, she has sparse, thin, and short hair on the scalp (Figure 1c). Under light microscopy, some of her hair shafts are twisted (Figure 1d), and the distal ends are tapered (Figure 1e), suggesting a certain hair growth defect. Her eyebrows, as well as her body hairs, were also sparse, whereas her eyelashes were not affected (not shown). By contrast, individual II-2 has shown a different clinical course with aging. At present, the hair density on his scalp is relatively normal. In addition, his hair shows a mildly wavy or nearly straight appearance, and grows relatively well (not shown). His facial and body hairs are not affected (not shown).
Figure 1. Pedigrees and clinical features of two Guyanese families with ARWH.
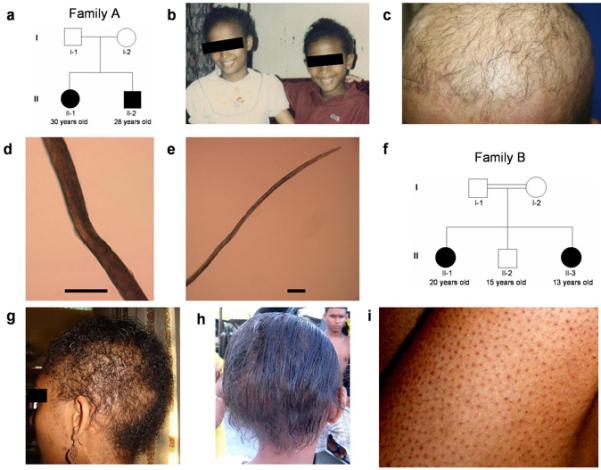
(a) Pedigree of Family A. (b) Clinical features of the affected individuals II-1 (left) and II-2 (right) in Family A when they were 12 and 10 years old, respectively. Note that both showed an obvious woolly hair phenotype. (c) Present clinical features of the affected individual II-1 in Family A. (d, e) Under light microscopy, hair shafts of the affected individual II-1 in Family A show twisting (d) and a tapered end (e). Scale bars: 100 μm. (f) Pedigree of Family B. (g, h) Hair phenotypes of the affected individuals in Family B. The affected individual II-1 show sparse and tightly curled hair (g), while the affected individual II-3 shows sparse and mildly wavy hair (h). (i) Diffuse keratotic follicular papules on legs of the affected individual II-1 in Family B.
Family B is a two-generation pedigree with two affected and one unaffected siblings (Figure 1f). The parents are first cousins, and neither is affected. Both affected individuals have had sparse hair and different degrees of WH since birth. Individual II-1 has shown tightly curled hair (Figure 1g), while the younger affected individual has showed slightly wavy hair (Figure 1h). Their eyebrows, eyelashes, and body hairs are also sparse. In addition, it is noteworthy that both affected individuals have showed diffuse keratotic follicular papules on their arms, legs, and abdomen since the age of one year (Figure 1i). Affected individuals in both families show normal facial features, teeth, nails, and sweating, and do not show palmoplantar hyperkeratosis. There was no family history of either heart disease, sudden death or neurologic abnormalities.
Identification of mutations in the LIPH gene
Since the clinical features of both families are consistent with ARWH, we first performed direct sequencing analysis of exons and exon-intron boundaries of the P2RY5 gene which is a causative gene for ARWH (Shimomura et al., 2008b), but did not find any sequence variants in P2RY5 in either family (data not shown). Next, we searched for mutations in the LIPH gene that we recently reported as a second causative gene for ARWH (Shimomura et al., 2008c). We sequenced all exons and exon-intron boundary sequences of the LIPH gene and identified pathogenic mutations in the LIPH in both families.
First, both affected individuals in Family A (II-1 and II-2) are heterozygous for a 7-nucleotide insertion at position 1309 in exon 10 of the LIPH gene, which is a tandem repeat of the sequences between positions 1303 and 1309, thus designated 1303_1309dupGAAAACG (Figure 2a). The mutation was not reported previously, and is predicted to cause a frameshift and a premature termination codon (PTC) (Val437GlyfsX4) (Figure 2a). Direct sequencing analysis showed that the mutation was inherited from their mother (I-2) (Figure 2a). Although we postulated the existence of another heterozygous mutation in the LIPH of this family, the initial direct sequencing analysis did not detect any other sequence variants. However, a large deletion mutation would not be detected by PCR from heterozygous individuals. We next performed PCR using primer pairs designed at intron 6 and intron 8 of the LIPH (E7F2 and E8R2; Figure 2b) to screen for the mutation Ex7_8del that we recently identified in several Pakistani families with ARWH (Shimomura et al., 2008c). A 183 bp fragment was amplified by this PCR from DNA of both affected individuals (II-1 and II-2) and their father (I-1), which was the same size as the fragment amplified from DNA of a Pakistani individual who is homozygous for the mutation Ex7_8del (lane PC, Figure 2b). Direct sequencing of the PCR product confirmed that Family A carries the identical deletion mutation that we identified in Pakistani families (Figure 2b) (Shimomura et al., 2009). Thus, affected individuals in Family A have compound heterozygous mutations 1303_1309dupGAAAACG and Ex7_8del, on the maternal and paternal alleles of the LIPH gene, respectively.
Figure 2. Identification of mutations in the LIPH gene.
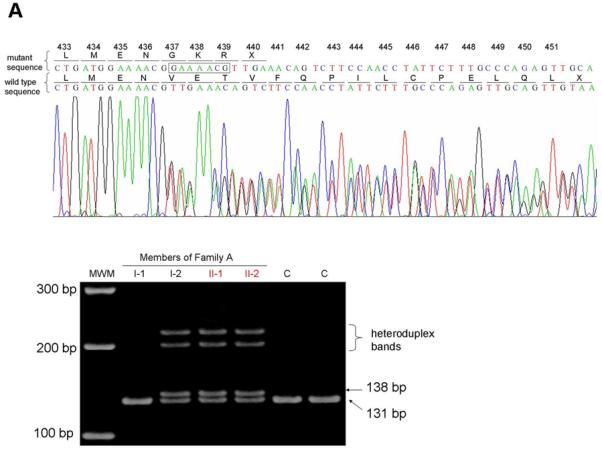
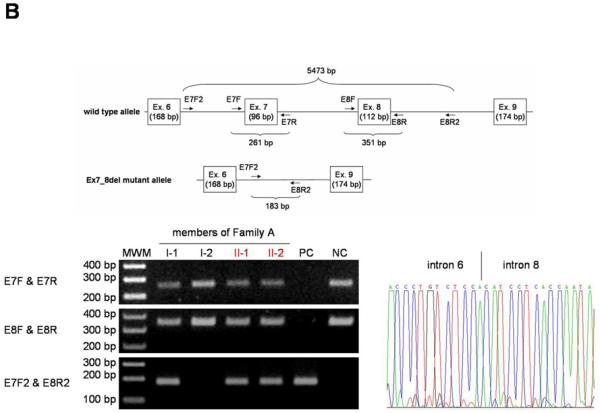
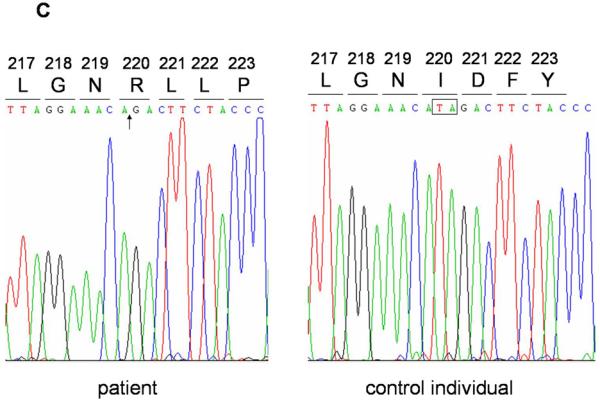
(a) Heterozygous mutation 1303_1309dupGAAAACG in the LIPH of Family A (top). The inserted nucleotides are boxed. Screening assay for the mutation 1303_1309dupGAAAACG (bottom) shows that the mutation was inherited from the mother (I-2) of the affected individuals (II-1 and II-2). C, control individuals. (b) Heterozygous mutation Ex7_8del in the LIPH of Family A. PCR with E7F2 and E8R2 primers amplified the 183 bp fragment from DNA of both affected individuals (II-1 and II-2) and their father (I-1) in Family A, as well as a Pakistani individual who is homozygous for the mutation Ex7_8del (PC), whereas this fragment was not amplified from DNA of either their mother (I-2) or a normal control individual (NC). Direct sequencing of this product clearly shows that Family A carries exactly the same deletion mutation Ex7_8del that was previously found in Pakistani families with ARWH. Note that the product from the wild type allele, 5473 bp in size, could not be amplified even from DNA of control individuals due to a short extension time of the PCR. Affected individuals are colored in red (a, b). MWM, molecular weight markers (a, b). (c) Homozygous mutation 659-660delTA in the LIPH gene of Family B. The position of the deletion is indicated by an arrow (left), and the deleted nucleotides are boxed (right).
In Family B, both affected individuals (II-1 and II-3) are homozygous for a 2-nucleotide deletion at positions 659-660 in exon 5 of the LIPH gene, designated 659-660delTA (Figure 2c), while the unaffected sibling (II-2) is homozygous for the wild type sequence (not shown). This mutation was previously identified in several Pakistani families with ARWH/hypotrichosis (Jelani et al., 2008; Shimomura et al., 2008c), and is predicted to result in a frameshift and a PTC at 25 amino acid residues downstream of the mutation (Ile220ArgfsX25). Screening assays showed that none of these mutations were found in 100 Pakistani control individuals (data not shown).
Evidence for founder mutations
To determine whether the mutations Ex7_8del and 659-660delTA were shared founder mutations between Pakistani and Guyanese families, we performed haplotype analysis using microsatellite markers within and around LIPH gene. The haplotype for the mutation Ex7_8del between LIPH-MS1 and LIPH-MS4 was the same between Family A and Pakistani families with this mutation (Figure 3; Shimomura et al., 2008c). In addition, affected individuals in Family B and Pakistani families with the mutation 659-660delTA shared the same haplotype between LIPH-MS1 and D3S1602 (Figure 3; Shimomura et al., 2008c).
Figure 3. Haplotype of members of Families A and B in the LIPH locus.
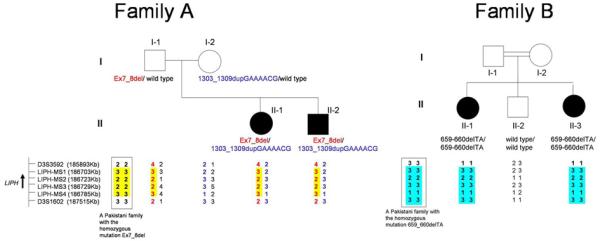
The position and the direction of transcription of the LIPH gene are indicated by an arrow. In Family A, the haplotypes for the mutations Ex7_8del and 1303_1309dupGAAAACG are colored in red and blue, respectively. The common haplotype for the mutation Ex7_8del between Family A and Pakistani families is highlighted in yellow, and that for the mutation 659-660delTA between Family B and Pakistani families is highlighted in blue. The haplotypes of a Pakistani family that is homozygous for either Ex7_8del or 659_660delTA are boxed.
Proteomic analysis
Hair samples were analyzed from the unaffected parents (M, mother; F, father) and two affected offspring (D, daughter; S, son) of Family A. From the unfractionated hair shaft, 41 proteins were identified in samples from both parents with no substantive differences between them being observed. The large majority (>95%) were keratins and keratin associated proteins (Figure S1). Since the high keratin content masks less abundant proteins in such analyses, isopeptide cross-linked protein (10-15% of the total hair shaft) was isolated and analyzed. Although major constituents of the cross-linked material were also keratins and keratin associated proteins (≈ 70%), the additional constituents included junctional and other membrane proteins (≈ 5%), histones (≈ 10%) and various proteins ordinarily found in the cytoplasm (≈ 10%), as previously observed (Lee et al., 2006). From this fraction, 53 proteins were identified, 29 of which were also detected in the unfractionated hair (Figure S2).
The 20 most abundant proteins for the total hair shaft and for the cross-linked fractions are shown in Table 1. The numbers of unique peptides attributed to each protein in the parental analyses were compared to those from the afflicted offspring. Because peptide identification by mass spectrometry is a stochastic process, numbers of unique peptides have some variability related to the complexity of the sample. Nevertheless, no large differences were evident between the parental and offspring samples. Table S1 gives the full listing of identified proteins, including those barely detectable. The present approach is not suited for determining the significance of the small differences observed in the latter.
Table 1. Number of unique peptides assigned to 20 most prevalent identified proteins among samples from the parents (M, mother; F, father) and afflicted offspring (D, daughter; S, son) in Family A.
| Total Hair Shaft | Cross-Linked Fraction | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein | D | S | M | F | emPAI | Protein | D | S | M | F | emPAI |
| KRT86 | 86 | 86 | 90 | 87 | 36.2 | KRT32 | 18 | 15 | 15 | 13 | 14.8 |
| KRT81 | 5 | 5 | 6 | 7 | 21.5 | KRT82 | 31 | 22 | 27 | 32 | 10.4 |
| KRT83 | 9 | 5 | 2 | 11 | 9.0 | KRT35 | 22 | 16 | 20 | 22 | 10.1 |
| KRT85 | 42 | 45 | 38 | 45 | 8.2 | KRT85 | 16 | 13 | 18 | 16 | 7.2 |
| KRT33B | 48 | 43 | 56 | 39 | 7.9 | KRT86 | 27 | 31 | 36 | 26 | 6.3 |
| KRT31 | 15 | 15 | 19 | 16 | 6.0 | HIST1H4 | 9 | 6 | 7 | 5 | 5.5 |
| KRT33A | 3 | 3 | 3 | 3 | 4.5 | SFN | 10 | 11 | 10 | 11 | 4.7 |
| KRT34 | 15 | 19 | 15 | 13 | 2.3 | H2AFX | 5 | 6 | 6 | 7 | 4.3 |
| KRTAP2-2 | 13 | 13 | 11 | 12 | 0.9 | KRT34 | 4 | 6 | 9 | 5 | 3.7 |
| KRT35 | 19 | 13 | 13 | 15 | 0.6 | KRT33B | 17 | 23 | 24 | 14 | 3.3 |
| KRT32 | 10 | 9 | 10 | 10 | 0.4 | KRT31 | 5 | 9 | 8 | 6 | 2.9 |
| S100A3 | 5 | 4 | 4 | 3 | 0.3 | HIST1H2BL | 8 | 6 | 7 | 4 | 2.7 |
| KRT82 | 21 | 15 | 11 | 17 | 0.3 | S100A3 | 3 | 4 | 4 | 3 | 2.6 |
| KRTAP3-1 | 4 | 5 | 6 | 4 | 0.3 | KRTAP11-1 | 13 | 13 | 10 | 3 | 2.5 |
| H2AFX | 6 | 6 | 6 | 5 | 0.2 | VSIG8 | 16 | 10 | 10 | 18 | 2.3 |
| HIST1H2BL | 5 | 3 | 4 | 4 | 0.2 | CALML3 | 5 | 4 | 5 | 4 | 2.0 |
| KRTAP3-2 | 2 | 3 | 3 | 2 | 0.2 | KRT36 | 5 | 4 | 3 | 3 | 1.1 |
| HIST1H4 | 6 | 5 | 3 | 4 | 0.1 | PKP1 | 10 | 12 | 12 | 10 | 1.0 |
| KRT36 | 2 | 2 | 2 | 2 | 0.1 | KRT40 | 6 | 5 | 6 | 7 | 0.9 |
| KRTAP11-1 | 4 | 4 | 3 | 5 | 0.1 | PRDX6 | 2 | 5 | 4 | 6 | 0.8 |
Data include emPAI semi-quantitative estimates of relative molar amounts normalized to 100 (Ishihama et al., 2005).
DISCUSSION
In this study, we analyzed two Guyanese families with ARWH and identified mutations in the LIPH gene in both families. Affected individuals in Family A carry compound heterozygous mutations Ex7_8del and 1303_1309dupGAAAACG in the LIPH gene, which were inherited on their paternal and maternal alleles, respectively (Figure 2a, b). These are the first compound heterozygous mutations identified in the LIPH gene. Secondly, affected individuals in Family B are homozygous for the mutation 659_660delTA in the LIPH gene. All three mutations result in a frameshift and downstream PTC. Most likely, aberrant transcripts from both the Ex7_8del and 659_660delTA alleles would be largely degraded due to nonsense-mediated mRNA decay (Maquat, 1996; Frischmeyer and Dietz, 1999), leading to loss of expression of LIPH protein. By contrast, because the mutation 1303_1309dupGAAAACG exists in the last exon of the LIPH gene, the mutant allele with this mutation is likely to generate a truncated LIPH protein which would lack only 15 amino acid residues in its C-terminus as compared with the wild type LIPH protein (Figure 2a). Nevertheless, the aberrant protein would not possess a cysteine residue at amino acid 446 which is considered to be important for the formation of a disulfide bond (Figure 2a) (Jin et al., 2002), and thus is predicted to severely affect the structure of LIPH protein.
The mutations Ex7_8del and 659-660delTA were previously identified in several Pakistani families with ARWH (Jelani et al., 2008; Shimomura et al., 2008c). Haplotype analysis suggests a common founder for these mutations between the Pakistani and Guyanese families, living in geographically distinct regions (Figure 3a). Interestingly, history reveals that more than 200,000 people emmigrated from India to Guyana between 1838 and 1917 (Bisnauth, 2000), and since Pakistan was separated from India in the 1940s, it is plausible that India is the common source for these chromosomes. Indeed, it is noteworthy that both families emigrated from India to Guyana about 100 years ago, and all members of the extended pedigrees of both families are of Indian descent.
Mutations in the LIPH gene were originally reported to underlie an autosomal recessive form of hypotrichosis (Kazantseva et al., 2006; Ali et al., 2007). Recently, we identified several pathogenic mutations in Pakistani families affected with ARWH (Shimomura et al., 2008c). During early childhood, all affected individuals in our families showed mainly WH, but then exhibited wide variability in the hypotrichosis phenotype with aging. While some affected individuals continued to show only WH, others suffered hair loss, leading not only to WH, but also hypotrichosis. In the most severe cases, the hypotrichosis became the only phenotype, leading even to complete lack of scalp hair. Furthermore, the severity of WH phenotype was also variable among individuals. Such variations in phenotype were detected even within a single family (Shimomura et al., 2008c). Similarly, although all affected individuals in the Guyanese families commonly had WH at birth, they show variations in severity with aging. In family A, the elder affected individual shows a severe hypotrichosis (Figure 1c), while the younger affected individual exhibits a relatively mild phenotype. In Family B, both affected individuals showed overlapping phenotypes between WH and hypotrichosis (Figure 1g, h), but the degrees of WH are different between them. Interestingly, both affected individuals in Family B also have keratosis pilaris-like eruption on their extremities and abdomen (Figure 1i), which may be a non-specific sign seen in many forms of hypotrichosis, atrichia, and fragile hair disorders (Zlotogorski et al., 2002; Weiss et al., 2004; Zlotogorski et al., 2006).
Inherited disorders of lipid metabolism leading to permeability barrier abnormalities of the skin drive pathophysiology of scaling disorders with effects on proliferation and inflammation (Elias et al., 2008). Whether lipid processing defects affect hair structure by altering protein expression is less well studied. The unusual physical properties of WH, including a reported characteristic shape, could plausibly reflect altered protein composition or structural organization. However, present data indicate that predominant proteins detected were affected little, if at all, by the LIPH gene defect. Thus, the unusual properties of the hair are likely due simply to defects in the lipid component.
Quantitating relative protein amounts in cross-linked complexes is an incompletely resolved challenge. Nevertheless, limited quantitative comparisons of given proteins among different samples appear feasible on the basis of normalized spectral abundance, where the numbers of peptides detected are anticipated to be proportional to protein length (Zybailov et al., 2006). While only a rough approximation, the exponentially modified protein abundance index (emPAI) approach used presently has an empirical basis (Ishihama et al., 2005; Ishihama et al., 2008). Such estimates are likely to improve as more targeted measurements of select “proteotypic” peptides of specific proteins are developed (Deutsch et al., 2008). While a more detailed study would be required to detect subtle differences in protein content or to identify changes among less prevalent proteins, our results suffice to rule out major changes in protein expression as a characteristic feature of the WH syndrome. By contrast, subtypes of WH connected with more serious defects could display such changes that reflect downstream effects of the genetic lesion. In that case, proteomic analysis could help in their classification.
We and others have recently shown that mutations in the P2RY5 gene underlie ARWH/hypotrichosis (Pasternack et al., 2008; Shimomura et al., 2008b). The clinical features of affected individuals with P2RY5 mutations are indistinguishable from those with LIPH mutations. The LIPH gene encodes a phospholipase A1 family member and is a key enzyme in the synthesis of lysophosphatidic acid (LPA) (Sonoda et al., 2002), which is an extracellular mediator of many biological functions and is known to promote hair growth in vivo (Takahashi et al., 2003). P2Y5 has recently been shown to be a LPA receptor (Pasternack et al., 2008), and furthermore, we have demonstrated that the expression of P2Y5 partially overlaps with that of LIPH in HFs in vivo (Shimomura et al., 2008c). These data underscore a crucial role of the LIPH/LPA/P2Y5 signaling pathway in hair growth in humans.
MATERIALS AND METHODS
Mutation analysis of the LIPH gene
Peripheral blood samples from the family members and 100 unrelated healthy control individuals of Pakistani origin were collected in EDTA-containing tubes following informed consent under institutional approval (IRB-AAAB4246) and in adherence to the Declaration of Helsinki Principles. We also collected blood samples from 100 unrelated healthy Pakistani individuals as controls because both Guyanese families are of Indian descent and Pakistani populations share the same genetic background with Indian populations. Genomic DNA was isolated from these samples using the PUREGENE DNA isolation kit (Gentra System, Minneapolis, MN). All exons and exon-intron boundaries of the P2RY5 and the LIPH genes were amplified by PCR using gene-specific primers and PCR conditions as described previously (Shimomura et al., 2008b; Shimomura et al., 2009). The amplified PCR products were directly sequenced in an ABI Prism 310 Automated Sequencer, using the ABI Prism Big Dye Terminator Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems, Foster City, CA).
For screening of the mutation 1303-1309dupGAAAACG, a part of exon 10 and 3′-noncoding sequence of the LIPH gene was PCR-amplified using a forward primer (5′- TGTCGGTATGATCTTGTCCTGAT -3′) and a reverse primer (5′-CCTGTGGTTGTAGCTTTCTTTCTA-3′). The amplification conditions were 94°C for 2 min, followed by 35 cycles of 94°C for 30 sec, 57°C for 30 sec, and 72°C for 30 sec, with a final extension at 72°C for 7 min. The amplified PCR products were run on 8% polyacrylamide gels. Screening assays for the mutations Ex7_8del and 659-660delTA were performed as described previously (Shimomura et al., 2009).
Genotyping and Haplotype Analysis
In order to analyze whether the mutations Ex7_8del and 659_660delTA are common founder mutations between Pakistan and Guyana, genomic DNA from members of families from both populations were amplified by PCR using primers for two microsatellite markers, D3S3592 and D3S1602, close to LIPH gene, as well as four additional markers (LIPH-MS1-4) around or within LIPH (Shimomura et al., 2008c). PCR products were run on 8% polyacrylamide gels and genotypes were assigned by visual inspection.
Proteomics analysis
Samples of hair shafts (6-8 mg) were rinsed briefly in 2% SDS to remove loosely adhering contaminants, incubated overnight at 70°C in 5 ml of 2% SDS − 0.1 M sodium phosphate (pH 7.8) − 20 mM DTE and pulverized by stirring for several hours with a small magnetic stirring bar. Insoluble material was recovered from parallel samples (15-30 mg of hair) by centrifugation, extracted 4 more times, with the protein content of the extract being monitored to ensure complete extraction. The unfractionated hair and the insoluble fractions were alkylated with iodoacetamide, digested for three days at room temperature with reductively methylated trypsin in fresh 0.1 M ammonium bicarbonate 10% acetonitrile. Our previous hair analysis subjected the digest to ion exchange and reverse phase column chromatography prior to mass spectrometric protein identification (Lee et al., 2006). For the present purpose, a streamlined approach using only the reverse phase separation has proven sufficient to identify numerous prominent constituents and to permit semi-quantiative comparison among the samples. Mass spectrometry of 15 μg of each digested sample was performed using an LTQ ion trap mass spectrometer (Thermo Finnigan, San Jose, CA) as described in Supplementary Methods. Database searching was performed on MASCOT against the IPI human database. Scaffold (Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Numbers of unique peptides were tabulated as a basis for selecting prominent proteins and compiling emPAI values from Mascot database reports (cut-off score 35). Estimates of relative molar amount were calculated by normalizing to the total emPAI values for the identified proteins based on the mean emPAI values for the parental samples (Ishihama et al., 2005).
Supplementary Material
The identified proteins were sorted into the categories of keratins and keratin associated proteins (Ker), other cytoplasmic proteins (Cyt), histones (His) and membrane and junctional proteins (Mem).
{kind=link}
Illustrated are those proteins identified only in the total hair samples, only in the non-solubilizable (cross-linked) samples, and in both samples (overlap) as indicated. The number in each category is listed at the bottom of the category. Relative amounts of the proteins (unique peptides, emPAI values) are given in Table 1 or Table S1.
{kind=link}
The proteins listed were identified in both parental samples (M, mother; F, father) and/or both of the offspring samples (D, daughter; S, son) in Family A with a minimum of two unique peptides per person. In cases of closely related proteins, grouped for parsimony, the number of proteins to which the peptides were matched by Scaffold are given (Matches). The database accession numbers of the matched proteins and the protein molecular weights (MW) in AMU are also given. The unique peptides identified in the total hair shaft and cross-linked fraction samples are listed for the given donors. The exponentially modified Protein Abundance Index (emPAI) values taken from the MASCOT output are listed for the parental samples (M, F) along with the average. For samples where MASCOT did not provide a value (*), it was assumed to be 0. These values were summed for the total hair and for the cross-linked fraction samples, and normalized to 100 to give estimates of the relative molar amounts. The cellular locations (Cell Loc) used in tabulating Figure S1 are also given.
ACKNOWLEDGEMENTS
We gratefully acknowledge the family members for having participated in this study. We thank Ha Mut Lam for excellent technical assistance. This work was supported by US Public Health Service National Institute of Health grant R01AR44924 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (A.M.C.), and grant P42 ES04699 from the National Institute of Environmental Health Sciences (R.R.).
Footnotes
Conflict of Interest Statement
All authors have no conflict of interest or financial interest in this work.
REFERENCES
- Ahmad W, Faiyaz ul Haque M, Brancolini V, Tsou HC, ul Haque S, Lam H, et al. Alopecia universalis associated with a mutation in the human hairless gene. Science. 1998;279:720–4. doi: 10.1126/science.279.5351.720. [DOI] [PubMed] [Google Scholar]
- Ahmad W, Zlotogorski A, Panteleyev AA, Lam H, Ahmad M, ul Haque MF, et al. Genomic organization of the human hairless gene (HR) and identification of a mutation underlying congenital atrichia in an Arab Palestinian family. Genomics. 1999;56:141–8. doi: 10.1006/geno.1998.5699. [DOI] [PubMed] [Google Scholar]
- Ali G, Chishti MS, Raza SI, John P, Ahmad W. A mutation in the lipase H (LIPH) gene underlie autosomal recessive hypotrichosis. Hum Genet. 2007;121:319–25. doi: 10.1007/s00439-007-0344-0. [DOI] [PubMed] [Google Scholar]
- Bisnauth D. The Settlement of Indians in Guyana 1890-1930. Peepal Tree Press; 2000. [Google Scholar]
- Chien AJ, Valentine MC, Sybert VP. Hereditary woolly hair and keratosis pilaris. J Am Acad Dermatol. 2006;54:S35–9. doi: 10.1016/j.jaad.2005.01.092. [DOI] [PubMed] [Google Scholar]
- Cichon S, Anker M, Vogt IR, Rohleder H, Pützstück M, Hillmer A, et al. Cloning, genomic organization, alternative transcripts and mutational analysis of the gene responsible for autosomal recessive universal congenital alopecia. Hum Mol Genet. 1998;7:1671–9. doi: 10.1093/hmg/7.11.1671. [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Reports. 2008;9:429–34. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PM, Williams ML, Holleran WM, Jiang YJ, Schmuth M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: inherited disorders of lipid metabolism. J Lipid Res. 2008;49:697–714. doi: 10.1194/jlr.R800002-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- Hutchinson PE, Cairns RJ, Wells RS. Woolly hair. Clinical and general aspects. Trans St Johns Hosp Dermatol Soc. 1974;60:160–77. [PubMed] [Google Scholar]
- Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Molec Cell Proteom. 2005;4:1265–72. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- Ishihama Y, Schmidt T, Rappsilber J, Mann M, Hartl FU, Kerner MJ, et al. Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics. 2008;9:102. doi: 10.1186/1471-2164-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelani M, Wasif N, Ali G, Chishti MS, Ahmad W. A novel deletion mutation in LIPH gene causes autosomal recessive hypotrichosis (LAH2) Clin Genet. 2008;74:184–8. doi: 10.1111/j.1399-0004.2008.01011.x. [DOI] [PubMed] [Google Scholar]
- Jin W, Broedl UC, Monajemi H, Glick JM, Rader DJ. Lipase H, a new member of the triglyceride lipase family synthesized by the intestine. Genomics. 2002;80:268–73. doi: 10.1006/geno.2002.6837. [DOI] [PubMed] [Google Scholar]
- Kamran-Ul-Hassan Naqvi S, Raza SI, Naveed AK, John P, Ahmad W. A novel deletion mutation in the phospholipase H (LIPH) gene in a consanguineous Pakistani family with autosomal recessive hypotrichosis (LAH2) Br J Dermatol. 2009;160:194–6. doi: 10.1111/j.1365-2133.2008.08822.x. [DOI] [PubMed] [Google Scholar]
- Kazantseva A, Goltsov A, Zinchenko R, Grigorenko AP, Abrukova AV, Moliaka YK, et al. Human hair growth deficiency is linked to a genetic defect in the phospholipase gene LIPH. Science. 2006;314:982–5. doi: 10.1126/science.1133276. [DOI] [PubMed] [Google Scholar]
- Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O’Shaughnessy R, Mahoney MG, et al. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell. 2003;113:249–60. doi: 10.1016/s0092-8674(03)00273-3. [DOI] [PubMed] [Google Scholar]
- Langbein L, Schweizer J. Keratins of the human hair follicle. Int Rev Cytol. 2005;243:1–78. doi: 10.1016/S0074-7696(05)43001-6. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Rice RH, Lee YM. Proteome analysis of human hair shaft: From protein identification to posttranslational modification. Molec Cell Proteom. 2006;5:789–800. doi: 10.1074/mcp.M500278-MCP200. [DOI] [PubMed] [Google Scholar]
- Levy-Nissenbaum E, Betz RC, Frydman M, Simon M, Lahat H, Bakhan T, et al. Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat Genet. 2003;34:151–3. doi: 10.1038/ng1163. [DOI] [PubMed] [Google Scholar]
- Maquat LE. Defects in RNA splicing and the consequence of shortened translational reading frames. Am J Hum Genet. 1996;59:279–86. [PMC free article] [PubMed] [Google Scholar]
- McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease) Lancet. 2000;355:2119–24. doi: 10.1016/S0140-6736(00)02379-5. [DOI] [PubMed] [Google Scholar]
- Nahum S, Pasternack SM, Pforr J, Indelman M, Wollnik B, Bergman R, et al. A large duplication in LIPH underlies autosomal recessive hypotrichosis simplex in four Middle Eastern families. Arch Dermatol Res. 2009;301:391–3. doi: 10.1007/s00403-008-0903-9. [DOI] [PubMed] [Google Scholar]
- Norgett EE, Hatsell SJ, Carvajal-Huerta L, Ruiz Cabezas JC, Common J, Purkis P, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9:2761–6. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- Pasternack SM, von Kugelgen I, Al Aboud K, Lee YA, Ruschendorf F, Voss K, et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet. 2008;40:329–34. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
- Salamon T. Über eine familie mit recessiver Kraushaarigkeit, hypotrichose und anderen anomalien. Hautarzt. 1963;14:540–4. [PubMed] [Google Scholar]
- Shimomura Y, Wajid M, Shapiro L, Christiano AM. P-cadherin is a p63 target gene with a crucial role in the developing human limb bud and hair follicle. Development. 2008a;135:743–53. doi: 10.1242/dev.006718. [DOI] [PubMed] [Google Scholar]
- Shimomura Y, Wajid M, Ishii Y, Shapiro L, Petukhova L, Gordon D, et al. Disruption of P2RY5, an orphan G protein-coupled receptor, underlies autosomal recessive woolly hair. Nat Genet. 2008b;40:335–9. doi: 10.1038/ng.100. [DOI] [PubMed] [Google Scholar]
- Shimomura Y, Wajid M, Petukhova L, Shapiro L, Christiano AM. Mutations in the Lipase H (LIPH) gene underlie autosomal recessive woolly hair/hypotrichosis. J Invest Dermatol. 2009;129:622–8. doi: 10.1038/jid.2008.290. [DOI] [PubMed] [Google Scholar]
- Sonoda H, Aoki J, Hiramatsu T, Ishida M, Bandoh K, Nagai Y, et al. A novel phosphatidic acid-selective phospholipase A1 that produces lysophosphatidic acid. J Biol Chem. 2002;277:34254–63. doi: 10.1074/jbc.M201659200. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kamimura A, Hamazono-Matsuoka T, Honda S. Phosphatidic acid has a potential to promote hair growth in vitro and in vivo, and activates mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in hair epithelial cells. J Invest Dermatol. 2003;121:448–56. doi: 10.1046/j.1523-1747.2003.12426.x. [DOI] [PubMed] [Google Scholar]
- Weiss G, Confino Y, Shemer A, Trau H. Cutaneous manifestations in the cardiofaciocutaneous syndrome, a variant of the classical Noonan syndrome. Report of a case and review of the literature. J Eur Acad Dermatol Venereol. 2004;18:324–7. doi: 10.1111/j.1468-3083.2004.00365.x. [DOI] [PubMed] [Google Scholar]
- Zlotogorski A, Panteleyev AA, Aita VM, Christiano AM. Clinical and molecular diagnostic criteria of congenital atrichia with papular lesions. J Invest Dermatol. 2002;118:887–90. doi: 10.1046/j.1523-1747.2001.01767.x. [DOI] [PubMed] [Google Scholar]
- Zlotogorski A, Marek D, Horev L, Abu A, Ben-Amitai D, Gerad L, et al. An autosomal recessive form of monilethrix is caused by mutations in DSG4: clinical overlap with localized autosomal recessive hypotrichosis. J Invest Dermatol. 2006;126:1292–6. doi: 10.1038/sj.jid.5700251. [DOI] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5:2339–47. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The identified proteins were sorted into the categories of keratins and keratin associated proteins (Ker), other cytoplasmic proteins (Cyt), histones (His) and membrane and junctional proteins (Mem).
Illustrated are those proteins identified only in the total hair samples, only in the non-solubilizable (cross-linked) samples, and in both samples (overlap) as indicated. The number in each category is listed at the bottom of the category. Relative amounts of the proteins (unique peptides, emPAI values) are given in Table 1 or Table S1.
The proteins listed were identified in both parental samples (M, mother; F, father) and/or both of the offspring samples (D, daughter; S, son) in Family A with a minimum of two unique peptides per person. In cases of closely related proteins, grouped for parsimony, the number of proteins to which the peptides were matched by Scaffold are given (Matches). The database accession numbers of the matched proteins and the protein molecular weights (MW) in AMU are also given. The unique peptides identified in the total hair shaft and cross-linked fraction samples are listed for the given donors. The exponentially modified Protein Abundance Index (emPAI) values taken from the MASCOT output are listed for the parental samples (M, F) along with the average. For samples where MASCOT did not provide a value (*), it was assumed to be 0. These values were summed for the total hair and for the cross-linked fraction samples, and normalized to 100 to give estimates of the relative molar amounts. The cellular locations (Cell Loc) used in tabulating Figure S1 are also given.